
Abstract
Biological nitrogen fixation contributes significantly to marine primary productivity. The current view depicts few cyanobacterial diazotrophs as the main marine nitrogen fixers. Here, we used 891 Tara Oceans metagenomes derived from surface waters of five oceans and two seas to generate a manually curated genomic database corresponding to free-living, filamentous, colony-forming, particle-attached, and symbiotic bacterial and archaeal populations. The database provides the genomic content of eight cyanobacterial diazotrophs including a newly discovered population related to known heterocystous symbionts of diatoms, as well as 40 heterotrophic bacterial diazotrophs that considerably expand the known diversity of abundant marine nitrogen fixers. These 48 populations encapsulate 92% of metagenomic signal for known nifH genes in the sunlit ocean, suggesting that the genomic characterization of the most abundant marine diazotrophs may be nearing completion. Newly identified heterotrophic bacterial diazotrophs are widespread, express their nifH genes in situ, and also occur in large planktonic size fractions where they might form aggregates that provide the low-oxygen microenvironments required for nitrogen fixation. Critically, we found heterotrophic bacterial diazotrophs to be more abundant than cyanobacterial diazotrophs in most metagenomes from the open oceans and seas, emphasizing the importance of a wide range of heterotrophic populations in the marine nitrogen balance.
Similar content being viewed by others


Introduction
Plankton communities in the sunlit ocean consist of numerous microbial lineages that influence global biogeochemical cycles and climate [1,2,3,4,5,6]. Phototrophic primary productivity is often constrained by the amount of bioavailable nitrogen [7, 8], a critical element for cellular growth and division. Only a few bacterial and archaeal populations within the large pool of marine microbial lineages are capable of performing nitrogen fixation, thereby providing an essential source of new nitrogen to phytoplankton [9,10,11]. These populations are known as diazotrophs and represent key marine players that sustain primary productivity in large oceanic regions [10]. Globally, marine nitrogen fixation is at least as important as the nitrogen fixation on land performed by Rhizobium bacteria in symbiosis with plants [12].
Cyanobacterial diazotrophs are abundant in open ocean surface waters and provide a substantial portion of bioavailable nitrogen [13,14,15]. They include populations within the genus Trichodesmium [16,17,18] and several lineages that enter symbiotic associations with eukaryotes (e.g., Richelia [19, 20], the Candidatus Atelocyanobacterium also labeled UCYN-A [21, 22]) or can exist as free-living cells such as Crocosphaera watsonii also labeled UCYN-B [23, 24]. A wide range of non-cyanobacterial diazotrophs has also been detected using amplicon surveys of the nifH gene required for nitrogen fixation. These molecular surveys showed non-cyanobacterial diazotrophs occurring in lower abundance compared to their cyanobacterial counterparts in various oceanic regions (e.g., [25,26,27,28,29,30,31]) but could also be relatively abundant in some samples (e.g., [32,33,34,35,36,37]). Overall, decades of Trichodesmium cultivation, flow cytometry, molecular surveys, imaging, and in situ nitrogen fixation rate measurements have led to the emergence of a view depicting cyanobacterial diazotrophs as the principal marine nitrogen fixers [38].
Recently, a genome-resolved metagenomic survey exposed free-living heterotopic bacterial diazotrophs (HBDs) abundant in the surface waters of large oceanic regions [39]. This first set of genome-resolved HBDs from the open ocean was subsequently found to express their nifH genes in situ using metatranscriptomics [40]. However, the sole focus on free-living bacterial cells in this survey excluded not only key cyanobacterial players but also other diazotrophs that might occur under the form of aggregates, preventing a comprehensive investigation of diazotrophs in the sunlit ocean. Here we used nearly nine hundred Tara Oceans metagenomes [41] to create a genomic database corresponding to free-living, as well as filamentous, colony-forming, particle-attached, and symbiotic bacterial and archaeal populations occurring in surface waters of the global ocean. Our genomic database includes dozens of previously unknown HBDs abundant in different size fractions and oceanic regions all of which express their nifH genes in situ. Most notably, we found HBDs to be more abundant compared to cyanobacterial diazotrophs in metagenomes covering most surface open oceans and seas, revealing their prevalence also under the form of putative large aggregates within plankton and suggesting they play a considerable role in the marine nitrogen balance.
Results and discussion
Part one: Genome-wide metagenomic analyses
Nearly 2,000 manually curated bacterial and archaeal genomes from the 0.8–2,000 µm planktonic cellular size fractions in the surface oceans and seas
We performed a comprehensive genome-resolved metagenomic survey of bacterial and archaeal populations from the euphotic zone of polar, temperate, and tropical oceans using 798 metagenomes derived from the Tara Oceans expeditions. They correspond to surface waters and deep chlorophyll maximum (DCM) layers from 143 stations covering the Pacific, Atlantic, Indian, Arctic, and Southern Oceans, as well as the Mediterranean and Red Seas, encompassing eight plankton size fractions ranging from 0.8 µm to 2000 µm (Table S1). These 280 billion reads were already used as inputs for 11 metagenomic co-assemblies using geographically bounded samples to recover eukaryotic metagenome-assembled genomes (MAGs) [42]. Here, we recovered nearly 2,000 bacterial and archaeal MAGs from these 11 co-assemblies.
We combined these MAGs with 673 MAGs previously generated from the 0.2 µm to 3 µm size fraction (93 metagenomes) [39] to create a culture-independent, non-redundant (average nucleotide identity <98%) genomic database for microbial populations consisting of 1,778 bacterial and 110 archaeal MAGs, all exhibiting >70% completion (average completion of 87.1% and redundancy of 2.5%; Table S2). We manually characterized and curated these 1,888 MAGs using a holistic framework within anvi’o [43, 44] that relied heavily on differential coverage across metagenomes within the scope of their associated co-assembly. This genomic database has a total size of 4.8 Gbp, with MAGs affiliated to Proteobacteria (n = 916), Bacteroidetes (n = 314), Planctomycetes (n = 154), Verrucomicrobia (n = 128), Euryarchaeota (n = 105), Actinobacteria (n = 68), Cyanobacteria (n = 51), Chloroflexi (n = 36), Candidatus Marinimicrobia (n = 30), Candidatus Dadabacteria (n = 10) and 24 other phyla represented less than 10 times (Table S1). We used their distribution and gene content to survey marine diazotrophs in the open ocean without relying on cultivation or nifH amplicon surveys.
A genomic collection of 48 marine diazotrophs abundant in the open ocean
While none of the 110 archaeal MAGs indicated a diazotrophic lifestyle, a total of 48 bacterial MAGs contained genes encoding the catalytic (nifHDK) and biosynthetic (nifENB) proteins required for nitrogen fixation (Table S3). Among these, only one MAG (Gammaproteobacterial) lacked the nifH gene, which is likely a result of the limitations inherent to genome-resolved metagenomics. Based on the taxonomic signal and the occurrence or absence of genes required for a photosynthetic lifestyle, these MAGs could be categorized into eight cyanobacterial diazotrophs and 40 HBDs. Their estimated completion averaged 93.4%, suggesting they correspond to near-complete environmental genomes (Fig. 1 and Table S4).

