
Abstract
Genome-wide association studies (GWAS) of major depression and its relevant biological phenotypes have been extensively conducted in large samples, and transcriptome-wide analyses in the tissues of brain regions relevant to pathogenesis of depression, e.g., dorsolateral prefrontal cortex (DLPFC), have also been widely performed recently. Integrating these multi-omics data will enable unveiling of depression risk genes and even underlying pathological mechanisms. Here, we employ summary data-based Mendelian randomization (SMR) and integrative risk gene selector (iRIGS) approaches to integrate multi-omics data from GWAS, DLPFC expression quantitative trait loci (eQTL) analyses and enhancer-promoter physical link studies to prioritize high-confidence risk genes for depression, followed by independent replications across distinct populations. These integrative analyses identify multiple high-confidence depression risk genes, and numerous lines of evidence supporting pivotal roles of the netrin 1 receptor (DCC) gene in this illness across different populations. Our subsequent explorative analyses further suggest that DCC significantly predicts neuroticism, well-being spectrum, cognitive function and putamen structure in general populations. Gene expression correlation and pathway analyses in DLPFC further show that DCC potentially participates in the biological processes and pathways underlying synaptic plasticity, axon guidance, circadian entrainment, as well as learning and long-term potentiation. These results are in agreement with the recent findings of this gene in neurodevelopment and psychiatric disorders, and we thus further confirm that DCC is an important susceptibility gene for depression, and might be a potential target for new antidepressants.
Similar content being viewed by others

Introduction
A primary current challenge in the psychiatry field is to dissect the underlying neurobiological basis of common mental illnesses such as major depression, which is said to be one of the ten most disabling conditions in the world1. Given the substantial heritability of major depression (~37%)2, the application of human genetic approaches is believed to both promote the understanding of its biological mechanisms and benefit discovery and development of effective clinical treatment strategies. Indeed, recent genome-wide association studies (GWAS) of depressive subjects and healthy controls have identified multiple statistically robust loci3,4, providing numerous candidates for in-depth exploration of its pathological mechanisms. Notably, there is growing consensus on such “in-depth functional exploration” of psychiatric disease-related loci, and several critical steps have been raised: (1) probe the risk genes from risk locus; (2) depict (novel or refined) disease mechanisms based on the risk genes; (3) assess the druggability of the risk gene product itself or a proximate pathway5,6. This research regimen has been widely applied in recent years, however, the fact that majority of the disease risk loci identified by GWAS reside in the noncoding genomic regions has significantly hampered the accomplishments in elucidating their biological and pathological impacts. Fortunately, accumulating studies have found that noncoding variations of complex diseases tend to be associated with mRNA expression7, and analyzing the expression quantitative trait loci (eQTL) effects of the risk alleles in relevant tissues is therefore a plausible strategy to probe the risk genes from risk locus8,9,10. In line with this, several integrative analyses using GWAS and brain eQTL data have revealed susceptibility genes and potential biological mechanisms for psychiatric disorders11,12,13.
For depression, recent genome-wide linkage disequilibrium (LD) score regression analyses have shown strong genetic correlations between this illness and multiple quantifiable behavioral phenotypes (e.g., emotional traits and cognitive functions)4. In addition, accumulating clinical and basic data derived from depression patients and inbred model mice have demonstrated that depression is associated with multiple levels of abnormalities of brain areas engaged in emotional and cognitive processes (e.g., dorsolateral prefrontal cortex (DLPFC) and hippocampus), including aberrant structure and function as well as neuronal atrophy and synaptic loss14,15,16,17. The observation that some antidepressants likely exert effects via blocking or reversing these aberrations further confirmed that such phenotypes might play pivotal roles in depression18,19. Therefore, translating genetic findings of depression may provide valuable insights into its pathological mechanisms and even facilitate therapeutic development, however, such translational approach requires thorough integrations of the data obtained from multiple perspectives including genomics, neuroscience, pharmacology, and biochemical20.
In the present study, with an aim to identify risk genes and relevant mechanisms of depression, we have employed summary data-based Mendelian randomization (SMR)21,22 and Bayesian integrative risk gene selector (iRIGS)23 to integrate omics data from GWAS, DLPFC eQTL, and genome-scale chromosome conformation capture (Hi-C). Multiple depression risk genes are identified after combined investigation of integrative results, among which the netrin 1 receptor (DCC) gene is prioritized as a high-confidence candidate. Risk alleles correlated with brain DCC mRNA levels show robust link with the onset of depression. We also find that DCC associating variants significantly predict depression relevant biological phenotypes, suggesting participation of this gene in the biological processes of depression pathogenesis. These results further confirm the previous functional analyses of DCC supporting the hypothesis that it is an authentic and important risk gene for depression.
Methods and materials
All the protocols and methods used in this study were approved by the institutional review board of the Kunming Institute of Zoology, Chinese Academy of Sciences.
Depression GWAS data
European GWAS data
Depression GWAS data was retrieved from that Howard et al. meta-analyses of UK Biobank, PGC2, and 23andMe GWAS datasets (a total of 246,363 cases and 561,190 controls)4. There were originally 102 independent loci identified as showing genome-wide significant associations with depression in Europeans4. Due to the restrictions on data share policy of 23andMe sample, the authors publicly deposited the genome-wide statistics combining UK Biobank and PGC2 GWAS datasets (170,756 cases and 329,443 controls) (https://datashare.is.ed.ac.uk/handle/10283/3203), which was utilized for the current analyses. Detailed information of sample characteristics, genotyping method, and statistical analyses of each GWAS dataset can be found in the original studies4.
Chinese GWAS data
Data of the GWAS of major depression in Han Chinese conducted by the CONVERGE Consortium was collected for the current study24. A total of 5303 patients and 5337 non-psychiatric controls after quality control were included. Data was accessed via the public sharing portal at https://doi.org/10.6084/m9.figshare.3840696. Details of the samples, genotyping method and statistical analyses can be found in the initial report24.
SMR integrative analyses
Mendelian randomization (MR) analyses utilize a genetic variation as the instrumental variable to examine causative effects of defined exposure variables (e.g., gene expression) on an outcome (e.g., illness)25. It is thus plausible to use MR analysis to identify the risk or even causal genes of complex illnesses through integrating the eQTL data. However, one potential handicap of MR analyses is that this method requires a large cohort of individuals with simultaneously available data on their phenotype, genotype and gene expression profiles, which is usually difficult to recruit. As a possible solution, Zhu et al. have developed a novel alternative method called summary data-based Mendelian randomization (SMR), which requires summary level statistical data from independent GWAS and eQTL datasets for the integration and prioritization of genes whose expression levels are relevant to the illness due to pleiotropic effects21. Based on SMR, the authors further developed a multi-SNP-based SMR test (--smr-multi) that considers multiple SNPs at a cis-eQTL locus in the SMR test22, and this method is applied in this study. We respectively integrated the brain eQTL datasets from BrainSeq Phase 226, CommonMind27, and PsychENCODE28 with the European depression GWAS4 to perform the SMR analyses. Details of the data and relevant procedure conducted are discussed below.
The BrainSeq Phase 2 is a RiboZero RNA-seq eQTL database of human brain tissues26. Data obtained from the DLPFC tissues of 397 individuals older than 13 were included, and gene-level expression eQTL was calculated based on the formula: log2(RPKM+1) ~SNP + diagnosis + sex + SNP PCs + expression PCs. The authors identified 1,577,964 eQTL associations at a false discovery rate (FDR) ≤ 1% between 945,693 genetic variants and 13,510 genes, and the eQTL summary data were downloaded from https://s3.us-east-2.amazonaws.com/libd-brainseq2/SupplementaryTable15_eQTL.tar.gz, and then transformed into SMR binary (BESD) file using SMR (version 1.02) for subsequent analyses21.
