
Abstract
The induced pluripotent stem cell (iPSC) technology has transformed in vitro research and holds great promise to advance regenerative medicine. iPSCs have the capacity for an almost unlimited expansion, are amenable to genetic engineering, and can be differentiated into most somatic cell types. iPSCs have been widely applied to model human development and diseases, perform drug screening, and develop cell therapies. In this review, we outline key developments in the iPSC field and highlight the immense versatility of the iPSC technology for in vitro modeling and therapeutic applications. We begin by discussing the pivotal discoveries that revealed the potential of a somatic cell nucleus for reprogramming and led to successful generation of iPSCs. We consider the molecular mechanisms and dynamics of somatic cell reprogramming as well as the numerous methods available to induce pluripotency. Subsequently, we discuss various iPSC-based cellular models, from mono-cultures of a single cell type to complex three-dimensional organoids, and how these models can be applied to elucidate the mechanisms of human development and diseases. We use examples of neurological disorders, coronavirus disease 2019 (COVID-19), and cancer to highlight the diversity of disease-specific phenotypes that can be modeled using iPSC-derived cells. We also consider how iPSC-derived cellular models can be used in high-throughput drug screening and drug toxicity studies. Finally, we discuss the process of developing autologous and allogeneic iPSC-based cell therapies and their potential to alleviate human diseases.
Similar content being viewed by others
Introduction
The development of induced pluripotent stem cell (iPSC) technology has opened vast opportunities for in vitro modeling of human biology and for cell therapy applications.1,2,3,4,5 Since the first reports of somatic cell reprogramming into mouse and human iPSCs in 2006 and 2007, respectively, iPSCs have been applied to model human development and diseases in vitro, screen drug candidates, and create cell therapies.1,2,3,4,5 Increasing understanding of the mechanisms that govern iPSC induction has shed light on cell fate decisions, accelerating the development of efficient iPSC derivation methods and protocols for iPSC differentiation into somatic cells.6 Modeling human biology with iPSCs and iPSC-derived cells is particularly attractive, given the human origin of iPSCs and the ability to derive patient-specific iPSCs with a disease-relevant genetic background.2 Indeed, iPSC-based cellular models may reveal human-specific phenotypes and molecular mechanisms that do not necessarily manifest in animal models.7,8,9 Furthermore, ever increasing complexity of iPSC-based cellular models has resulted in the development of sophisticated human-like tissues, such as organoids, that contain multiple cell types, exhibit primitive human tissue-like architecture and enable modeling of higher order cell-cell interactions.10 Various iPSC-derived cellular models can be applied to probe disease mechanisms, evaluate drug activity and toxicity, and develop next-generation cell therapies. Given that iPSCs can be genetically modified and differentiated into otherwise inaccessible cell types, autologous and allogeneic cell therapies are being actively developed using the iPSC technology and hold a great promise to provide new approaches for treating complex diseases.11
In this review, we begin by outlining the historical development of the iPSC technology, including the key discoveries that led to the breakthrough of somatic cell reprogramming to iPSCs in 2006 and 2007.3,4,5 Subsequently, we summarize the key molecular and cellular events governing iPSC induction as well as the methods for somatic cell reprogramming to iPSCs. We then discuss the versatile applications of iPSCs, including in vitro modeling of human development and diseases, drug discovery, and cell therapy applications.
Historical overview of somatic cell reprogramming to iPSCs
Today, it is well established that most somatic cells harbor complete genetic information required for the development of an entire organism, whereas phenotypic diversity is achieved by epigenetic mechanisms that define gene expression potential in each cell.12,13,14 However, prior to such modern understanding of animal development, various hypotheses to explain how immense physiological complexity of an adult animal could emerge were contemplated. Popular in the 17th and 18th centuries, a theory of preformationism posited that animals would grow from miniature versions of themselves; the imagined homunculi were microscopic preformed human beings that would simply grow into their adult versions.15 As pioneering work in embryology accumulated and microscopy power improved, preformationism was gradually replaced by the theory of epigenesis, postulating sequential cell differentiation and organ development from an egg.16,17 Yet, it remained unclear how an egg cell could give rise to the breathtaking phenotypic diversity of somatic cells.