Top panel displays a phylogenomic tree of the 48 diazotroph MAGs using 37 gene markers and visualized with anvi’o [43]. Additional layers of information display the length of MAGs alongside environmental signal computed using genome-wide metagenomic read recruitments across 937 metagenomes, and nifH primer compatibilities (only full length and non-fragmented nifH genes were considered). For each MAG, the “maximal percent of mapped reads” layer displays the percent of mapped reads corresponding to the sample for which this metric was the highest among all 937 metagenomes. Thus, this sample is MAG dependent. In contrast, the “relative abundance” layers display for each MAG the average number of mapped reads across samples corresponding to the same size fraction. Bottom panel displays the ratio of cumulative genome-scale mean coverage between eight cyanobacterial diazotrophs (green) and 40 HBDs (red) across 385 metagenomes we organized into five size fractions.
The reconstructed cyanobacterial MAGs recapitulated findings of major marine diazotrophs previously discovered within this phylum and for which a genome (partial or complete) had been characterized previously using either available cultures or sorted cells from flow cytometry: UCYN-A1 (ANI of 99.3%) and UCYN-A2 (ANI of 99.6%), Crocosphaera watsonii (strain WH-8501; ANI of 99.4%), Richelia intracellularis (strain RintHH01; ANI of 99.5%), Trichodesmium erythraeum (strain IMS101; ANI of 99%), and Trichodesmium thiebautii (strain H9-4; n = 2 with ANI of 98.7% and 98%). Interestingly, while the two Trichodesmium thiebautii populations displayed high genomic similarity (ANI of 97.9%) and correlated across 81 metagenomes with signal for this lineage (R2 = 0.93), the mean coverage ratio revealed one population that was dominant at three sites of the North Atlantic Ocean while the second population was relatively more abundant in the Indian Ocean, Pacific Ocean and Red Sea (Fig. S1). In addition, one MAG corresponded to an unknown population we tentatively named ‘Candidatus Richelia exalis’ given its close evolutionary relationship with R. intracellularis (e.g., ANI of 87.3% when compared to the strain RintHH01; see Table S3 for more comparisons) (Fig. 1). The strong signal of ‘Candidatus Richelia exalis’ in the large size fractions, similar to R. intracellularis, and their comparable functional traits (see following section) suggest this species also leads a symbiotic lifestyle.
Compared to the cyanobacterial diazotrophs that were already well characterized prior to this genome-resolved metagenomic survey, the HBDs we recovered substantially increase the number of known diazotrophic populations. In addition to eight previously characterized HBDs reconstructed from the 0.2–3 μm size fraction [39] (five of which were replaced by MAGs characterized from the larger size fractions that displayed improved completion statistics), the genomic database includes 32 additional HBDs belonging to the phyla Deltaproteobacteria (eight HBDs; six new nifH genes when compared to a comprehensive set of reference databases [18], see methods), Gammaproteobacteria (16 HBDs; four new nifH genes), Planctomycetes (three HBDs; one new nifH gene), Alphaproteobacteria (eight HBDs; three new nifH genes), Epsilonproteobacteria (2 HBDs; two new nifH genes), and Verrucomicrobia (three HBDs; three new nifH genes) (Fig. 1 and Table S5). Interestingly, some of the newly identified nifH gene sequences are incompatible with the design of several primers frequently used in nifH gene amplicon surveys (Fig. S2 and Table S6). This was especially true of the “nifH4” primer (round one of widely utilized nested primers [34, 45,46,47]) (Fig. 1) that appears incompatible with most HBDs identified in this study.
The emergence of three main functional groups for marine HBDs
In order to provide a global view of functional capabilities among the 48 diazotrophs, we accessed functions in their gene content using COG20 functions, categories and pathways [48], KOfam [49], KEGG modules, and classes [50] from within the anvi’o genomic workflow [43] (Table S7). Genomic clustering based on the completeness of 322 functional modules exposed four distinct groups: (1) the cyanobacterial diazotrophs, (2) HBDs dominated by Alphaproteobacteria, (3) HBDs associated with Gammaproteobacteria, and finally (4) HBDs organized in closely related subgroups corresponding to Deltaproteobacteria, Epsilonproteobacteria, Verrucomicrobia and Planctomycetes (Fig. 2). Several HBDs have the metabolic capacity to generate energy using pathways other than aerobic respiration. One population associated with Alphaptroteobacteria (genus Marinibacterium) for example encodes anoxygenic photosystem II as well as all pathways required for aerobic respiration, thiosulfate oxidation and dissimilatory nitrate reduction to ammonia. Within the HBD group affiliated with Alphaproteobacteria, the majority of populations encode the SOX complex necessary for thiosulfate oxidation (Table S7) and one population encodes the genes required for denitrification. Among the HBDs affiliated with Deltaptroteobacteria, a large majority encodes the pathway for dissimilatory sulfate reduction and mostly lack metabolic pathways required for aerobic respiration. Four representatives of the Gammaproteobacteria have the metabolic potential for denitrification and one population can generate energy via thiosulfate oxidation, a capacity that is also encoded in one of the HBDs affiliated with Epsilonproteobacteria. The metabolic pathway for dissimilatory nitrate reduction to ammonia can be found in all taxonomic groups (occurrence: 20–100%) (Table S7). This intriguing metabolic diversity among HBDs indicates their potential importance in major biogeochemical cycles. All deltaproteobacterial HBDs encode the complex biosynthesis pathway for cobalamin, also found in a majority of cyanobacterial diazotrophs (including the symbionts) (Table S7). Only the final 5–6 steps of cobalamin synthesis are also encoded in HBD populations associated with Gamma- and Alphaproteobacteria (Table S7). Overall, we found the HBDs to be functionally more diverse compared to their cyanobacterial counterparts.

The figure displays a heatmap of the completeness of 322 functional modules across the 48 diazotrophic MAGs. Clustering of MAGs and modules is based on completeness values (Euclidean distance and ward linkage) and the data were visualized using anvi’o [43]. The cosmopolitan score corresponds to the number of stations in which a given MAG was detected (cut-off: >25% of the MAG is covered by metagenomic reads).
HBDs are generally more abundant compared to cyanobacterial diazotrophs
The 48 diazotrophs occurred at up to 49 stations (out of 119 stations considered to compute this cosmopolitan score) and recruited up to 3.7% of metagenomic reads (Figs. 1, 2, and Table S2) when considered individually. Yet, the locally most abundant diazotrophs were not the most widespread (R2 of 0.007 when comparing the maximal number of recruited reads and cosmopolitan score). We detected no diazotrophs in the Arctic Ocean or the Red Sea, only a single HBD in the Southern Ocean [39] and very few representatives in the Mediterranean Sea. Within temperate and tropical open ocean regions, marine diazotrophs affiliated with Epsilonproteobacteria, Deltaproteobacteria and Verrucomicrobia mostly occurred in the Pacific Ocean. The remaining diazotrophic lineages occurred in the Pacific, Indian, and Atlantic Oceans. Within the group of cyanobacterial diazotrophs, the two populations of Trichodesmium thiebautii were highly abundant in some of the large size fractions and generally prevailed in the Indian Ocean (Fig. 1). The overall geographic distribution of diazotrophs indicates that the Pacific Ocean is dominated by HBDs, corroborating previously observed trends [18, 34, 39].