The CommonMind dataset contains polyA+ RNA-seq eQTL data of DLPFC tissues collected from 467 European donors (age > 17 years old)27. The mRNA levels of genes were normalized using log (CPM) (read counts per million per reads) and adjusted for diagnosis, sex, institution, AOD, PMI, RIN, RIN2, clustered LIB, and 20 SVs. The expression levels were then quantified using a linear eQTL analysis on the imputed genotype dosages based on the formula: adjusted gene expression ~SNP + 5 ancestry vectors + diagnosis. This dataset identified 3,725,946 significant cis-eQTL at a FDR ≤ 5% between 16,089 genes and 2,072,425 SNPs. We downloaded the eQTL summary data from Synapse (https://www.synapse.org/#!Synapse:syn5652278) and transformed them into BESD file using SMR (version 1.02) for the integrative analyses21.
A recent study provided the PsychENCODE eQTL summary data at http://resource.psychencode.org/28. Briefly, the authors calculated the eQTL association results after correcting for 100 hidden covariate (HCP) factors28, and the data was generated based on 1387 individuals recruited by the research projects of CommonMind, CommonMind-HBCC, BrainGVEX, LIBD, and BipSeq etc.29. Therefore, although duplicative individuals exist between PsychENCODE and CommonMind and BrainSeq datasets, the PsychENCODE is a larger eQTL dataset with a stronger detection power. The PsychENCODE eQTL summary data in BESD format was directly downloaded from http://resource.psychencode.org/28.
For SMR analysis, firstly, these eQTL summary statistics were transformed to the binary format files named BESD, as the key input files of SMR program. In addition, the other major input file was the genome-wide statistics of depression GWAS. SNPs and genes in the major histocompatibility complex (MHC) extended region were removed before SMR analyses due to the complexity of this genomic region (chr6:26M-34M, hg19). The threshold of eQTL p-value in the SMR analysis was set to be 1.00 × 10−5 (--peqtl-smr 0.00001) and the default values of other parameters were used. The genes with p-value less than 5.00 × 10−4 of multi-SNP-based SMR test and passed the HEIDI test (PHEIDI > 0.005) were considered as susceptibility genes, whose mRNA expression alterations associated with risk SNPs of depression.
Integrative risk gene selector (iRIGS) analyses
The growing knowledge of the importance of physical interactions between distal regulatory elements (DREs) and target promoters has promoted recent development of multiple technologies (e.g., Hi-C) capable of detecting such interactions30,31. Through integrating results of studies applying these approaches, Wang et al.23 developed a Bayesian framework, named iRIGS, to probabilistically rank high-confidence risk genes at each GWAS locus of schizophrenia. Following their method, we herein focused on the 102 independent lead risk SNPs from previous European depression GWAS4, and used the iRIGS analyses to estimate whether these index SNPs interacted with promoters of particular genes based on the omics data of short-range and long-range enhancer-promoter links. These omics data were derived from four sources in three independent published studies as previously described23. The first study is the Hi-C analyses of the cortical/subcortical plate and the germinal zone of human cerebral cortex conducted by Won et al.31. This study reported 221,069 and 228,323 DRE-promoter links in the cortical/subcortical plate and the germinal zone, respectively. The second study is a capture Hi-C analysis of the cell line GM12878, a human Epstein-Barr virus (EBV)-transformed lymphoblastoid cell line30 and identified 1,618,000 DRE-promoter links. The third study is conducted by the FANTOM5 project to infer the enhancer-promoter links across multiple human tissues, and eventually obtained 66,899 enhancer-promoter links32. The data of these studies were placed at https://www.vumc.org/cgg/irigs in a ready-to-use format for iRIGS analyses23. The R code of iRIGS were run using the default pipeline. The Bayesian posterior probability of observing the link between a SNP and a particular gene higher than 0.8 was considered to be strong, in which case the gene is a potential risk gene regulated by the SNP.
Gene-wise MAGMA analyses
The gene-wise p-values were respectively calculated using MAGMA (v1.07b)33 based on the depression GWAS statistics from Europeans and Han Chinese4,24. MAGMA applies Brown’s method to combine SNP p-values which will consider LD, and the window size of each gene is defined as the region spanning 35-kb upstream and 10-kb downstream of this gene as previously described3. The snp-wise = mean gene analysis model was used in the present study, which tests the mean SNP association for each gene. For the LD reference, we utilized European-ancestry and Han Chinese-ancestry individuals from the 1000 Genomes Project (Phase 3)34.
LD analyses
The Haploview program (version 4.1)35 was utilized to estimate LD between paired SNPs using the r2 algorithm, and to determine the haplotype blocks based on the SNP data from the 1000 Genomes Project34. The regional association results are plotted using LocusZoom (http://locuszoom.sph.umich.edu/locuszoom/)36.
GWAS of depressive symptoms, neuroticism, life satisfaction, positive affect, and well-being spectrum
Phenotypes such as neuroticism, life satisfaction, and positive affect are generally believed to be associated with depression. Specifically, neuroticism refers to a personality trait characterized by significantly instable mood in response to stress-inducing events37, and is presumed to be a risk factor for depression38. Phenotypes of the well-being spectrum, including depressive symptoms, neuroticism, life satisfaction, and positive affect, have all been found genetically correlated with depression38. We therefore collected GWAS resources of these biological phenotypes from a recent study39, in which the authors measured the well-being spectrum using survey questions on depressive symptoms, neuroticism, life satisfaction, and positive affect, and applied multivariate genome-wide-association meta-analysis (GWAMA) on univariate GWAMAs of depressive symptoms (N = 1,295,946), neuroticism (N = 582,989), life satisfaction (N = 80,852), and positive affect (N = 410,603), as well as the combinatory well-being spectrum (N = 2,370,390).
GWAS of cognitive performance and intelligence
The data of cognitive performance were retrieved from a recent GWAS of 257,828 individuals40. In their study, the authors conducted a meta-analysis of the general cognitive ability GWAS by the COGENT consortium41 and additional results of the recent cognitive performance analyses in UK Biobank42. Meanwhile, the authors also conducted GWAS analysis of educational attainment (N = 766,345)40, a proxy phenotype of cognitive abilities that is believed to also reflect some personality traits. In addition, we also retrieved the results of intelligence from a GWAS of 269,867 individuals into the current study43. The authors calculated and applied a common latent g factor underlying multiple dimensions of cognitive functioning during statistical analyses given the distinct approaches of intelligence measurement in each sample.
GWAS of brain imaging phenotypes
Statistics of brain imaging analyses were extracted from a recent GWAS of 3144 functional and structural brain imaging phenotypes (e.g., hippocampal volume, putamen volume, task functional MRI median BOLD faces) in 8,428 subjects by the UK Biobank (accessed at http://big.stats.ox.ac.uk/)44. In this website, GWAS results of other related phenotypes were also deposited (i.e., GWAS of a total of 3999 UK Biobank brain imaging phenotypes and other traits). We also collected the independent data from the ENIGMA Consortium GWAS of different subcortical brain volumes (accumbens, amygdala, caudate, hippocampus, pallidum, putamen, and thalamus)45. According to the previous study44, there was a strong correlation between the UK Biobank and ENIGMA imaging samples, suggesting that methodologies applied in the measurement and statistical analyses of these phenotypes were relatively consistent. In addition, a more recent GWAS has been conducted to meta-analyze imaging data from CHARGE, ENIGMA and UK Biobank, resulting in a total of 38,851 subjects46.