In 1892, the German evolutionary biologist August Weismann (1834–1914) proposed the germ plasm theory, also known as the Weismann barrier, postulating that germ cells alone were used to transmit heritable information, whereas acquisition of somatic cell fate involved irreversible modification of heritable information, enabling phenotypic diversity to emerge.18 The idea of irreversible restriction of a differentiated somatic cell state during development was reiterated by the British developmental biologist Conrad Waddington (1905–1975) in 1957.19 Waddington proposed a model that would become known as the Waddington’s epigenetic landscape, suggesting that cell differentiation resembled a ball rolling downhill towards a more and more restricted and irreversible state.19 However, it remained elusive whether somatic cell differentiation truly required irreversible mutational events to occur or whether it could be achieved by some other means, such as by reversible epigenetic mechanisms.14 A year later, the American geneticist David Nanney (1925–2016) proposed that while the DNA sequence conferred gene expression potential, phenotypic differences in cells sharing the same genome could arise because of gene expression “specificities” regulated by epigenetic systems.20 Indeed, the reversibility of the mechanisms governing somatic cell specification was demonstrated by the British developmental biologist John Gurdon (b. 1933), who performed somatic cell nuclear transfer (SCNT) experiments (Fig. 1a, b).21,22,23,24,25 In 1962, using a model of the Xenopus laevis frog, Gurdon demonstrated that a nucleus isolated from a terminally differentiated somatic cell and transplanted into an enucleated egg harbored all the genetic information required to give rise to germline-competent organisms.21,22,23,24 Therefore, the SCNT experiments revealed that genetic information was preserved during differentiation, whereas phenotypic diversity of somatic cells was likely achieved by reversible epigenetic mechanisms. What kind of epigenetic mechanisms could enable such elaborate yet reversible phenotypic diversity? Among the many layers of epigenetic regulation known today, DNA methylation is a prominent example of stable, yet reversible epigenetic memory acquired along the course of cell fate specification.26,27,28,29 For a historical review of discovering DNA methylation as a central mechanism of gene expression regulation and maintenance over mitotic divisions, the readers are referred to Tompkins, 2022.14

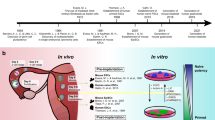
Development of the induced pluripotent stem cell (iPSC) technology. a A timeline of key breakthroughs related to the iPSC technology. b (Top) Somatic cell nuclear transfer (SCNT) experiments were pioneered by John Gurdon in the African clawed frog. Gurdon demonstrated that somatic cells retained all the genetic information necessary to give rise to a germline-competent organism. Successful SCNT in mammals was demonstrated by Keith Campbell, Ian Wilmut, and colleagues who cloned Dolly the sheep. (Bottom) Masako Tada and colleagues demonstrated that pluripotency can also be achieved by fusing a somatic cell with an embryonic stem cell, leading to the formation of a hybrid tetraploid cell. 4N, tetraploid. c The groundbreaking experiments of fibroblast reprogramming to pluripotency were pioneered by Kazutoshi Takahashi and Shinya Yamanaka. The researchers selected 24 factors as candidates for reprogramming and delivered these factors into mouse fibroblasts in various combinations by retroviral transduction. Eventually, Takahashi and Yamanaka identified a combination of 4 reprogramming factors—Oct4, Sox2, Klf4, and Myc—that was sufficient to reprogram mouse fibroblasts into embryonic stem cell-like pluripotent cells, known as iPSCs. Subsequently, Yamanaka and James Thomson independently reprogrammed human fibroblasts into iPSCs in 2007