The majority of the 48 diazotrophs were associated with the 0.2–5 µm size fraction that covers most of the free-living bacterial cells, while the remaining diazotrophs were detected in the 5–20 µm (n = 15) and 20–180 µm (n = 2; Richelia intracellularis and ‘Candidatus Richelia exalis’) size fractions (Fig. 1, Table S4). We then computed the ratio of cumulative mean coverage (i.e., number of times a genome is sequenced) between the eight cyanobacterial diazotrophs and 40 HBDs across 385 metagenomes organized by size fraction (552 metagenomes with no signal for any of the 48 diazotrophs were not considered here). Overall, HBDs displayed a cumulative mean coverage superior to that of cyanobacterial diazotrophs in 250 metagenomes, compared to 135 for the latter. Furthermore, a clear signal emerged in which HBDs were more abundant in most metagenomes representing the 0.2–5 μm (86.5%) and 0.8–2000 μm (92.6%) size fractions while cyanobacterial diazotrophs predominated in the 20–180 μm (92.3%) and 180–2000 μm (86.2%) size fractions (Fig. 1, bottom panel). Finally, the 5–20 µm size fraction was more balanced between HBDs and cyanobacterial diazotrophs.
The 0.8–2000 μm size fraction was not collected in the Mediterranean Sea, Red Sea and Indian Ocean, but became an integral part of Tara Oceans sampling efforts in the other oceans [51]. This broad size range fraction provides a valuable metric to compare the relative abundance of diazotrophs that otherwise would be separated between the different size fractions. In other words, this size fraction could be used to effectively compare the genomic signal of diazotrophs corresponding to free-living, particle-attached, filamentous, colony-forming, and symbiotic cells, provided they (or their hosts) pass through 2 mm filter pores, either undamaged or fragmented (e.g., Trichodesmium colonies are known to be fragile). While uncertainty remains in the Indian Ocean, trends from metagenomes corresponding to the 0.8–2000 μm size fraction in other regions largely mirrored the free-living size fraction and were typically dominated by HBD signal. Metagenomes representing microbial populations from the 0.2–3 μm and 0.8–2000 μm size fractions indicate that HBDs are more abundant compared to their cyanobacterial counterparts in most of the surface oceans investigated here.
Co-occurrence of HBDs in large size fractions from a Pacific Ocean station
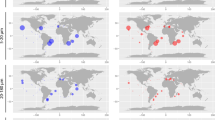
We detected a considerable metagenomic signal for HBDs at Station 98 in the South Pacific Ocean (Fig. 3; Table S4), which was also found using reference nifH genes [18]. Station 98 includes five surface and three DCM metagenomes covering all size fractions except for 0.8–2000 μm. The only cyanobacterial diazotroph we detected in this metagenomic set was ‘Candidatus Richelia exalis’ with a mean coverage of just 0.4X in the 20–180 μm size fraction of the surface layer. The 40 HBDs remained undetected in the DCM and only two HBDs were marginally detected in the 0.2–3 μm size fraction of the surface layer. In marked contrast, 14 HBDs were detected in the 5–20 μm, 20–180 μm, and 180–2000 μm size fractions of surface waters with a cumulative mean coverage reaching 1,106X (i.e., their genomes were sequenced cumulatively more than one thousand times in this particular metagenome), 15X and 283X, respectively. Such a high genomic coverage for bacterial populations in large size fractions is unusual and exceeded the maximum signal associated with UCYN-A and Trichodesmium in this study (Fig. 3; Table S4). The 14 HBDs were affiliated with Deltaproteobacteria (n = 5), Alphaproteobacteria (n = 2), Gammaproteobacteria (n = 2), Epsilonproteobacteria (n = 2), Planctomycetes (n = 2) and Verrucomicrobia (n = 1). Surface waters at Station 98 were nitrogen depleted (nitrate near the detection limit at 0.001 μM; Table S1), likely providing favorable conditions for a diverse assemblage of HBDs that were particularly abundant within the large size fractions. Lack of signal in the small size fraction suggests that similar populations might be missed in oceanic sampling that typically restricts bacterial analyses to free-living cells. Mechanisms maintaining diazotrophs in large plankton size fractions have yet to be fully elucidated [34, 52,53,54,55,56]. Our results nonetheless support recent observations in coast and estuary linking active HBDs to large aggregates [57, 58]. Exopolymer particles and aggregates might create low-oxygen microenvironments favorable for nitrogen fixation in marine environments [59], as observed in laboratory cultures [58, 60]. Thus, we suggest that HBDs formed a considerable number of large aggregates (up to >180 μm in size) at Station 98 in order to optimize their nitrogen fixation capabilities.

The world map provides coordinates for 15 Tara Oceans metagenomes (10 stations) displaying cumulative genomic coverage >100X for MAGs affiliated to diazotrophic Trichodesmium, UCYN-A or the HBDs. The bottom panel summarizes multi-omic signal (including at the level of nifH genes) statistics for those 15 metagenomes.
Part two: Gene-centric multi-omic analyses (nifH gene)
48 diazotrophic MAGs may cover >90% of cells containing known nifH genes
In order to analyze the significance of 48 diazotrophic MAGs with regard to other marine diazotrophic populations, we combined their nifH gene sequences with a comprehensive set of nifH sequences obtained from cultures, metagenomic assemblies, clones and amplicon surveys (see Methods). We used this extended nifH database (n = 328; redundancy removal at 98% identity over 90% of the length) to recruit metagenomic reads from Tara Oceans (Table S8). Strikingly, nifH genes corresponding to the eight cyanobacterial diazotrophs and 40 HBDs recruited 42.3% and 49.1% of all mapped metagenomic reads, respectively, with just 8.7% of the signal corresponding to 280 orphan nifH genes for which the genomic content within plankton has not yet been characterized (Fig. 4 and Table S8). These include a well-known diazotroph that awaits genomic characterization, the Gamma-A lineage [61], which accounted for 0.4% of mapped reads. Overall, this nifH centric metagenomic survey indicates that the 48 bacterial diazotrophic MAGs characterized in this study encapsulate 90% of read recruitment signal for known nifH genes in the surface oceans and seas investigated during Tara Oceans. One remaining uncertainty is the extent of abundant marine heterotrophic bacterial nifH genes that have yet to be discovered. These might further swell the ranks of HBDs in years to come.

The figure displays the proportion of metagenomic and metatranscriptomic reads mapping onto nifH genes as a function of ranges in two size fractions. Target genes correspond to the extended nifH gene database of 328 sequences including 280 orphan genes. The mapped samples (781 metagenomes and 520 metatranscriptomes) correspond to the surface and deep chlorophyll maximum layers of all oceans and two seas. For each size fraction range, the number of cumulated mapped reads represents each diazotrophic lineage (seven categories) across all samples. Results are displayed in relative proportion. The >0.8 μm size fraction range includes up to five size fractions: 0.8–5 μm, 5–20 μm, 20–180 μm, 180–2000 μm, and 0.8–2000 μm.