RNA-seq, mRNA correlation, and pathway analyses in human brains
We downloaded the aligned (hg19 as reference genome) RNA-seq data (bam files) of the DLPFC tissue from three independent sample pools (BrainGVEX, CommonMind, and LIBD)27,29, and only non-psychiatric controls were utilized to prevent effects of confounders relevant to medical treatment. Based on this criterion, 59 controls from BrainGVEX, 50 controls from CommonMind, and 70 controls from LIBD were included. We applied the same procedures and criteria for the quality control across the three RNA-seq datasets as previously described47,48. The counts of aligned reads at the gene level were calculated using featureCounts according to the annotation file of GRCh37p1349. We calculated the transcripts per million reads (TPM) of each gene using R program following a previous study28, and only kept the protein-coding genes with their average TPM ≥ 1.0 for the following analyses. The TPM of each gene was log2 transformed followed by Pearson analyses to assess their correlations with DCC. We used clusterProfiler50 to analyze whether the “DCC-correlated” genes were significantly enriched in specific molecular pathways and biological processes via Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) annotations based on the integrative database called org.Hs.eg.db, and FDR q-value less than 0.1 was set to be reliable. We then performed semantic similarity analyses with GOSemSim51 to narrow down those GO terms based on their similarity between each other (r > 0.5 was considered highly similar).
Results
Integrative analyses of multi-omics data identified high-confidence risk genes for depression in Europeans
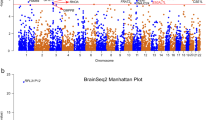
We applied multi-SNP-based SMR method to test the associations between risk SNPs of depression identified in the GWAS study4 and mRNA expression based on two independent DLPFC eQTL datasets from BrainSeq Phase 2 (N = 397)26 and CommonMind (N = 467)27, respectively. Subsequently, we performed additional analyses to confirm these findings using a larger eQTL dataset from PsychENCODE29, which consisted of more individuals (N = 1387) that were also partly overlapped with those in CommonMind and BrainSeq. The number of genes included in the SMR analysis were 5502 (BrainSeq Phase 2), 6036 (CommonMind) and 13,567 (PsychENCODE), respectively. The remaining genes were 5,455, 5,981 and 13,372 after ignoring the genes failing to pass the HEIDI test and 3239 genes were overlapped in all datasets. Finally, 16, 18, and 30 susceptibility genes (PSMR-multi ≤ 5.00 × 10−4) were identified in the three eQTL datasets, respectively. As expected, SMR analyses using this larger eQTL samples replicated most of the risk genes identified in the earlier datasets, and a total of 10 risk genes exhibited statistical significance throughout all the SMR analyses (Fig. 1; Table 1).

Ten genes of PSMR-multi less than 5.00 × 10−4 in all three eQTL datasets and sixteen genes of posterior probability more than 0.8 in iRIGS were marked in red.
We then conducted iRIGS analyses to identify potential risk genes through evaluating the physical interactions between enhancers and promoters. Briefly, whether the 102 independent lead risk SNPs from previous European depression GWAS were involved in such interactions were examined4. SNPs and genes in the MHC extended region were removed before iRIGS analyses, and the enhancer-promoter links between 1229 genes and these depression risk SNPs were then captured. Our results revealed that the posterior probabilities of 16 genes were higher than 0.8, indicating that they are potentially high-confidence depression risk genes under the influences of the risk SNPs (Fig. 1 and Table S1).
Through the SMR and iRIGS analyses, two genes were identified in both approaches (marked in bold and underlined in Table 1). The first gene, LRFN5 at 14q23.1, showed significant associations in SMR (PSMR-multi = 4.05 × 10−6 in BrainSeq Phase 2, PSMR-multi = 1.96 × 10−4 in CommonMind, PSMR-multi = 4.11 × 10−4 in PsychENCODE) and iRIGS analyses (posterior probability = 1.000). This gene encodes a protein of leucine rich repeat and fibronectin type III domain containing 5, and previous studies implicated its crucial roles in synapse formation and differentiation52,53,54,55,56.
Another high-confidence risk gene is DCC at 18q21.3 which encodes a transmembrane receptor for the protein netrin-1. DCC exhibited significant association signals in both SMR (PSMR-multi = 6.58 × 10−5 in BrainSeq Phase 2, PSMR-multi = 1.27 × 10−4 in CommonMind, PSMR-multi = 2.78 × 10−5 in PsychENCODE) and iRIGS analyses (posterior probability = 0.828). DCC has been extensively described in a recent review regarding its potential roles in the central nervous systems and psychiatric disorders57, in brief, this gene is highly expressed in dendritic spines of pyramidal neurons, and exerts pivotal regulatory effects on synaptic function and plasticity in adult brain58. The Dcc protein is known to be a cell adhesion molecule that mediates the effects of netrin-1 on axon outgrowth59,60,61, and deficits of Dcc in the adult forebrain neurons results in aberrant long-term potentiation (LTP), long-term depression (LTD) and dendritic spine morphology, as well as impaired spatial and recognition57,58. Intriguingly, recent studies have shown that DCC mRNA levels in the prefrontal cortex of antidepressant-free depression patients who committed suicide were significantly elevated comparing with control subjects (i.e., DCC mRNA levels were 48% higher in cases compared to control subjects in the first cohort by Manitt et al. (p < 0.026)62, and were ~40% higher in cases in comparison with controls in the second independent cohort by Torres-Berrío et al. (p = 0.02)63). Moreover, in a blood transcriptome analysis, DCC was again significantly upregulated in depression patients compared to controls in two independent samples (GSK-HiTDIP cohort, fold change = 1.085, p = 0.0231; Janssen-BRC cohort, fold change = 1.137, p = 0.000655)64. In addition, in the mPFC of mice exhibiting chronic social defeat stress (CSDS) exposure-induced depressive-like symptoms, expression level of Dcc is also increased63; on the other hand, decreased Dcc expression in the mouse PFC pyramidal neurons produces resilience against stress-induced depression-like phenotypes63.
Independent replications across populations further confirmed the association of DCC with depression
Our analyses so far suggest pivotal roles of the SNPs spanning LRFN5 and DCC in the risk of depression in Europeans4. We then performed the gene-level analyses via MAGMA33, which primarily consider LD structures. Intriguingly, the significant associations between these two genes with the risk of depression were again observed in Europeans (PMAGMA = 1.01 × 10−11 for LRFN5, PMAGMA = 2.85 × 10−12 for DCC, Table 1). Given the emerging evidence supporting the notion that vital genetic markers for psychiatric disorders are normally associated with the disease across different ethnic populations65 (e.g., psychiatric risk loci in ZNF804A, FADS1, and VRK2 show significant associations in both Europeans and East Asians66,67,68,69,70,71), we then examined whether LRFN5 and DCC were also associated with depression in Han Chinese subjects through MAGMA gene-level analyses using a published Han Chinese GWAS dataset (5303 cases and 5337 controls)24. Notably, DCC was associated with depression as well in Han Chinese despite the lower level of statistical significance compared with that in Europeans (PMAGMA = 3.21 × 10−4, Table 1), but LRFN5 was not associated with depression in Han Chinese (PMAGMA = 0.406, Table 1). A detailed examination found that none of the SNPs spanning LRFN5 were significantly associated with depression Han Chinese (all p > 0.01, Fig. S1).