In 1981, British biologists Martin Evans (b. 1941) and Matthew Kaufman (1942–2013) as well as the American biologist Gail Martin (b. 1944) isolated mouse embryonic stem cells (ESCs) that would serve as a reference point for subsequent somatic cell reprogramming experiments.30,31 Human ESCs were isolated by the American developmental biologist James Thomson (b. 1958) and colleagues in 1998.32 Cell fusion experiments of mouse33 and human34 ESCs with somatic cells revealed the capacity of the resulting heterokaryon for reprogramming to pluripotency, thus reaffirming the notion of cellular plasticity and somatic cell fate reversibility observed by Gurdon (Fig. 1b). Transdifferentiation experiments by ectopic expression of transcriptions factors further revealed the importance of transcription factors in establishing cell fate; for example, overexpression of the C/EBPα/β transcription factors was found to promote B cell reprogramming into macrophages.35,36,37,38 With ESCs as a reference point for features of pluripotency and an emerging understanding of how transcription factors orchestrated gene expression, the Japanese stem cell biologist Shinya Yamanaka (b. 1962) together with his postdoctoral fellow Kazutoshi Takahashi designed a series of somatic cell reprogramming experiments that would lead to the breakthrough development of mouse iPSCs in 2006 (Fig. 1c).4 Aiming to induce pluripotency in mouse embryonic fibroblasts (MEFs), Takahashi and Yamanaka selected 24 potential reprogramming factors that included transcription factors known to be important for the ESC state and other effectors. The reprogramming factors were cloned into retroviral vectors for MEF transduction, whereas MEFs were engineered to carry β-galactosidase and neomycin resistance encoding genes under a pluripotency-specific promoter of the Fbxo15 gene. Screening different combination of the 24 reprogramming factors, Takahashi and Yamanaka narrowed down the list to four transcription factors that were sufficient to induce pluripotency in MEFs: Oct4, Sox2, Klf4, and Myc (together known as OSKM or Yamanaka factors).4 Remarkably, these mouse iPSCs resembled the biological potency, gene expression, and the epigenetic landscape of ESCs.39 A year later, Yamanaka and Thomson independently demonstrated that human fibroblasts could also be reprogrammed into iPSCs; Yamanaka used the same OSKM factors, whereas Thomson used OCT4, SOX2, NANOG, and LIN28.3,5 These combinations of reprogramming factors remain widely used today, whereas Gurdon and Yamanaka were awarded the 2012 Nobel Prize in Physiology or Medicine for their discoveries. Since 2007, various modifications to the original cocktail of reprogramming factors have been developed. For example, small-molecule assisted somatic cell reprogramming was first reported in 2008,40,41 whereas fully chemical reprogramming of murine fibroblasts using seven small-molecule compounds was achieved in 2013.42
Molecular mechanisms of somatic cell reprogramming to iPSCs
When pluripotent stem cells undergo differentiation into somatic cells, they acquire epigenetic memory and undergo global changes to their chromatin conformation, resulting in inactivation of pluripotency-specific genes and activation of somatic cell-specific genes.43 Reprogramming of somatic cells back to the pluripotency state involves the erasure of many of these somatic cell signatures; therefore, induction of pluripotency has been proposed to partially resemble the a sequence of developmental events in reverse.6,44,45,46 Broadly, reprogramming occurs in two phases, early and late. During the early phase, somatic genes are silenced, whereas early pluripotency-associated genes are activated; during the late phase, late pluripotency-associated genes are activated. Early events of reprogramming are largely stochastic, presumably owing to the inefficient access of closed chromatin by OSKM and other transcription factors, whereas late events appear to be more deterministic.6 Universal aspects of reprogramming, such as two transcriptional waves, are accompanied by somatic cell type-specific reprogramming trajectories and transient events.47 Overall, reprogramming entails profound remodeling of the chromatin structure and the epigenome as well as changes to almost every aspect of cell biology, including metabolism, cell signaling, intracellular transport, proteostasis, and others.48,49,50,51,52 Given that iPSCs are most often derived from fibroblasts, mesenchymal-to-epithelial transition (MET) is another critical event that occurs during reprogramming.53
Uncovering the molecular mechanisms of iPSC induction facilitates the development of novel reprogramming approaches and reveals the underlying principles of cell fate transitions and cell fate determination. This knowledge can subsequently be used to design rational strategies for iPSC differentiation towards the desired cell types in an efficient manner. In this section, we focus on the roles of transcription factors as well as chromatin and DNA methylation dynamics in reprogramming.