HBD populations express their nifH genes
We mapped hundreds of Tara Oceans metatranscriptomes against the extended nifH database to gain some insights into the potential for nitrogen fixation activity of cyanobacterial diazotrophs and HBDs. Specifically, we recruited “bacteria-compatible” metatranscriptomic reads from the free-living bacterial size fraction (0.2–3 µm), as well as poly-A enriched metatranscriptomic reads from larger size fractions ranging from 0.8 µm to 2,000 µm that was produced primarily to explore the transcriptomic diversity of microbial eukaryotes [62]. Bacterial transcripts are rarely polyadenylated, and even when it occurs, polyadenylation is often a degradation signal [63]. Importantly, all of the HBD nifH genes recruited reads, indicating at the very least a basal expression of genes encoding the nitrogen fixation apparatus (Table S8). Furthermore, the considerable genomic signal for HBDs at station 98 was reflected in the metatranscriptomic signal, demonstrating the expression of nifH genes by HBDs in these waters.
Given the methodological differences in RNA sequencing and other factors that may influence the observed signal (e.g., RNA stability across the bacterial tree of life, time intervals from sampling to RNA storage across stations and size fractions), we present global trends for the free-living bacterial size fraction (0.2–3 µm) and the larger size fractions as a combined pool (Fig. 4). When considering the extended nifH database as a whole, most of the signal among metatranscriptomes corresponded to UCYN-A1, followed by UCYN-A2, HBDs, and Trichodesmium (Table S8). The predominance of UCYN-A signal (including in the 0.2–3 µm size fraction) was driven by the high nitrogen fixation activity for UCYN-A1 at Stations 78 and 80 in the South West region of the Atlantic Ocean in which hundreds of thousands of metatranscriptomic reads corresponded to its nifH gene alone (Fig. 3, Table S8), as reported previously [64]. Metatranscriptomic read recruitments suggest that the UCYN-A1 symbiont drives a substantial portion of nitrogen fixation at the critical interface between oceans and atmosphere, which is quantitatively not reflected in the metagenomic signal (this genome was detected in just 13 stations). This metatranscriptomic analysis at large scale substantiates the importance of UCYN-A as previously observed with in situ nitrogen fixation surveys (e.g., [21]). A trend emerged in which the nifH genes for symbiotic diazotrophs (UCYN-A, Richelia) were more significantly detected relative to their metagenomic signal compared to non-symbiotic diazotrophs, corroborating previous studies (e.g., [65, 66]). These symbiotic relationships appear highly successful, and likely have an improved nitrogen-fixing capacity in contrast to free-living cells [22, 64, 67]. At the same time, the high abundance of nifH transcripts related to diazotrophic symbionts may partially reflect a protective effect of the host cell resulting in a sampling bias. Given that bacterial RNA molecules are highly unstable, marine metatranscriptomes should be interpreted with caution. Nevertheless, the relatively low signal for Trichodesmium and HBDs was surprising but might partially be related to the exclusion of bacterial transcripts from the larger size fractions.
For now, the nitrogen fixation activity of HBDs versus cyanobacterial diazotrophs remains unclear. HBDs may contribute very little to nitrogen fixation rates among plankton, in particular as compared to UCYN-A, Richelia, and Trichodesmium populations. For instance, the streamlined genomes of UCYN-A populations and beneficial interactions with their hosts have created highly effective nitrogen fixation machineries [22, 64, 67] compared to what HBDs can do by themselves and without ATP production from photosynthesis. Yet metatranscriptomic surveys cannot be trusted to the same extent as metagenomes for semi-quantitative investigations, and do not equate to activity. Our only certitude at this point is that HBDs (1) are widespread and sufficiently abundant to make a real difference in the oceanic nitrogen balance, and (2) regularly transcribe their nifH gene in the sunlit ocean, including when co-occurring in large size fractions. These environmental genomic insights indicate that HBDs should not be excluded from the restricted list of most relevant marine nitrogen fixers (currently only represented by cyanobacterial lineages [10]), at least until extensive studies of putative aggregates in the field as well as culture conditions shed light on their functional lifestyle and metabolic activities.
A simple nomenclature to keep track of genome-resolved marine HBDs
As an effort to maintain some continuity between studies, here we suggest applying a simple nomenclature to name with a numerical system the non-redundant HBD MAGs with sufficient completion statistics as a function of their phylum-level affiliation (historic NCBI naming). For example, HBDs affiliated to Alphaproteobacteria and discovered thus far were named HBD Alpha 01 to HBD Alpha 08. Table S3 describes the 40 HBDs using this nomenclature, which could easily be expanded moving forward. To this point, only MAGs with completion >70% are part of this environmental genomic database, and the redundancy removal was set to ANI of 98%. Their genomic content can be accessed from https://figshare.com/articles/dataset/Marine_diazotrophs/14248283.
Conclusion
Our genome-resolved metagenomic survey of plankton in the surface of five oceans and two seas covering organismal sizes ranging from 0.2 μm to 2,000 μm has allowed us to go beyond cultivation and nifH amplicon surveys to characterize the genomic content and geographic distribution of key diazotrophs in the ocean. Briefly, we identified eight cyanobacterial diazotrophs, seven of which were already known at the species level, and 40 HBDs, 32 of which were first characterized in this study. The 40 HBDs are functionally diverse and expand the known diversity of abundant marine nitrogen fixers within Proteobacteria and Planctomycetes while also covering Verrucomicrobia. Overall, the collection of 48 diazotrophs we characterized here encapsulates 90% of metagenomic signal for known nifH genes in the sunlit ocean. In other words, the genomic search for the most abundant diazotrophs at the surface of the open ocean may be nearing completion.
Nitrogen fixers in the sunlit ocean have long been categorized into two main taxonomic groups: few cyanobacterial diazotrophs contributing most of the fixed nitrogen input [14, 19, 21, 68], and a wide range of non-cyanobacterial diazotrophs considered to have little impact on the marine nitrogen balance, in part due to their very low abundances within plankton as seen from several nifH based amplicon surveys [25,26,27,28,29,30,31,32]. Here we provide three results contrasting with this paradigm. First, we found that a wide range of HBDs can occasionally co-occur under nitrate-depleted conditions in large size fractions, with metagenomic signals exceeding what was observed for UCYN-A and Trichodesmium lineages in other oceanic regions. Critically, insights from estuaries [57, 60] may offer an explanation for the presence of HBDs in large size fractions of the open ocean, indicating their ability to form aggregates that provide low-oxygen microenvironments favorable for nitrogen fixation. These insights could explain, at least to some extent, high nitrogen fixation rates previously observed in parts of the Pacific Ocean that are depleted in cyanobacterial diazotrophs, which at the time was referred to as a paradox [46]. But most importantly, genome-wide metagenomic read recruitments for the 48 diazotrophs indicated that HBDs are more abundant than their cyanobacterial counterparts in most regions of the surface ocean. Metagenomes covering a wide size range of plankton (the 0.8–2000 μm size fraction) were critical to reach this conclusion. Mismatches between the widely used “nifH4” primer and the nifH genes of most HBDs might partially explain the growing gap between prior nifH based sequence surveys and genome-resolved metagenomics studies. Finally, we found that all HBDs express their nifH genes, including when co-occurring in large size fractions, expanding on previous observations based on a subset of the lineages in the 0.2–3 μm size fraction [40]. As a result, a new understanding is emerging from large-scale multi-omic surveys that depict nitrogen fixers in the sunlit ocean as the sum of few cyanobacterial diazotrophs and a wide range of HBDs, all capable of using their nitrogen fixation machinery while thriving in specific size fractions and oceanic regions. Surveying HBD aggregates, including their nitrogen-fixing activity, might represent a new key asset in understanding the marine nitrogen cycle and its balance.