Identification of SNPs in DCC showing significant associations with depression in both populations
It is generally acknowledged that potential causal variants usually exhibit consistent associations with the illnesses across populations, whereas the associations for other linked variants may be significantly affected by the different LD structures between populations72,73. To further explore the genetic architecture of complex illnesses, cross-population replication analysis is usually necessary. In this study, our detailed examinations found that although multiple SNPs spanning DCC showed genome-wide significant associations with depression in Europeans, their associations with the illness in Han Chinese were distinct possibly due to variations in LD (Fig. 2). For example, rs7227069 showed genome-wide significant association with depression in 170,756 European cases and 329,443 matching controls collected by UK Biobank and PGC2 (rs7227069, Pdepression = 4.64 × 10−9, odds ratio (OR) = 1.026 for A-allele, Fig. 2), and when the 23andMe GWAS dataset was also added, the association between rs7227069 and depression was strengthened in the new sample pool of 246,363 cases and 561,190 controls (Pdepression = 1.50 × 10−11, OR = 1.024 for A-allele)4. The risk association was further confirmed in an independent cohort of 414,055 cases and 892,299 controls (Pdepression = 8.89 × 10−19, OR = 1.025 for A-allele)4. While in Han Chinese subjects, rs7227069 was only marginally associated with depression (Pdepression = 0.071, OR = 1.127 for A-allele in 5303 cases and 5337 controls, Fig. 2)24. By comparing the allele distributions of rs7227069 in Europeans and Chinese, we found that the frequency of A-allele at rs7227069 was 0.425 in Europeans, but it is not a common SNP in Han Chinese (frequency of A-allele is 0.038), which likely contributed to the different levels of association significance of this SNP with depression between populations.

A physical map of the region is given and depicts known genes within the region, and the LD is defined based on the SNP rs1367635. The LD between rs7227069 and rs1367635 in both populations are also shown.
Notably, we found another SNP rs1367635 in DCC, which was genome-wide significantly associated with depression in Europeans (Pdepression = 4.35 × 10−9, OR = 1.026 for C-allele in 170,756 cases and 329,443 controls, Fig. 2), was also highly associated with this illness in Han Chinese (Pdepression = 1.21 × 10−5, OR = 1.173 for C-allele in 5303 cases and 5337 controls, Fig. 2). Rs1367635 and rs7227069 lie more than 129.0-kb apart in different introns of the DCC gene. They are in moderate LD in Europeans (r2 = 0.61), while low LD in Han Chinese (r2 = 0.14), highlighting the genetic heterogeneity in this locus between populations. Rs1367635 is a common SNP in both Europeans and Han Chinese despite different allelic frequencies between populations (C-allele, 0.501 in Europeans versus 0.183 in Han Chinese), and rs1367635 exhibited a stronger association signal with depression in Han Chinese than rs7227069 probably due to its higher minor allele frequency (MAF) in this population. Although the relatively smaller sample size of the current depression GWAS of Han Chinese (5303 cases and 5337 controls)24 has limited the level of statistical significance of the association signal of rs1367635 with depression, the effect size of rs1367635 on the risk of depression is comparable to other risk loci identified in genome-wide analyses3. We also performed a power analysis of rs1367635 according to its effect size on the risk of depression in Han Chinese and its allelic frequency in this population. This analysis estimated that at least 39,628 cases and controls in total were needed to reach the 80% power of detecting genome-wide significant association. Further analyses of the association between rs1367635 and depression in a large enough cohort will likely reveal a genome-wide level significant association.
The depression risk alleles indicated higher DCC mRNA level in DLPFC
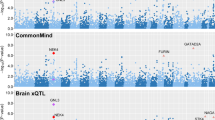
We examined whether the depression risk SNPs (e.g., rs7227069 and rs1367635) spanning DCC were also associated with DCC mRNA expression in DLPFC. Rs7227069 showed significant association with mRNA expression of DCC in BrainSeq Phase 2 eQTL dataset, which included the RiboZero RNA-seq results of DLPFC tissues from 397 individuals (PeQTL = 5.36 × 10−5, Fig. 3); in the CommonMind polyA+ RNA-seq eQTL dataset of 467 individuals, rs7227069 again exhibited significant association with DCC mRNA levels (PeQTL = 4.41 × 10−6, Fig. 3); while rs7227069 was not covered in the PsychENCODE dataset of 1,387 subjects, its complete LD SNP rs8086812 (r2 = 1.000 in Europeans) was significantly associated with DCC mRNA expression (PeQTL = 7.05 × 10−15, eQTL plot in PsychENCODE dataset was not available since we do not have access to individual-level genotype and expression data).

In the CommonMind eQTL dataset, rs1367635 was not directly genotyped or imputed, and we therefore used its high LD SNP rs4940252 as a proxy readout (r2 = 0.880 between rs1367635-T/C and rs4940252-A/G in Europeans).
Rs1367635 also showed significant association with mRNA expression of DCC in BrainSeq Phase 2 and PsychENCODE eQTL datasets (PeQTL = 5.86 × 10−6 in BrainSeq Phase 2, PeQTL = 1.76 × 10−8 in PsychENCODE; Fig. 3). In the CommonMind eQTL dataset, rs1367635 was not genotyped or imputed, and we therefore used its high LD SNP rs4940252 as a proxy readout (r2 = 0.880 in Europeans). Again, rs4940252 was significantly associated with DCC mRNA expression in the CommonMind dataset (PeQTL = 6.82 × 10−3, Fig. 3). Therefore, the depression risk alleles at rs7227069 and rs1367635 consistently indicated higher DCC expression in the three selected eQTL datasets. This result is consistent with the published diagnostic analyses of DCC mRNA level in humans and rodent models62,63, suggesting that increased expression of DCC in the brain likely serves as a risk factor of depression.
We also explored the expression pattern of DCC in diverse human tissues using GTEx (Genotype-Tissue Expression project; https://www.gtexportal.org/) dataset74, and found that DCC was preferably expressed in frontal cortex, caudate, nucleus accumbens, putamen and hippocampus compared with most other peripheral tissues (only except testis) (Fig. S2). Further spatio-temporal expression pattern analyses using data of the developing prefrontal cortex from the BrainCloud dataset (http://braincloud.jhmi.edu/)75 showed that the mRNA expression levels of DCC were higher at early developmental stages (i.e., fetal stages) compared with the later childhood and adulthood stages (Fig. S3). We then performed the same analyses in data of multiple human brain tissues (e.g., dorsolateral prefrontal cortex, hippocampus, cerebellar cortex) from the BrainSpan dataset (http://www.brainspan.org/static/home)76. Again, we observed that the mRNA levels of DCC were higher in prenatal brain tissues than in postnatal brain tissues. Notably, the expression of DCC was the highest during early or mid prenatal stages (9–13 post-conceptional weeks (pcw)) (Fig. S4), at which the developing brain starts to undergo striking changes such as the formation of gyri and sulci77. This result supports the putative molecular origins of psychiatric disorders arise from the early developmental events78,79. Given the high expression levels of DCC in prenatal brains, using eQTL datasets including samples from only postnatal donors (e.g., CMC samples > 17 years old; BrainSeq2 samples > 13 years old) may not fully reveal eQTL association signals involved in psychiatric disorders. We therefore also explored the eQTL associations of risk SNPs with DCC mRNA expression in fetal brains using recently published data in European populations80,81. Briefly, data of 120 prenatal human brain samples (second trimester of gestation) included in the Human Developmental Biology Resource (http://www.hdbr.org)80 was retrieved and analyzed. The risk A-allele at rs7227069 again predicted higher DCC expression (p = 0.01) (Table S2). There were 427 additional SNPs significantly associated with the expression of DCC (p < 0.05), and many of these SNPs also showed genome-wide significant associations with depression (Table S2). However, rs1367635 was not associated with DCC expression in this dataset. In another 201 prenatal human brain samples (mid-gestational human brains) from the UCLA Gene and Cell Therapy core81, neither rs7227069 nor rs1367635 showed significant association with DCC expression, but there were other SNPs significantly associated with both DCC expression and risk of depression (Table S3). Overall, these results support the hypothesis that certain genetic variations influencing DCC expression in human brains also affect risk of depression.