Transcription factors
OSKM and other transcription factors orchestrate somatic cell reprogramming to pluripotency.54,55 Through concerted action, OSKM expel somatic cell-specific transcription factors from somatic enhancers and activate pluripotency enhancers; silencing of somatic cell-specific enhancers is initiated early in reprogramming, whereas activation of pluripotency-specific enhancers occurs later in reprogramming.56,57 Notably, the chromatin and DNA methylation landscape is restrictive early in reprogramming, requiring pioneering activity of the OSKM factors to access closed chromatin and initiate gene expression.58 Oct4, Klf4, and Sox2 target partial motifs in the nucleosome-enriched loci, indicating their pioneering activity,55 whereas Sox2 has even been proposed to be a super pioneer due to its ability to induce DNA demethylation and overcome repressive epigenome.59 Multiple studies have revealed the dynamics of OSKM binding to DNA and their mode of action. For example, Oct4 dynamics exhibit a hierarchical sequence of events, with Oct4 targeting epigenetically primed states and then maintaining stable DNA occupancy for the duration of reprogramming.56 Mutagenesis-based analysis of Oct4 protein domains has revealed dynamic DNA and nucleosome binding kinetics and highlights the importance of stable Oct4 interactions with nucleosomes to maintain chromatin accessibility of pluripotency enhancers.60 Klf4 facilitates topological enhancer-promoter connectivity and organization required for reprogramming to pluripotency,61 whereas Myc targets open promoter regions to facilitate cell cycle progression.6,62,63 Importantly, OSKM closely cooperate with each other to exert global reprogramming of gene expression, which can be illustrated by the concerted action of OSKM to drive MET: Oct4 and Sox2 suppress Snail expression, Klf4 promotes Chd1 expression (encoding E-cadherin), and Myc suppresses the TGFβ signaling axis.64 In addition to OSKM, multiple other transcription factors play important roles in reprogramming downstream of OSKM and can partially substitute certain OSKM factors.65,66,67,68,69 For example, Klf4 and Sox2 can be substituted by their close homologs,6,70 whereas NKX3-1 or a dominant-negative variant of c-Jun can substitute Oct4.67,71 Notably, certain cell types that endogenously express SKM, such as neural progenitor cells, can be reprogrammed into iPSCs with exogenous expression of Oct4 alone.72,73,74 Overall, transcription factors are the drivers of somatic cell reprogramming to pluripotency that coordinate the rewiring of gene expression as well as the remodeling of chromatin and DNA methylation as discussed next.
Chromatin dynamics and histone remodeling
Chromatin remodeling represents another layer of dynamic changes that occur during reprogramming.44,75,76 Although pioneer transcription factors can access closed chromatin, the ability of non-pioneer transcription factors to exert gene expression programs requires extensive chromatin remodeling. Given that chromatin becomes progressively restricted during cell differentiation to establish somatic cell-specific gene expression programs,43 decompaction and remodeling of chromatin is essential for induction of pluripotency. Chromatin remodeling often precedes changes in gene expression and is required for establishing pluripotency-supporting spatial organization of DNA regulatory elements as well as for enabling access of transcription factors to DNA during reprogramming.45,77 Chromatin remodeling occurs in waves as loci enriched for somatic genes transition from open to closed early in reprogramming, whereas loci enriched for OSK motifs transition from closed to open late in reprogramming.75,78
Chromatin dynamics are highly influenced by nucleosome remodeling and histone modifications that modulate chromatin compaction and transcription factor accessibility to DNA. Nucleosome remodeling factors, such as the NuRD complex and the histone chaperone CAF-1, exert context-dependent regulation of gene expression in somatic cells and during induction of pluripotency.79,80 For example, CAF-1 is required for maintaining somatic cell identity, whereas suppression of CAF-1 facilitates chromatin opening at enhancer regions and promotes Sox2-mediated activation of pluripotency genes.79 Various histone modifiers are also involved in reprogramming; for example, the histone methyltransferase EZH2 is a positive regulator of reprogramming, presumably required to silence somatic cell-specific genes.81 On the other hand, histone methyltransferase DOT1L is a negative regulator of reprogramming because it maintains permissive chromatin in fibroblast-specific genes associated with the epithelial-to-mesenchymal transition.81 Changes in global levels of specific histone modifications have also been documented in reprogramming. For example, H3K9 methylation is depleted in iPSCs, and suppression of the H3K9 reader heterochromatin protein Cbx3 promotes fibroblast reprogramming to pluripotency.82,83 Global remodeling of histone modifications can be driven by metabolic reprogramming during the induction of pluripotency. For example, the transcription factor Glis1 targets glycolytic genes to enhance glycolytic flux during reprogramming, leading to increased production of acetyl-CoA and lactate intermediates required for histone acetylation and lactylation at pluripotency genes.84 Given the roles of histone modifiers in chromatin compaction and reprogramming, small-molecule compounds targeting histone modifiers are often used to promote chromatin decompaction during chemical or transcription factor-mediated reprogramming. For example, the histone deacetylase inhibitor valproic acid as well as the Dot1l inhibitor SGC0946 promote somatic cell reprogramming to pluripotency.40,85,86
DNA methylation