Now that genome-resolved metagenomics has shed light on dozens of abundant marine HBDs, first within the scope of free-living cells [39], and now by covering a much wider plankton size range of plankton, it becomes apparent how little we know about their ecology and role in supporting oceanic primary productivity via nitrogen fixation. As a starting point, genomic analyses exposed three main functional groups of HBDs that might denote distinct diazotrophic lifestyles. Moving forward, it will be critical to enrich or cultivate these HBDs, as done for some of the key cyanobacterial diazotrophs decades ago [69] or HBDs from the coast or estuaries more recently [58, 60]. Experiments with HBDs in cell culture conditions and in situ investigations could shed light on HBD nitrogen fixation rates and elucidate the conditions that elicit nitrogen-fixing activity by these populations. These lines of research should strongly benefit our understanding of nitrogen budgets in the open ocean.
Material and methods
Tara Oceans metagenomes
We analyzed a total of 937 Tara Oceans metagenomes available at the EBI under project PRJEB402. Table S1 reports general information (including the number of reads and environmental metadata) for each metagenome.
Genome-resolved metagenomics
The 798 metagenomes corresponding to size fractions ranging from 0.8 µm to 2 mm were previously organized into 11 ‘metagenomic sets’ based upon their geographic coordinates [42]. Those 0.28 trillion reads were used as inputs for 11 metagenomic co-assemblies using MEGAHIT [70] v1.1.1, and the scaffold header names were simplified in the resulting assembly outputs using anvi’o [43] v.6.1. Co-assemblies yielded 78 million scaffolds longer than 1,000 nucleotides for a total volume of 150.7 Gbp. Here, we performed a combination of automatic and manual binning on each co-assembly output, focusing only on the 11.9 million scaffolds longer than 2,500 nucleotides, which resulted in 1,925 manually curated bacterial and archaeal metagenome-assembled genomes (MAGs) with a completion >70%. Briefly, (1) anvi’o profiled the scaffolds using Prodigal [71] v2.6.3 with default parameters to identify an initial set of genes, and HMMER [72] v3.1b2 to detect genes matching to bacterial and archaeal single-copy core gene markers, (2) we used a customized database including both NCBI’s NT database and METdb to infer the taxonomy of genes with a Last Common Ancestor strategy [62] (results were imported as described in http://merenlab.org/2016/06/18/importing-taxonomy), (3) we mapped short reads from the metagenomic set to the scaffolds using BWA v0.7.15 [73] (minimum identity of 95%) and stored the recruited reads as BAM files using samtools [74], (4) anvi’o profiled each BAM file to estimate the coverage and detection statistics of each scaffold, and combined mapping profiles into a merged profile database for each metagenomic set. We then clustered scaffolds with the automatic binning algorithm CONCOCT [75] by constraining the number of clusters per metagenomic set to a number ranging from 50 to 400 depending on the set. Each CONCOCT clusters (n = 2,550, ~12 million scaffolds) was manually binned using the anvi’o interactive interface. The interface considers the sequence composition, differential coverage, GC-content, and taxonomic signal of each scaffold. Finally, we individually refined each bacterial and archeal MAG with >70% completion as outlined in Delmont and Eren [76], and renamed scaffolds they contained according to their MAG ID. Table S2 reports the genomic features (including completion and redundancy values) of the bacterial and archaeal MAGs.
MAGs from the 0.2–3 μm size fraction
We incorporated into our database 673 bacterial and archaeal MAGs with completion >70% and characterized from the 0.2–3 μm size fraction [39], providing a set of MAGs corresponding to bacterial and archaeal populations occurring in size fractions ranging from 0.2 µm to 2 mm.
Characterization of a non-redundant database of SMAGs
We determined the average nucleotide identity (ANI) of each pair of MAGs using the dnadiff tool from the MUMmer package [77] v.4.0b2. MAGs were considered redundant when their ANI was >98% (minimum alignment of >25% of the smaller SMAG in each comparison). We then selected the MAG with the best statistics (highest value when computing completion minus redundancy) to represent a group of redundant MAGs. This analysis provided a non-redundant genomic database of 1,888 MAGs.
Taxonomical inference of MAGs
We determined the taxonomy of MAGs using both ChekM [78] and GTDB version 86 [79]. However, we used NCBI taxonomy from the GTDB output to describe the phylum of MAGs in the results and discussion sections, in order to be in line with the literature.
Biogeography of MAGs
We performed a final mapping of all metagenomes to calculate the mean coverage and detection of the MAGs. Briefly, we used BWA v0.7.15 (minimum identity of 90%) and a FASTA file containing the 1,888 non-redundant MAGs to recruit short reads from all 937 metagenomes. We considered MAGs were detected in a given filter when >25% of their length was covered by reads to minimize non-specific read recruitments [39]. The number of recruited reads below this cut-off was set to 0 before determining vertical coverage and percent of recruited reads.
Cosmopolitan score
Using metagenomes from the Station subset 1 (n = 757; excludes the 0.8–2000 µm size fraction lacking in the first leg of the Tara Oceans expeditions), MAGs were assigned a “cosmopolitan score” based on their detection across 119 stations, as previously quantified for eukaryotes [42].
Identification of diazotroph MAGs
In a first step, we used three HMM models from Pfam [80] within anvi’o (e-value citoff of e-15) and targeting the catalytic genes (nifH, nifD, nifK) and biosynthetic genes (nifE, nifN, nifB) for nitrogen fixation. We then ran Interproscan [81] on genes with a HMM hit and used TIGRFAMs [82] results (we found those to be the most relevant for nitrogen fixation) to identify diazotroph MAGs. Finally, we used RAST [83] as a complementary approach to identify nitrogen-fixing genes the HMM/Inteproscan approach failed to characterize. Among the 48 diazotroph MAGs, only one single gene (nifH) was not recovered with this approach. The most likely explanation is that the gene is simply missing from the MAG.
Functional inferences of diazotroph MAGs
We inferred functions among the genes of diazotrophic MAGs using COG20 functions, categories, and pathways [48], KOfam [49], KEGG modules, and classes [50] within the anvi’o genomic workflow [43]. Regarding the KOfam modules, we calculated their level of completeness in each genomic database using the anvi’o program “anvi-estimate-metabolism” with default parameters. The URL https://merenlab.org/m/anvi-estimate-metabolism describes this program in more detail.