DCC was associated with well-being spectrum, cognitive function, and putamen structure
Given that several biological and psychological phenotypes such as mood instability, depressive symptoms and aberrant cognitive functions have been common observed in depression patients38,82, we hypothesized that these phenotypes were also associated with the depression risk alleles at DCC. We utilized the published GWAS resources and found that DCC SNPs showed significant associations with depressive symptoms (rs7227069, p = 6.18 × 10−16; rs1367635, p = 8.57 × 10−15), and were also highly associated with neuroticism (rs7227069, p = 4.73 × 10−16; rs1367635, p = 8.23 × 10−15), well-being spectrum (rs7227069, p = 2.12 × 10−16; rs1367635, p = 8.58 × 10−15), life satisfaction (rs7227069, p = 4.92 × 10−11; rs1367635, p = 1.91 × 10−11) and positive affect (rs7227069, p = 4.36 × 10−15; rs1367635, p = 5.25 × 10−13)39, among which the depression risk allele carriers tended to show increased vulnerable personality traits compared with the protective allele carriers. We also found that rs7227069 and rs1367635 showed strong associations with cognitive performance (rs7227069, p = 2.15 × 10−6; rs1367635, p = 1.48 × 10−6)40, educational attainment (rs7227069, p = 5.95 × 10−13; rs1367635, p = 1.10 × 10−9)40, and intelligence (rs7227069, p = 1.34 × 10−6; rs1367635, p = 2.58 × 10−8)43, while carriers of the depression risk alleles tended to exhibit worse cognitive abilities compared with the non-risk allele carriers.
We then took one step further to delve into potential neural mechanisms underlying this risk gene. In a phewas analysis including GWAS of 3999 UK Biobank brain imaging phenotypes and other traits (http://big.stats.ox.ac.uk/), rs7227069 and rs1367635 were again primarily associated with depression related traits (such as mood swings and frequency of depressed mood in last 2 weeks, Fig. S5)44, further highlighting the involvement of DCC in this illness. More intriguingly, in this explorative analysis, we found that DCC was suggestively genome-wide significantly associated with putamen volume in the UK Biobank dataset (N = 8428 individuals; rs7227069, p = 9.90 × 10−7; rs1367635, p = 8.90 × 10−6; Fig. S5)44. We then tried to replicate this observation in an independent GWAS of brain imaging phenotypes from ENIGMA consortium45, and again observed suggestive genome-wide association between DCC and putamen volume in the same direction of allelic effects (N = 13,145 subjects; rs7227069, p = 3.32 × 10−7; rs1367635, p = 6.64 × 10−7)45. The imaging samples from UK Biobank and ENIGMA were totally independent. More intriguingly, in a latest meta-analysis of imaging data from CHARGE, ENIGMA and UK Biobank (N = 38,851), both SNPs showed genome-wide significant associations with putamen volume (rs7227069, p = 4.64 × 10−10; rs1367635, p = 8.72 × 10−10)46.
DCC participated in the pathways and biological processes relevant to depression pathogenesis
Previous neurological studies using cellular and molecular technologies have suggested that DCC might be involved in the regulation of synaptic plasticity and relevant brain functions58,59,83,84. Given the importance of these biological processes in cognition and emotion, we sought to further explore the possibility that DCC participates in depression via affecting these processes using population-level expression data. We hypothesized that genes significantly correlated with the mRNA expression of DCC should belong to the molecular pathways and biological processes in which DCC was involved. To test this hypothesis, the global mRNA expression data in three independent human brain RNA-seq datasets (i.e., BrainGVEX, CommonMind, and LIBD)27,29 was retrieved and the Pearson analysis was conducted. We ranked the top 800 genes (~top 5% among all protein-coding genes) in each dataset according to the corresponding significance levels of correlations between their expression and DCC mRNA levels. These top 800 genes from all three datasets then underwent an overlapping analysis, which yielded 145 genes consistently showing high correlations with DCC expression. There genes were significantly enriched in the pathways and biological processes related to synaptic plasticity, axon guidance, circadian entrainment, learning, and long-term potentiation (Fig. 4). Therefore, our analyses using population-level expression data also supports the putative roles of DCC in synapses and brain functions, providing hints for the molecular mechanisms explaining the participation of DCC in depression pathogenesis.

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) biological processes enrichment analyses of DCC correlated genes in the human DLPFC brain tissues.
Discussion
Great efforts have been spent to elucidate genetic and biological basis of depression in the past few decades85,86, however, the high prevalence, great phenotypic heterogeneity, and limited penetrance of known genetic risk markers of this illness have together significantly hampered success in this field. Nevertheless, multiple breakthroughs have been made through genetic studies of depression since the application of recent large GWAS, and integration of the massive data to dissect mechanisms underlying depression pathogenesis has been widely called on. In the present study, we utilize data of depression GWAS, and conduct the genome-wide integrative analyses through combining multiple brain eQTL and Hi-C datasets, followed by independent replications across populations and explorative analyses of relevant biological phenotypes. Through this stepwise analysis, we find that the gene DCC confers risk of depression in both Europeans and Han Chinese. Besides, the risk alleles predict higher DCC mRNA expression in the DLPFC, which is also proven to affect depression-relevant personality traits, cognitive function and putamen volumes in independent samples. The current study reveals that the depression risk alleles indicate larger putamen volume. Putamen is a brain region known to influence motor behaviors and learning abilities, and has also been reported to involve in the “hate circuit”87 that was lately proven vital in depression88. Although previous studies did not identify significant putamen volume differences between depression patients and healthy controls89, its involvement in this illness is appealing for further analyses with larger samples of different ethnic backgrounds to gain insights into the sophisticated mechanisms of depression.
Notably, a recent meta-analysis of 232,964 cases and 494,162 controls across eight psychiatric illnesses (anorexia nervosa, attention-deficit/hyperactivity disorder, autism spectrum disorder, bipolar disorder, major depression, obsessive-compulsive disorder, schizophrenia, and Tourette syndrome) detected 109 loci associated with at least two of them. Intriguingly, the DCC genomic region featured the most pleiotropic association (Pmeta = 4.26 × 10−12) in this meta-analysis, exhibited significant associations with all eight diseases90. Additionally, previous studies have also highlighted DCC in mood instability and the risk of depression57,91,92,93 as well as schizophrenia94, and multiple in vitro and murine analyses have proven its potential impact on synaptic function. For example, the protein netrin-1 and its receptor (encoded by Dcc) are widely expressed in cortical neurons during synapse formation, with significant enrichment at synapses83. Another study showing enrichment of Dcc in dendritic spines of pyramidal neurons further supports its involvement in synaptic physiology, and selective deletion of Dcc leads to loss of LTP, intact LTD, reduced spine length, and impaired spatial and recognition memory in mice58,95. The Dcc deficient/haploinsufficient mice study found that alterations in dcc expression resulted in selective alternations in dopaminergic function (e.g., exaggerated mPFC dopamine concentrations), differences in dopaminergic related behaviors during adulthood, and blunted behavioral responses to amphetamine96,97,98,99,100. From the neurodevelopmental perspective, Dcc likely controls the growth of dopamine axon targeting in adolescence, and thereby affects the development and function of prefrontal cortex84,101,102, resulting in aberrant cognitive processes found in depression. In agreement with this hypothesis, a previous study found that the DCC haploinsufficient adult Quebecers showed similar phenotypic features with adult Dcc haploinsufficient mice103.