Given the critical role of DNA methylation in establishing epigenetic memory during cell differentiation, active remodeling of DNA methylation is another essential part of reprogramming. In development, DNA cytosine methylation is orchestrated by de novo DNA methyltransferases DNMT3A/B that guide DNA methylation at regulatory regions, thus modulating transcription factor accessibility and downstream gene expression.87,88 During reprogramming, such somatic cell-specific DNA methylation patterns are reversed by active DNA demethylation mediated by ten-eleven translocation (Tet) enzymes.89,90,91 Indeed, waves of global DNA demethylation during reprogramming result in the loss of DNA methylation at regulatory regions that become enriched for 5-hydroxymethylcytosine (5hmC), an intermediate of Tet-mediated DNA demethylation.92,93,94,95 These actions of Tet enzymatic activity not only facilitate pluripotency-specific gene expression, but also drive other events required for reprogramming, including MET.96 Furthermore, Tet enzymes target specific loci to facilitate reprogramming; for example, Tet1 demethylates the endogenous Oct4 locus to reactivate Oct4 expression.97,98 Tet1 can even substitute exogenous Oct4 during reprogramming, indicating a central role for active DNA demethylation in reprogramming to pluripotency.98 Tet enzymes cooperate with pluripotency-specific transcription factors to reactivate pluripotency-specific genes. For example, Nanog physically interacts with Tet1 and Tet2, whereas cooperative binding of Nanog and Tet1 to loci of pluripotency-specific genes primes their expression during reprogramming.97 Tet1 activity is also influenced by exogenous vitamin C, indicating that small-molecule compounds can influence active DNA demethylation and epigenetic remodeling during reprogramming.99 Overall, remodeling of chromatin accessibility and DNA methylation erases somatic cell identity and creates a permissive epigenetic landscape for the pluripotency state during reprogramming.
Population-level dynamics during iPSC induction
The dynamics of cell fate transitions at the population level reveal a stochastic and heterogenous nature of iPSC induction.76 Somatic cells transition through a continuum of reprogramming intermediates that bifurcate into intermediates that will successfully complete reprogramming and those that will acquire an alternative fate.100 Most cells do not complete reprogramming, whereas clonal competition leads to the emergence of dominant clones that overtake the culture during reprogramming.101 Clonal competition is also fueled by the heterogeneity of the starting somatic cell population, the extent of which may be dependent on the somatic cell source.101 There is a great interest in isolating rare intermediates that complete reprogramming more efficiently than do other cells, so that molecular mechanisms governing productive reprogramming could be elucidated.102 For example, rare intermediates that exhibit chromatin hyperaccessibility at pluripotency-specific genes and distinct DNA methylation profiles have been isolated based on the presence of pluripotency-specific surface markers.103 We anticipate that improving high-throughput profiling of gene expression and chromatin accessibility at single cell level will continue to provide new insights into cell fate transitions and reprogramming trajectories during iPSC induction.
Residual somatic cell memory and reprogramming cell source
Although iPSCs resemble primary ESCs in terms of their cellular characteristics and the potential for differentiation into all lineages, limitations associated with reprogramming and persistent features of somatic cell identity render iPSCs distinct. Reprogramming of various somatic cell types reveals persistence of somatic cell transcriptional, DNA methylation, and chromatin accessibility signatures.104,105,106,107 Incomplete removal of somatic cell-specific epigenetic signatures as well as aberrant de novo DNA methylation associated with reprogramming can affect the status and the differentiation potential of iPSCs.105,107,108 Adding small-molecule compounds that target chromatin modifiers to the reprogramming cocktail can facilitate the erasure of the residual chromatin signatures and increase the differentiation potential of iPSCs into alternative lineages.108 On the other hand, persistence of somatic cell-specific epigenetic signatures can be exploited to enhance iPSC differentiation into the desired cell type by deriving iPSCs from the same somatic cell type. For example, iPSCs derived from pancreatic beta cells retain open chromatin signatures at loci important for beta cell identity; consequently, beta cells can be differentiated more efficiently from beta cell-derived iPSCs as compared to non-beta-cell-derived iPSCs.104
The cell source used for reprogramming can also influence the heterogeneity and the mutational burden of the resulting iPSCs. iPSCs derived from skin fibroblasts contain common ultraviolet (UV) light-related mutations and exhibit genomic heterogeneity, likely arising from the already heterogenous fibroblast population of the skin.109 On the contrary, iPSCs derived from peripheral blood mononuclear cells (PBMCs) do not exhibit UV-related damage and may have fewer mutations than do iPSCs derived from skin fibroblasts. Nonetheless, PBMC-derived iPSCs may contain other mutations that are selected for during reprogramming, such as oncogenic mutations in the BCOR gene encoding the BCL-6 corepressor.109 Age-related heteroplasmic variants of mitochondria can also influence the mitochondrial genetic makeup of iPSCs derived from different donors.110 Furthermore, spontaneous mutations that arise in the mitochondrial genome during reprogramming could result in the production of novel immunogenic epitopes; new iPSC-specific mitochondrial DNA mutations have been observed in >70% of iPSC lines.110,111 Overall, iPSCs exhibit increased heterogeneity as compared to ESCs due to persistent somatic cell signatures and mutational burden.112 Such heterogeneity can influence the quality of iPSCs, including their differentiation potential and the immunogenicity of iPSC-derived cellular products, among other features.