Sequence novelty for the nifH genes
The 47 nifH genes identified in the MAGs were considered novel if their sequence identity scores never exceeded 98% identity over an alignment of al least 200 nucleotides, when compared to a recently built nifH gene catalog by Pierella Karlusich et al. [18] using blast [84]. Briefly, the nifH gene catalog consists of sequences from Zehr laboratory (mostly diazotroph isolates and environmental clone libraries; https://www.jzehrlab.com), sequenced genomes, and additional sequences retrieved from Tara Oceans metagenomic assemblies co-assemblies [39] and the OM-Reference Gene Catalog version 2 [40]).
A new database of nifH genes including diazotroph MAGs
We created a database of nifH genes covering the diazotroph MAGs as well as few hundred sequences from Pierella Karlusich et al. [18] with signal in Tara Oceans metagenomes. We removed redundancy (cut-off=98% identity) between the diazotroph MAGs and the Pierella Karlusich database, except for Trichodesmium thiebautii due to the occurrence of multiple populations (and slight differences between MAGs and culture representatives) that stressed the need to further explore nifH gene microdiversity within this species. We performed a mapping of metagenomes and metatranscriptomes to calculate the mapped reads and mean coverage of sequences in the extended nifH gene database. Briefly, we used BWA v0.7.15 (minimum identity of 90%) and a FASTA file containing the sequences to recruit short reads.
Phylogenetic analyses of diazotroph MAGs
We used PhyloSift [85] v1.0.1 with default parameters to infer associations between MAGs in a phylogenomic context. Briefly, PhyloSift (1) identifies a set of 37 marker gene families in each genome, (2) concatenates the alignment of each marker gene family across genomes, and (3) computes a phylogenomic tree from the concatenated alignment using FastTree [86] v2.1. We used anvi’o to visualize the phylogenomic tree in the context of additional information and root it at the level of the phylum Cyanobacteria.
Metatranscriptomic read recruitment for nifH genes
We performed a mapping of 587 Tara Oceans metatranscriptomes to calculate the mean coverage of sequences in the extended nifH gene database. Briefly, we used BWA v0.7.15 (minimum identity of 90%) and a FASTA file containing the nifH gene sequences to recruit short reads from all 587 metatranscriptomes.
Data availability
All data our study generated are publicly available at http://www.genoscope.cns.fr/tara/ (metagenomic co-assemblies, FASTA files) or https://figshare.com/articles/dataset/Marine_diazotrophs/14248283 for the supplemental tables and information, as well as the genomic content of 48 marine diazotrophs using the new nomenclature (diazotrophic genomic database).
Change history
20 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41396-021-01173-9
References
Boyd PW. Toward quantifying the response of the oceans’ biological pump to climate change. Front Mar Sci. 2015. https://doi.org/10.3389/fmars.2015.00077.
Charlson RJ, Lovelock JE, Andreae MO, Warren SG. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature. 1987;326:655–61.
Falkowski PG, Barber RT, Smetacek V. Biogeochemical controls and feedbacks on ocean primary production. Science (80-). 1998;281:200–6.
Arrigo KR. Marine microorganisms and global nutrient cycles. Nature. 2005;437:349–55.
Sanders R, Henson SA, Koski M, De La Rocha CL, Painter SC, Poulton AJ, et al. The biological carbon pump in the North Atlantic. Prog Oceanogr e-pub print. 2014. https://doi.org/10.1016/j.pocean.2014.05.005.
De Vargas C, Audic S, Henry N, Decelle J, Mahé F, Logares R, et al. Eukaryotic plankton diversity in the sunlit ocean. Science. 2015. https://doi.org/10.1126/science.1261605.
Moore CM, Mills MM, Arrigo KR, Berman-Frank I, Bopp L, Boyd PW, et al. Processes and patterns of oceanic nutrient limitation. Nat Geosci. 2013;6:701–10.
Tyrrell T. The relative influences of nitrogen and phosohorus on oceanic primary production. Nature. 1999;400:525–31.
Dos Santos PC, Fang Z, Mason SW, Setubal JC, Dixon R. Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics. 2012;13:162.
Zehr JP, Capone DG. Changing perspectives in marine nitrogen fixation. Science. 2020. https://doi.org/10.1126/science.aay9514.
Zehr JP, Jenkins BD, Short SM, Steward GF. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Env Microbiol. 2003;5:539–54.
Galloway JN, Dentener FJ, Capone DG, Boyer EW, Howarth RW, Seitzinger SP, et al. Nitrogen cycles: Past, present, and future. Biogeochemistry. 2004. https://doi.org/10.1007/s10533-004-0370-0.
Carpenter EJ, Capone DG, Rueter JG. Marine pelagic cyanobacteria: trichodesmium and other diazotrophs. Boston: Kluwer Academic Publishers; 1992.
Carpenter EJ, Romans K. Major role of the cyanobacterium trichodesmium in nutrient cycling in the north atlantic ocean. Science. 1991;254:1356–8.
Karl D, Letelier R, Tupas L, Dore J, Christian J, Hebel D. The role of nitrogen fixation in biogeochemical cycling in the subtropical North Pacific Ocean. Nature. 1997;388:533–8.
Capone DG. Trichodesmium, a globally significant marine cyanobacterium. Science (80-). 1997;276:1221–9.
Dyhrman ST, Chappell PD, Haley ST, Moffett JW, Orchard ED, Waterbury JB, et al. Phosphonate utilization by the globally important marine diazotroph. Trichodesmium Nat. 2006;439:68–71.
Pierella Karlusich JJ, Pelletier E, Lombard F, Carsique M, Dvorak E, Colin S, et al. Global distribution patterns of marine nitrogen-fixers by imaging and molecular methods. Nat Commun 2021 121. 2021;12:1–18.
Gómez F, Furuya K, Takeda S. Distribution of the cyanobacterium Richelia intracellularis as an epiphyte of the diatom Chaetoceros compressus in the western Pacific Ocean. J Plankton Res. 2005. https://doi.org/10.1093/plankt/fbi007.
Hilton JA, Foster RA, James Tripp H, Carter BJ, Zehr JP, Villareal TA. Genomic deletions disrupt nitrogen metabolism pathways of a cyanobacterial diatom symbiont. Nat Commun. 2013. https://doi.org/10.1038/ncomms2748.
Martínez-Pérez C, Mohr W, Löscher CR, Dekaezemacker J, Littmann S, Yilmaz P, et al. The small unicellular diazotrophic symbiont, UCYN-A, is a key player in the marine nitrogen cycle. Nat Microbiol 2016. https://doi.org/10.1038/nmicrobiol.2016.163.
Tripp HJ, Bench SR, Turk KA, Foster RA, Desany BA, Niazi F, et al. Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature. 2010;464:90–94.
Moisander PH, Beinart RA, Hewson I, White AE, Johnson KS, Carlson CA, et al. (2010). Unicellular cyanobacterial distributions broaden the oceanic N2 fixation domain. Science (80-). https://doi.org/10.1126/science.1185468.
Montoya JP, Holl CM, Zehr JP, Hansen A, Villareal TA, Capone DG (2004). High rates of N2 fixation by unicellular diazotrophs in the oligotrophic Pacific Ocean. Nature. https://doi.org/10.1038/nature02824.
Church MJ, Short CM, Jenkins BD, Karl DM, Zehr JP. Temporal patterns of nitrogenase gene (nifH) expression in the oligotrophic North Pacific Ocean. Appl Environ Microbiol. 2005;71:5362–70.