Earlier studies using postmortem brain tissues have revealed neuronal loss, reduced synapse density and abnormal expression of synaptic markers in the DLPFC of depression patients14,15, and in vitro and in vivo studies also found that risk factors of depression (e.g., stress and genetic effects) usually result in disruption of synaptic morphology and function as well as brain circuits that are essential for mood regulation and cognition104,105, and thereby eventually lead to the onset of depression. Moreover, both established antidepressants used in clinical settings (e.g., serotonin-reuptake inhibitors) and molecules recently found to alleviate depressive symptoms (e.g., ketamine) confer protective effects on synaptic deficits related to depression18,19. Therefore, it is widely accepted that synaptic dysfunctions play determinant roles in the pathogenesis of depression16,19. Recent depression GWAS have also supported this contention, as genes involved in synaptic structure and neurotransmission related pathways have been repeatedly highlighted4. DCC is also an example of such genes, and further functional studies are urgently needed to gain mechanistic insights into whether and how it affects synaptic functions, brain circuits and behaviors in a disease-specific manner. Whether this gene or its proximate signaling pathway might serve as potential therapeutic targets should also be analyzed.
From the Fig. 2, we can see that sequence variations spanning TCF4 gene were also genome-wide significantly associated with depression in European populations4, and the magnitude is even stronger than those spanning DCC. However, TCF4 was not associated with depression in Han Chinese, and the current study therefore did not include this gene in the subsequent analyses. Nevertheless, the potential importance of this gene in depression or other relevant illnesses/phenotypes should not be denied. In fact, TCF4 has gained considerable attention from researchers due to its significant associations with depression, schizophrenia, cognitive processing106, and Pitt-Hopkins Syndrome107. In a recent schizophrenia GWAS108, numerous SNPs spanning TCF4 showed genome-wide significance, and the involvement of this gene in synaptic plasticity109,110, CNS development111 and neuronal activity regulatory network112,113 has been defined by functional analyses. We also sought to identify whether the association of DCC with depression in Europeans were a reflection of its potential LD with TCF4, and examined the distance and LD among SNPs spanning the two genes. We found that DCC and TCF4 were ~2.0 Mb away from each other on the genome, the risk signals of these two genes were independent in Europeans according to the LD structure (Fig. 2). Therefore, SNPs in DCC are likely genetic risk markers of depression independent of those in TCF4.
Despite the above implications brought by this study, there are limitations to be interpreted. First, our study mainly focuses on variants affecting expression of particular genes, but a number of the genome-wide risk alleles for depression were not associated with expression of any genes in our analyses. While the principal purpose of our study is to prioritize high-confidence risk genes through integrative stepwise analyses, the possibility that the variants not highlighted in eQTL analyses also contribute to the progression of depression should not be denied. In fact, the characteristics of the eQTL datasets utilized in the current study might have “twisted our perceptions” of these variants. For example, the eQTL data we utilized was obtained from DLPFC homogenates rather than from specific types of cells, in the event that some variants and genes exert function in particular types of cells, the current datasets would be inappropriate for detecting such signals. Besides, the individuals in the eQTL datasets were postnatal in a wide range of different ages, making it an ideal source for exploring genes functioning throughout the lifespan but not those participate in depression in a spatio-temporal specific manner. In fact, previous studies have shown that the some psychiatric risk eQTLs are detectable only in a specific stage of life81,114. Further comprehensive functional annotations of other depression risk alleles are necessary to gain a better knowledge of the genetic risk architecture of depression. In addition, despite we found that multiple risk SNPs were significantly associated with mRNA expression of DCC, all of them were in the intron regions. Although we attempted to make functional predictions of the risk SNPs using HaploReg v4.1115 and GWAVA116 through assessing whether they reside in the binding regions of H3K4me1, H3K4me3, H3K9ac, H3K27ac, or transcription factors, we were unable to identify any SNPs showing potential regulatory functions. Therefore, a more complicated regulatory mechanism might underlie the association between the risk SNPs and DCC mRNA expression. One possibility might be that the risk SNPs reflect a causal structural variation beyond the detection scope of the current GWAS approach. While the importance of DCC in depression should be acknowledged, further studies revealing the causal variant in this genomic region are needed.
References
Kupfer, D. J., Frank, E. & Phillips, M. L. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet 379, 1045–1055 (2012).
Sullivan, P. F., Neale, M. C. & Kendler, K. S. Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiatry 157, 1552–1562 (2000).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Wendland, J. R. & Ehlers, M. D. Translating neurogenomics into new medicines. Biol. Psychiatry 79, 650–656 (2016).
Gandal, M. J., Leppa, V., Won, H., Parikshak, N. N. & Geschwind, D. H. The road to precision psychiatry: translating genetics into disease mechanisms. Nat. Neurosci. 19, 1397–1407 (2016).
Edwards, S. L., Beesley, J., French, J. D. & Dunning, A. M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797 (2013).
Li, M. et al. A human-specific AS3MT isoform and BORCS7 are molecular risk factors in the 10q24.32 schizophrenia-associated locus. Nat. Med. 22, 649–656 (2016).
Liu, W. et al. The depression GWAS risk allele predicts smaller cerebellar gray matter volume and reduced SIRT1 mRNA expression in Chinese population. Transl. Psychiatry 9, 333 (2019).
Yang, Z. et al. Identification of a functional 339-bp Alu polymorphism in the schizophrenia-associated locus at 10q24.32. Zool. Res. 41, 84–89 (2020).
Wu, Y. et al. Identification of the primate-specific gene BTN3A2 as an additional schizophrenia risk gene in the MHC loci. EBioMedicine 44, 530–541 (2019).
Yang, C. P. et al. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat. Commun. 9, 838 (2018).
Yang, Z. et al. The genome-wide risk alleles for psychiatric disorders at 3p21.1 show convergent effects on mRNA expression, cognitive function and mushroom dendritic spine. Mol. Psychiatry 25, 48–66 (2020).
Kang, H. J. et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413–1417 (2012).
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E. & Woolfrey, K. M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 (2011).
Forrest, M. P., Parnell, E. & Penzes, P. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 19, 215–234 (2018).
Russo, S. J. & Nestler, E. J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625 (2013).
Duman, R. S., Aghajanian, G. K., Sanacora, G. & Krystal, J. H. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249 (2016).
Duman, R. S. & Aghajanian, G. K. Synaptic dysfunction in depression: potential therapeutic targets. Science 338, 68–72 (2012).
Niculescu, A. B. & Le-Niculescu, H. Convergent functional genomics: what we have learned and can learn about genes, pathways, and mechanisms. Neuropsychopharmacology 35, 355–356 (2010).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Wang, Q. et al. A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data. Nat. Neurosci. 22, 691–699 (2019).
Converge consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591 (2015).
Smith, G. D. & Ebrahim, S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 32, 1–22 (2003).