Methods of iPSC induction
Since the groundbreaking experiments of fibroblast reprogramming into iPSCs, various approaches to deliver reprogramming factors into somatic cells and induce pluripotency have been developed.113,114,115 Viral vectors carrying OSKM expression cassettes are commonly used for reprogramming due to their high efficiency of infection and the capacity to transduce various somatic cell types.3,4,5,113,115,116,117,118,119 Viral vectors can be classified as either integrating or non-integrating vectors; lentiviral or retroviral delivery of the reprogramming factors leads to their integration into the genome and thus stable expression for iPSC induction.3,4,5 However, viral vector integration into the genome may result in insertional mutagenesis and undesired transgene reactivation beyond the duration of reprogramming. An alternative approach is to use non-integrating viral vectors, such as adenovirus, adeno-associated virus, or Sendai virus.115,119 Non-integrating viral vectors are gradually cleared from proliferating iPSCs, resulting in reprogramming without permanent OSKM integration or disruption of the genome. OSKM factors can also be delivered using non-viral vectors, such as transposons,120,121 episomal plasmids,122,123 mRNA,124 and others.115 For example, plasmid-based episomal vectors are commonly used to derive iPSCs for clinical development; reprogramming efficiency when using episomal vectors is comparable to that of Sendai virus-mediated reprogramming, but the cost is much lower.122,123,125 Somatic cells can also be reprogrammed into iPSCs without OSKM overexpression. Various combinations of miRNAs can be used to activate the endogenous pluripotency gene networks.126,127 For example, human and mouse iPSCs can be derived by overexpression of miR-200c, miR-302s, and miR-369s.127 Alternatively, pluripotency can be induced using a cocktail of small-molecule compounds that modulate various signaling pathways and epigenetic modifiers.128 Small-molecule-based chemical reprogramming is highly attractive due to its simplicity and potential for scalability.128,129,130 Combining transcription factors and small-molecule compounds may further accelerate reprogramming.131,132,133 Overall, the desired method is often selected based on its efficiency, feasibility, safety, and cost.115
It should be noted that new insights into the molecular mechanisms of reprogramming using the methods described above are constantly emerging. For example, chemical reprogramming is associated with distinct cell fate transitions and chromatin accessibility dynamics as compared to transcription factor-mediated reprogramming, but it remains unclear if such differences affect the status of the derived iPSCs.134,135 Furthermore, aberrant Oct4 off-target activity has been linked to changes in gene expression and epigenetic profiles that may alter the iPSC differentiation potential.136 Therefore, newly developed reprogramming methods should be rigorously assessed for their effects on the iPSC status, quality, and differentiation potential.
Applications of iPSCs
Development of the iPSC technology has transformed in vitro research and therapeutic development.2,137 iPSCs can proliferate almost indefinitely and be differentiated into the diversity of human cell types, but with reduced ethical constraints as compared to using human ESCs.138,139 As a result, iPSC-derived cells are widely used for modeling human development and diseases, performing high-throughput drug screening, and developing autologous and allogeneic cell therapies, among other applications. In the rest of the review, we discuss the diverse applications of iPSCs, their key advantages, as well as the limitations that remain to be overcome.
iPSC-derived cellular models
Assembling cellular models of human development and diseases in vitro requires access to large quantities of cells that faithfully recapitulate human biology. Although various primary cell types, such as skin, blood, and cancer cells, can be easily isolated from living donors, other cell types, such as brain and heart cells, are largely unavailable. An alternative approach is to use rodent cells; however, animal models exhibit substantial species divergence and may not recapitulate certain human-specific phenotypes.7,8,9 The iPSC technology can be used to overcome both limitations: iPSCs can be readily differentiated into hard-to-access cell types, whereas their human origin and relevant genetic background enable robust modeling of human biology in vitro.