Church MJ, Björkman KM, Karl DM, Saito MA, Zehr JP. Regional distributions of nitrogen-fixing bacteria in the Pacific Ocean. Limnol Oceanogr. 2008;53:63–77.
Zehr JP, Montoya JP, Jenkins BD, Hewson I, Mondragon E, Short CM, et al. Experiments linking nitrogenase gene expression to nitrogen fixation in the North Pacific subtropical gyre. Limnology and Oceanography. 2007;52:169–83.
Fong AA, Karl DM, Lukas R, Letelier RM, Zehr JP, Church MJ. Nitrogen fixation in an anticyclonic eddy in the oligotrophic North Pacific Ocean. ISME J. 2008;2:663–76.
Moisander PH, Beinart RA, Voss M, Zehr JP. Diversity and abundance of diazotrophic microorganisms in the South China Sea during intermonsoon. ISME J. 2008;251:954–67.
Benavides M, Moisander PH, Daley MC, Bode A, Arístegui J (2016). Longitudinal variability of diazotroph abundances in the subtropical North Atlantic Ocean. J Plankton Res. https://doi.org/10.1093/plankt/fbv121.
Langlois RJ, LaRoche J, Raab PA (2005). Diazotrophic diversity and distribution in the tropical and subtropical Atlantic Ocean. Appl Environ Microbiol. https://doi.org/10.1128/AEM.71.12.7910-7919.2005.
Man-Aharonovich D, Kress N, Zeev EB, Berman-Frank I, Béjà O. Molecular ecology of nifH genes and transcripts in the eastern Mediterranean Sea. Environ Microbiol. 2007;9:2354–63.
Bombar D, Paerl RW, Riemann L. Marine non-cyanobacterial diazotrophs: moving beyond molecular detection. Trends Microbiol. 2016;24:916–27.
Farnelid H, Andersson AF, Bertilsson S, Al-Soud WA, Hansen LH, Sørensen S, et al. Nitrogenase gene amplicons from global marine surface waters are dominated by genes of non-cyanobacteria. PLoS One 6. 2011. https://doi.org/10.1371/journal.pone.0019223.
Riemann L, Farnelid H, Steward GF. Nitrogenase genes in non-cyanobacterial plankton: prevalence, diversity and regulation in marine waters. Aquat Micro Ecol. 2010;61:235–47.
Moisander PH, Benavides M, Bonnet S, Berman-Frank I, White AE, Riemann L. Chasing after non-cyanobacterial nitrogen fixation in marine pelagic environments. Front Microbiol. 2017. https://doi.org/10.3389/fmicb.2017.01736.
Moreira-Coello V, Mouriño-Carballido B, Marañón E, Fernández-Carrera A, Bode A, Sintes E, et al. Temporal variability of diazotroph community composition in the upwelling region off NW Iberia. Sci Rep. 2019. https://doi.org/10.1038/s41598-019-39586-4.
Luo YW, Doney SC, Anderson LA, Benavides M, Berman-Frank I, Bode A, et al. Database of diazotrophs in global ocean: abundance, biomass and nitrogen fixation rates. Earth Syst Sci Data. 2012. https://doi.org/10.5194/essd-4-47-2012.
Delmont TO, Quince C, Shaiber A, Esen ÖC, Lee ST, Rappé MS, et al. Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes. Nat Microbiol. 2018;3:804–13.
Salazar G, Paoli L, Alberti A, Huerta-Cepas J, Ruscheweyh HJ, Cuenca M, et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell. 2019. https://doi.org/10.1016/j.cell.2019.10.014.
Sunagawa S, Acinas SG, Bork P, Bowler C, Eveillard D, Gorsky G, et al. Tara Oceans: towards global ocean ecosystems biology. Nat Rev Microbiol. 2020;18:428–45
Delmont TO, Gaia M, Hinsinger DD, Fremont P, Fernandez Guerra A, Murat et al. Functional repertoire convergence of distantly related eukaryotic plankton lineages revealed by genome-resolved metagenomics. bioRxiv. 2020. 2020.10.15.341214.
Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319.
Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat Microbiol 2020;6:3–6.
Gaby JC, Buckley DH (2012). A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One. https://doi.org/10.1371/journal.pone.0042149.
Turk-Kubo KA, Karamchandani M, Capone DG, Zehr JP. The paradox of marine heterotrophic nitrogen fixation: abundances of heterotrophic diazotrophs do not account for nitrogen fixation rates in the Eastern Tropical South Pacific. Environ Microbiol. 2014;16:3095–114.
Zehr JP, Turner PJ. Nitrogen fixation: nitrogenase genes and gene expression. METHODS Microbiol. 2001;30:271–86.
Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021. https://doi.org/10.1093/nar/gkaa1018.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020. https://doi.org/10.1093/bioinformatics/btz859.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017. https://doi.org/10.1093/nar/gkw1092.
Pesant S, Not F, Picheral M, Kandels-Lewis S, Le Bescot N, Gorsky G, et al. Open science resources for the discovery and analysis of Tara Oceans data. Sci Data 2015 21. 2015;2:1–16.
Farnelid H, Tarangkoon W, Hansen G, Hansen PJ, Riemann L. Putative N2-fixing heterotrophic bacteria associated with dinoflagellate-cyanobacteria consortia in the low-nitrogen Indian Ocean. Aquat Microb Ecol. 2010. https://doi.org/10.3354/ame01440.
Farnelid H, Turk-Kubo K, Ploug H, Ossolinski JE, Collins JR, Van Mooy BAS, et al. Diverse diazotrophs are present on sinking particles in the North Pacific Subtropical Gyre. ISME J. 2019. https://doi.org/10.1038/s41396-018-0259-x.
Foster RA, Carpenter EJ, Bergman B. Unicellular cyanobionts in open ocean dinoflagellates, radiolarians, and tintinnids: ultrastructural characterization and immuno-localization of phycoerythrin and nitrogenase. J Phycol. 2006. https://doi.org/10.1111/j.1529-8817.2006.00206.x.
Scavotto RE, Dziallas C, Bentzon-Tilia M, Riemann L, Moisander PH. Nitrogen-fixing bacteria associated with copepods in coastal waters of the North Atlantic Ocean. Environ Microbiol. 2015. https://doi.org/10.1111/1462-2920.12777.
Zani S, Mellon MT, Collier JL, Zehr JP. Expression of nifH genes in natural microbial assemblages in Lake George, New York, detected by reverse transcriptase PCR. Appl Environ Microbiol. 2000;66:3119–24.
Geisler E, Bogler A, Rahav E, Bar-Zeev E. Direct Detection of Heterotrophic Diazotrophs Associated with Planktonic Aggregates. Sci Rep. 2019. https://doi.org/10.1038/s41598-019-45505-4.
Martínez-Pérez C, Mohr W, Schwedt A, Dürschlag J, Callbeck CM, Schunck H, et al. Metabolic versatility of a novel N2-fixing Alphaproteobacterium isolated from a marine oxygen minimum zone. Environ Microbiol. 2018. https://doi.org/10.1111/1462-2920.14008.