Collado-Torres, L. et al. Regional heterogeneity in gene expression, regulation, and coherence in the frontal cortex and hippocampus across development and Schizophrenia. Neuron 103, 203–216 (2019).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, 6420 (2018).
PsychEncode Consortium et al. The PsychENCODE project. Nat. Neurosci. 18, 1707–1712 (2015).
Mifsud, B. et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606 (2015).
Won, H. et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Lahey, B. B. Public health significance of neuroticism. Am. Psychol. 64, 241–256 (2009).
Kendler, K. S., Gatz, M., Gardner, C. O. & Pedersen, N. L. Personality and major depression: a Swedish longitudinal, population-based twin study. Arch. Gen. Psychiatry 63, 1113–1120 (2006).
Baselmans, B. M. L. et al. Multivariate genome-wide analyses of the well-being spectrum. Nat. Genet. 51, 445–451 (2019).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Trampush, J. W. et al. GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol. Psychiatry 22, 336–345 (2017).
Davies, G. et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun. 9, 2098 (2018).
Savage, J. E. et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 50, 912–919 (2018).
Elliott, L. T. et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216 (2018).
Hibar, D. P. et al. Common genetic variants influence human subcortical brain structures. Nature 520, 224–229 (2015).
Satizabal, C. L. et al. Genetic architecture of subcortical brain structures in 38,851 individuals. Nat. Genet. 51, 1624–1636 (2019).
Li, H. et al. Integrative analyses of major histocompatibility complex loci in the genome-wide association studies of major depressive disorder. Neuropsychopharmacology 44, 1552–1561 (2019).
Li, H. et al. Interactome analyses implicated CAMK2A in the genetic predisposition and pharmacological mechanism of bipolar disorder. J. Psychiatr. Res. 115, 165–175 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Yu, G. et al. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978 (2010).
Lin, Z., Liu, J., Ding, H., Xu, F. & Liu, H. Structural basis of SALM5-induced PTPdelta dimerization for synaptic differentiation. Nat. Commun. 9, 268 (2018).
Mah, W. et al. Selected SALM (synaptic adhesion-like molecule) family proteins regulate synapse formation. J. Neurosci. 30, 5559–5568 (2010).
Zhu, Y. et al. Neuron-specific SALM5 limits inflammation in the CNS via its interaction with HVEM. Sci. Adv. 2, e1500637 (2016).
Goto-Ito, S. et al. Structural basis of trans-synaptic interactions between PTPdelta and SALMs for inducing synapse formation. Nat. Commun. 9, 269 (2018).
Choi, Y. et al. SALM5 trans-synaptically interacts with LAR-RPTPs in a splicing-dependent manner to regulate synapse development. Sci. Rep. 6, 26676 (2016).
Vosberg, D. E., Leyton, M. & Flores, C. The Netrin-1/DCC guidance system: dopamine pathway maturation and psychiatric disorders emerging in adolescence. Mol. Psychiatry 25, 297–307 (2020).
Horn, K. E. et al. DCC expression by neurons regulates synaptic plasticity in the adult brain. Cell Rep. 3, 173–185 (2013).
Keino-Masu, K. et al. Deleted in colorectal cancer (DCC) encodes a netrin receptor. Cell 87, 175–185 (1996).
Brankatschk, M. & Dickson, B. J. Netrins guide Drosophila commissural axons at short range. Nat. Neurosci. 9, 188–194 (2006).
Ly, A. et al. DSCAM is a netrin receptor that collaborates with DCC in mediating turning responses to netrin-1. Cell 133, 1241–1254 (2008).
Manitt, C. et al. dcc orchestrates the development of the prefrontal cortex during adolescence and is altered in psychiatric patients. Transl. Psychiatry 3, e338 (2013).
Torres-Berrio, A. et al. DCC confers susceptibility to d epression-like behaviors in humans and mice and is regulated by miR-218. Biol. Psychiatry 81, 306–315 (2017).
Leday, G. G. R. et al. Replicable and coupled changes in innate and adaptive immune gene expression in two case-control studies of blood microarrays in major depressive disorder. Biol. Psychiatry 83, 70–80 (2018).
Li, Z. et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 49, 1576–1583 (2017).
Chang, H., Xiao, X. & Li, M. The schizophrenia risk gene ZNF804A: clinical associations, biological mechanisms and neuronal functions. Mol. Psychiatry 22, 944–953 (2017).
Li, M. & Yue, W. VRK2, a candidate gene for psychiatric and neurological disorders. Mol. Neuropsychiatry 4, 119–133 (2018).
Wang, L. et al. Further evidence of an association between NCAN rs1064395 and bipolar disorder. Mol. Neuropsychiatry 4, 30–34 (2018).
Ikeda, M. et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry 23, 639–647 (2018).
Zhao, L. et al. Replicated associations of FADS1, MAD1L1, and a rare variant at 10q26.13 with bipolar disorder in Chinese population. Transl. Psychiatry 8, 270 (2018).
Xiao, X. et al. The Gene Encoding Protocadherin 9 (PCDH9), a novel risk factor for major depressive disorder. Neuropsychopharmacology 43, 1128–1137 (2018).
Kichaev, G. & Pasaniuc, B. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet. 97, 260–271 (2015).
Li, Y. R. & Keating, B. J. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 6, 91 (2014).
GTEx Consortium. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Colantuoni, C. et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523 (2011).
Miller, J. A. et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014).
Stiles, J. & Jernigan, T. L. The basics of brain development. Neuropsychol. Rev. 20, 327–348 (2010).
Birnbaum, R. et al. Investigation of the prenatal expression patterns of 108 schizophrenia-associated genetic loci. Biol. Psychiatry 77, e43–e51 (2015).
Birnbaum, R., Jaffe, A. E., Hyde, T. M., Kleinman, J. E. & Weinberger, D. R. Prenatal expression patterns of genes associated with neuropsychiatric disorders. Am. J. Psychiatry 171, 758–767 (2014).
O’Brien, H. E. et al. Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 19, 194 (2018).
Walker, R. L. et al. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell 179, 750–771 e722 (2019).
Hinkelmann, K. et al. Cognitive impairment in major depression: association with salivary cortisol. Biol. Psychiatry 66, 879–885 (2009).
Goldman, J. S. et al. Netrin-1 promotes excitatory synaptogenesis between cortical neurons by initiating synapse assembly. J. Neurosci. 33, 17278–17289 (2013).
Reynolds, L. M. et al. DCC receptors drive prefrontal cortex maturation by determining dopamine axon targeting in adolescence. Biol. Psychiatry 83, 181–192 (2018).
Cai, N. et al. Molecular signatures of major depression. Curr. Biol. 25, 1146–1156 (2015).
Bigdeli, T. B. et al. Genetic effects influencing risk for major depressive disorder in China and Europe. Transl. Psychiatry 7, e1074 (2017).
Zeki, S. & Romaya, J. P. Neural correlates of hate. PLoS ONE 3, e3556 (2008).
Tao, H. et al. Depression uncouples brain hate circuit. Mol. Psychiatry 18, 101–111 (2013).
Schmaal, L. et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol. Psychiatry 21, 806–812 (2016).
Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482 (2019).
Zeng, Y. et al. A combined pathway and regional heritability analysis indicates NETRIN1 pathway is associated with major depressive disorder. Biol. Psychiatry 81, 336–346 (2017).
Arnau-Soler, A. et al. Genome-wide by environment interaction studies of depressive symptoms and psychosocial stress in UK Biobank and Generation Scotland. Transl. Psychiatry 9, 14 (2019).