To date, hundreds of protocols to differentiate iPSCs into various cell types have been developed. This is often achieved by mimicking developmental signaling cues in vitro with relevant proteins and small-molecule compounds or by overexpression of cell fate-determining transcription factors to instruct the desired gene expression programs. Certain cell types, such as neurons or cardiomyocytes, can be differentiated with limited resources and training required in about one week.140,141 Other cell types, such as oligodendrocytes or T cells are more difficult to differentiate and require extensive technical expertise.142,143,144 For example, differentiation of oligodendrocytes, which arise late in human brain development, involves multiple stages, requires several different media formulations, and can take several months.143,144,145 Approaches for uncovering key effectors required for efficient cell differentiation include CRISPR/Cas9-based screens, temporal high-throughput profiling of differentiation trajectories, and comprehensive annotation of transcription factor activity, among others.146,147,148,149 In-depth understanding of developmental trajectories facilitates rational design of differentiation protocols to derive specific cell types and subtypes. For example, hematopoietic lineage cells can be derived by sequential specification of the mesoderm and the hemogenic endothelium to obtain hematopoietic progenitor cells followed by terminal differentiation of lymphoid and myeloid lineages in the presence of relevant cytokines.150,151 Neural cells can be derived by dual SMAD inhibition that promotes neuroectoderm specification and the emergence of neural progenitor cells (Fig. 2a).152,153 Furthermore, various morphogens can be applied to instruct regional identity of the differentiating neural cells to obtain specialized cell subtypes; for example, inhibition of the WNT signaling pathways specifies forebrain identity of neural cells.153 iPSC differentiation can also be considerably accelerated by ectopic expression of cell fate-determining transcription factors. For example, overexpression of six microglia fate-determining transcription factors facilitates rapid differentiation of iPSCs into microglia in as few as 8 days, as compared to several weeks required for microglia differentiation without the use of transcription factors.154

Induced pluripotent stem cell (iPSC)-derived cellular models. The iPSC technology can be applied to derive cellular models of varying complexity, ranging from two-dimensional mono-cultures to three-dimensional multicellular assemblies. Various neural cellular models are shown as an example. a Differentiation of neural progenitor cells (NPCs) from iPSCs is achieved by promoting neuroectoderm specification by dual SMAD inhibition. Subsequently, NPCs can be differentiated into terminal neural lineage cells, such as neurons and astrocytes. b iPSC-derived cells can be maintained in a mono-culture or together with other cell types in a co-culture. Different cell types can also be assembled into an organ-on-a-chip that contains separate compartments and enables modeling of complex tissue architecture. Alternatively, iPSC-derived cells can be transplanted in vivo to expose the cells to a complex tissue environment. c iPSCs can be differentiated into three-dimensional self-organizing organoids that partially resemble endogenous tissue architecture and contain several cells types. Organoids can also be transplanted in vivo to promote their vascularization and maturation. d Different types of organoids can be fused together into assembloids for the study of higher-order tissue interactions, such as long-distance innervation and cell migration
Cellular models of varying complexity can be assembled from iPSC-derived cells (Fig. 2b). A particular cell type can be studied in mono-culture experiments to evaluate the cellular response to experimental perturbations and uncover cell autonomous molecular mechanisms and phenotypes. Due to its simplicity, mono-culture is also often used to perform high-throughput screens, such as CRISPR/Cas9-based screens, high-content imaging, and drug screening.155,156,157 However, the mono-culture environment lacks heterotypic paracrine signaling and cell-cell interactions that are indispensable in vivo. To increase the complexity of iPSC-derived in vitro models, different cell types can be co-cultured together. Co-culture not only enables the study of cell-cell communication, but also promotes cell maturation. For example, co-culturing neurons with astrocytes enhances neuron maturation and survival because astrocytes provide neurotrophic factors required for neuron maintenance.140 Tri-culture of neurons, astrocytes, and microglia further increases the physiological relevance of the in vitro brain model, enabling complex phenotypes to emerge.158,159 Yet, co-culture experiments still lack the three-dimensional (3D) complexity and organization of human tissues. Remarkably, iPSCs have the capacity to self-organize into 3D tissues, known as organoids, if appropriate differentiation conditions are provided (Fig. 2c).10,160,161,162,163,164 Organoids are often comprised of several cell types and partially recapitulate the complexity of human tissues, enabling the study of context-dependent cell function, organogenesis, and organ-specific diseases. The organoid field has