Rahav E, Bar-Zeev E, Ohayon S, Elifantz H, Belkin N, Herut B, et al. Dinitrogen fixation in aphotic oxygenated marine environments. Front Microbiol. 2013. https://doi.org/10.3389/fmicb.2013.00227.
Bentzon-Tilia M, Severin I, Hansen LH, Riemann L. Genomics and ecophysiology of heterotrophic nitrogen-fixing bacteria isolated from estuarine surface water. MBio 6. 2015. https://doi.org/10.1128/mBio.00929-15.
Cornejo-Castillo FM, Zehr JP. Intriguing size distribution of the uncultured and globally widespread marine non-cyanobacterial diazotroph Gamma-A. ISME J. 2021. https://doi.org/10.1038/s41396-020-00765-1.
Carradec Q, Pelletier E, Da Silva C, Alberti A, Seeleuthner Y, Blanc-Mathieu R, et al. A global ocean atlas of eukaryotic genes. Nat Commun. 2018. https://doi.org/10.1038/s41467-017-02342-1.
Güell M, Yus E, Lluch-Senar M, Serrano L. Bacterial transcriptomics: what is beyond the RNA horiz-ome? Nat Rev Microbiol. 2011. https://doi.org/10.1038/nrmicro2620.
Cornejo-Castillo FM, Cabello AM, Salazar G, Sánchez-Baracaldo P, Lima-Mendez G, Hingamp P, et al. Cyanobacterial symbionts diverged in the late Cretaceous towards lineage-specific nitrogen fixation factories in single-celled phytoplankton. Nat Commun. 2016. https://doi.org/10.1038/ncomms11071.
Needoba JA, Foster RA, Sakamoto C, Zehr JP, Johnson KS. Nitrogen fixation by unicellular diazotrophic cyanobacteria in the temperate oligotrophic North Pacific Ocean. Limnol Oceanogr. 2007. https://doi.org/10.4319/lo.2007.52.4.1317.
Foster RA, Paytan A, Zehr JP. Seasonality of N2 fixation and nifH gene diversity in the Gulf of Aqaba (Red Sea). Limnol Oceanogr. 2009. https://doi.org/10.4319/lo.2009.54.1.0219.
Thompson AW, Foster RA, Krupke A, Carter BJ, Musat N, Vaulot D, et al. Unicellular cyanobacterium symbiotic with a single-celled eukaryotic alga. Science. 2012;337:1546–50.
Zehr JP, Waterbury JB, Turner PJ, Montoya JP, Omoregie E, Steward GF, et al. Unicellular cyanobacteria fix N2 in the subtropical North Pacific Ocean. Nature. 2001;412:635–8.
Ohki K, Zehr JP, Fujita Y. Trichodesmium: establishment of culture and characteristics of N2- fixation. Mar pelagic cyanobacteria. 1992. https://doi.org/10.1007/978-94-015-7977-3_20.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2014;31:1674–6.
Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 2010;11:119.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6.
Delmont TO, Eren AM. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 2016;4:e1839.
Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–83.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2020. https://doi.org/10.1093/bioinformatics/btz848.
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer ELL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–6.
Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–8.
Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–3.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Darling AE, Jospin G, Lowe E, Matsen FA, Bik HM, Eisen JA. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014;2:e243.
Price MN, Dehal PS, Arkin AP. FastTree 2 — Approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490.
Acknowledgements
Our survey was made possible by two scientific endeavors: the sampling and sequencing efforts by the Tara Oceans Project, and the bioinformatics and visualization capabilities afforded by anvi’o. We are indebted to all who contributed to these efforts, as well as other open-source bioinformatics tools for their commitment to transparency and openness. Tara Oceans (which includes the Tara Oceans and Tara Oceans Polar Circle expeditions) would not exist without the leadership of the Tara Oceans Foundation and the continuous support of 23 institutes (https://oceans.taraexpeditions.org/). Some of the computations were performed using the platine, titane and curie HPC machine provided through GENCI grants (t2011076389, t2012076389, t2013036389, t2014036389, t2015036389, and t2016036389). This work has been supported by the FFEM - French Facility for Global Environment, French Government ‘Investissements d’Avenir’ programs OCEANOMICS (ANR-11-BTBR- 0008), FRANCE GENOMIQUE (ANR-10-INBS-09-08), MEMO LIFE (ANR-10-LABX- 54), and PSL Research University (ANR-11-IDEX-0001-02). C.B. additionally acknowledges funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Diatomic; grant agreement No. 835067) and Agence Nationale de la Recherche “Phytomet” (ANR-16-CE01-0008) projects. Finally, RAF is supported by the Knut and Alice Wallenberg Foundation. JJPK acknowledges postdoctoral funding from the Fonds Français pour l’Environnement Mondial and Iva Veseli acknowledges support by the National Science Foundation Graduate Research Fellowship under Grant No. 1746045.
Author information
Authors and Affiliations
Contributions
T. O. Delmont conducted the study and performed the primary data analysis. Eric Pelletier and Juan Pierella Karlusich performed analyses regarding the abundance of MAGs and nifH genes (including helping creating the extended nifH gene database) across Tara Oceans metagenomes and metatranscriptomes. Iva Veseli and Jessika Fuessel performed functional analyses of the diazotrophic MAGs. A. M. Eren computed to compatibility between nifH genes and widely used primers. All authors helped interpret the data. T. O. Delmont wrote the manuscript, with critical inputs from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised:
In this article the References 30 – 32 were not given in the correct order in List of References: The correct order is given below
30. Benavides M, Moisander PH, Daley MC, Bode A, Arístegui J (2016). Longitudinal variability of diazotroph abundances in the subtropical North Atlantic Ocean. J Plankton Res. https://doi.org/10.1093/plankt/fbv121.
31. Langlois RJ, LaRoche J, Raab PA (2005). Diazotrophic diversity and distribution in the tropical and subtropical Atlantic Ocean. Appl Environ Microbiol. https://doi.org/10.1128/AEM.71.12.7910-7919.2005.
32. Man-Aharonovich D, Kress N, Zeev EB, Berman-Frank I, Béjà O. Molecular ecologyof nifH genes and transcripts in the eastern Mediterranean Sea. Environ Microbiol.2007;9:2354–63.
The original article has been corrected.
Rights and permissions
About this article

Cite this article
Delmont, T.O., Pierella Karlusich, J.J., Veseli, I. et al. Heterotrophic bacterial diazotrophs are more abundant than their cyanobacterial counterparts in metagenomes covering most of the sunlit ocean. ISME J 16, 927–936 (2022). https://doi.org/10.1038/s41396-021-01135-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01135-1
This article is cited by
-
Mirusviruses link herpesviruses to giant viruses
Nature (2023)
-
Molecular hydrogen in seawater supports growth of diverse marine bacteria
Nature Microbiology (2023)
-
Co-diversification of an intestinal Mycoplasma and its salmonid host
The ISME Journal (2023)
-
Symbiotic UCYN-A strains co-occurred with El Niño, relaxed upwelling, and varied eukaryotes over 10 years off Southern California
ISME Communications (2023)
-
Nitrogen fixation and diazotroph diversity in groundwater systems
The ISME Journal (2023)

{kind=link}
{kind=link}