Ward, J. et al. Genome-wide analysis in UK Biobank identifies four loci associated with mood instability and genetic correlation with major depressive disorder, anxiety disorder and schizophrenia. Transl. Psychiatry 7, 1264 (2017).
Grant, A., Fathalli, F., Rouleau, G., Joober, R. & Flores, C. Association between schizophrenia and genetic variation in DCC: a case-control study. Schizophr. Res 137, 26–31 (2012).
Manitt, C. et al. The netrin receptor DCC is required in the pubertal organization of mesocortical dopamine circuitry. J. Neurosci. 31, 8381–8394 (2011).
Flores, C. et al. Netrin receptor deficient mice exhibit functional reorganization of dopaminergic systems and do not sensitize to amphetamine. Mol. Psychiatry 10, 606–612 (2005).
Pokinko, M., Moquin, L., Torres-Berrio, A., Gratton, A. & Flores, C. Resilience to amphetamine in mouse models of netrin-1 haploinsufficiency: role of mesocortical dopamine. Psychopharmacology 232, 3719–3729 (2015).
Kim, J. H. et al. Netrin-1 receptor-deficient mice show age-specific impairment in drug-induced locomotor hyperactivity but still self-administer methamphetamine. Psychopharmacology 230, 607–616 (2013).
Reynolds, L. M., Gifuni, A. J., McCrea, E. T., Shizgal, P. & Flores, C. dcc Haploinsufficiency results in blunted sensitivity to cocaine enhancement of reward seeking. Behav. Brain Res. 298, 27–31 (2016).
Grant, A. et al. Netrin-1 receptor-deficient mice show enhanced mesocortical dopamine transmission and blunted behavioural responses to amphetamine. Eur. J. Neurosci. 26, 3215–3228 (2007).
Flores, C. Role of netrin-1 in the organization and function of the mesocorticolimbic dopamine system. J. Psychiatry Neurosci. 36, 296–310 (2011).
Hoops, D. & Flores, C. Making dopamine connections in adolescence. Trends Neurosci. 40, 709–719 (2017).
Vosberg, D. E. et al. Mesocorticolimbic connectivity and volumetric alterations in DCC mutation carriers. J. Neurosci. 38, 4655–4665 (2018).
Ota, K. T. et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat. Med. 20, 531–535 (2014).
Duric, V. et al. A negative regulator of MAP kinase causes depressive behavior. Nat. Med. 16, 1328–1332 (2010).
Zhu, X. et al. Associations between TCF4 gene polymorphism and cognitive functions in schizophrenia patients and healthy controls. Neuropsychopharmacology 38, 683–689 (2013).
Blake, D. J. et al. TCF4, schizophrenia, and Pitt-Hopkins Syndrome. Schizophr. Bull. 36, 443–447 (2010).
Pardinas, A. F. et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389 (2018).
Kennedy, A. J. et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep. 16, 2666–2685 (2016).
Rannals, M. D. et al. Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of SCN10a and KCNQ1. Neuron 90, 43–55 (2016).
Li, H. et al. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol. Psychiatry 24, 1235–1246 (2019).
Forrest, M. P. et al. The psychiatric risk gene transcription factor 4 (TCF4) regulates neurodevelopmental pathways associated with schizophrenia, autism, and intellectual disability. Schizophr. Bull. 44, 1100–1110 (2018).
Wittmann, M. T. & Haberle, B. M. Linking the neuropsychiatric disease gene TCF4 to neuronal activity-dependent regulatory networks. J. Neurosci. 38, 2653–2655 (2018).
Tao, R. et al. Expression of ZNF804A in human brain and alterations in schizophrenia, bipolar disorder, and major depressive disorder: a novel transcript fetally regulated by the psychosis risk variant rs1344706. JAMA Psychiatry 71, 1112–1120 (2014).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Ritchie, G. R., Dunham, I., Zeggini, E. & Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 11, 294–296 (2014).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81722019 to M.L., 81871067 to H.C., 81771439 to L.H.); Hubei Province health and family planning scientific research project (WJ2015Q033 to N.Q.); Population and Family Planning Commission of Wuhan (WX14B34 to N.Q.). X.X. was supported by the Chinese Academy of Sciences Western Light Program, and Youth Innovation Promotion Association, CAS. M.L. was supported by CAS Pioneer Hundred Talents Program and the 1000 Young Talents Program. Data were generated as part of the PsychENCODE Consortium, supported by: U01MH103392, U01MH103365, U01MH103346, U01MH103340, U01MH103339, R21MH109956, R21MH105881, R21MH105853, R21MH103877, R21MH102791, R01MH111721, R01MH110928, R01MH110927, R01MH110926, R01MH110921, R01MH110920, R01MH110905, R01MH109715, R01MH109677, R01MH105898, R01MH105898, R01MH094714, P50MH106934, U01MH116488, U01MH116487, U01MH116492, U01MH116489, U01MH116438, U01MH116441, U01MH116442, R01MH114911, R01MH114899, R01MH114901, R01MH117293, R01MH117291, and R01MH117292 awarded to: Schahram Akbarian (Icahn School of Medicine at Mount Sinai), Gregory Crawford (Duke University), Stella Dracheva (Icahn School of Medicine at Mount Sinai), Peggy Farnham (University of Southern California), Mark Gerstein (Yale University), Daniel Geschwind (University of California, Los Angeles), Fernando Goes (Johns Hopkins University), Thomas M. Hyde (Lieber Institute for Brain Development), Andrew Jaffe (Lieber Institute for Brain Development), James A. Knowles (University of Southern California), Chunyu Liu (SUNY Upstate Medical University), Dalila Pinto (Icahn School of Medicine at Mount Sinai), Panos Roussos (Icahn School of Medicine at Mount Sinai), Stephan Sanders (University of California, San Francisco), Nenad Sestan (Yale University), Pamela Sklar (Icahn School of Medicine at Mount Sinai), Matthew State (University of California, San Francisco), Patrick Sullivan (University of North Carolina), Flora Vaccarino (Yale University), Daniel Weinberger (Lieber Institute for Brain Development), Sherman Weissman (Yale University), Kevin White (University of Chicago), Jeremy Willsey (University of California, San Francisco), and Peter Zandi (Johns Hopkins University). Data were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138, and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories and the NIMH Human Brain Collection Core. CMC Leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), Thomas Lehner, Barbara Lipska (NIMH).
Author information
Authors and Affiliations
Contributions
X.X., Y.L., and M.L. designed the study and interpreted the results. H.J.L., N.Q., and L.H. conducted the primary analysis. X.C., C.Y.Z., B.L.Z., S.F.Z., J.C., B.X., W.L., L.W., Q.F.J., and H.C. contributed to literature search and assistance in analysis. H.L., X.X., and M.L. drafted the manuscript, and all authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, HJ., Qu, N., Hui, L. et al. Further confirmation of netrin 1 receptor (DCC) as a depression risk gene via integrations of multi-omics data. Transl Psychiatry 10, 98 (2020). https://doi.org/10.1038/s41398-020-0777-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-0777-y
This article is cited by
-
Prioritization of risk genes in colorectal cancer by integrative analysis of multi-omics data and gene networks
Science China Life Sciences (2024)
-
Mendelian randomization studies of depression: evidence, opportunities, and challenges
Annals of General Psychiatry (2023)
-
Identifying causal genes for depression via integration of the proteome and transcriptome from brain and blood
Molecular Psychiatry (2022)
-
Multi-omic analysis unveils biological pathways in peripheral immune system associated to minimal hepatic encephalopathy appearance in cirrhotic patients
Scientific Reports (2021)