grown extensively in recent years, and dozens of protocols have been developed to derive organoids representing major human organs.10,160,161,162,163,164 Importantly, organoids can develop impressive complexity; brain organoids patterned by Sonic hedgehog (SHH) signaling exhibit human-like topographical specification with neocortical, ganglionic eminence, and hypothalamic regions.165 Kidney organoids contain nephron-like segments, including the Bowman’s capsule, proximal tubules, the loop of Henle, and distal convoluted tubules in a continuous arrangement reflective of the human kidney architecture.166 Increasing sophistication of organoid differentiation protocols also enables derivation of organoids resembling specific organ regions. For example, exposure of developing neural organoids to various combinations of patterning morphogens yields cortical,167,168 midbrain,169,170 hippocampal,171 cerebellar,172,173 retinal,174,175,176 and other specialized brain organoids.177,178,179,180 Similarly, fundic and antral gastric organoids recapitulate distinct epithelial lining of the corpus and antrum regions of the stomach, respectively.181,182 Organoid complexity can be further increased by developing multi-lineage organoids or fusing heterotypic organoids to form assembloids (Fig. 2d).183,184,185 For example, multi-lineage neuromuscular organoids contain both neurons and skeletal muscle cells and thus form functional neuromuscular junctions.186 Similarly, fusing cortical organoids with spinal cord organoids and skeletal muscle spheroids results in the formation of corticofugal projections and innervation of the muscle tissue.187
An alternative platform to self-organizing organoids is the organ-on-a-chip (OoC), a biomimetic assembly of tissue-relevant cell types into a microfluidics device to recapitulate certain aspects of tissue architecture.188,189,190,191,192,193,194 OoCs have separate compartments and are constantly perfused, enabling controlled tissue assembly, exposure to shear fluid forces, and separation of culture medium reservoirs. OoCs can be used to model tissue interfaces, such as the blood-brain barrier (BBB)195,196 or the airway epithelium,197 where compartment separation is critical. Assembling iPSC-derived neural cells and brain microvascular endothelial-like cells (BMECs) into a BBB-on-a-chip yields a BBB model that exhibits in vivo-like transendothelial electrical resistance and restricted permeability.198 As a result, the BBB-on-a-chip can be perfused with whole human blood at the BMEC interface without inducing toxicity in the neural cell compartment.198 Microfluidics devices can also be designed to incorporate other functional elements, such as valves to support the mechanical function of cardiac tissue. Fabrication of a microfluidics system with valves has been used to establish an iPSC-derived heart-on-a-chip with unidirectional fluid flow and a closed pressure-volume loop.199 Heart-on-a-chip devices can record various parameters of cardiac function, including contractile dynamics, active force, tension, and electrical properties of the engineered tissue.200
iPSC-derived cells and organoids can also be transplanted in vivo to obtain humanized animal models (Fig. 2c).201,202,203,204,205 In this way, the advantages of iPSC-derived cells, including their human origin and donor-specific genetic background, can be combined with the advantages of animal models, such as their physiological complexity, ability to exhibit cognitive phenotypes, and others. For example, transplantation of iPSC-derived microglia into the mouse brain leads to even distribution of microglia in the brain parenchyma, improved maturation, and long-term survival of microglia.206,207,208,209,210 Similarly, blood vessel organoids form perfusable vascular networks upon transplantation, which is challenging to achieve in vitro.211 Overall, iPSC-derived cellular models of varying complexity can be generated to address specific hypotheses of cellular function, cell-cell interactions, and tissue-level activity.
Maturation of iPSC-derived cells
Differentiation of iPSCs into various cellular models, especially in mono-culture, occurs with limited exposure of the differentiating cells to a physiologically-relevant tissue microenvironment and at an accelerated rate as compared to cell differentiation in vivo. As a result, iPSC-derived cells are often immature, which is a significant limitation of the iPSC technology to disease modeling and cell therapy applications. Immature cells lack complete functionality of their in vivo counterparts and thus may not reveal important phenotypes when used for disease modeling or be as efficacious as primary cells when used in cell therapy. For example, immature iPSC-derived spinal motor neurons exhibit fetal-like signatures, whereas expression of gene networks relevant to amyotrophic lateral sclerosis (ALS) correlates with motor neuron maturation and aging; these observations suggest that immature iPSC-derived neurons may not fully recapitulate ALS pathology.212 Therefore, achieving robust maturation of iPSC-derived cells is an important consideration before downstream applications are pursued.
Somatic cells differentiate and mature in the context of their tissue microenvironment that provides signaling cues, metabolites, and cell-cell contacts required for maturation. Reconstituting a physiologically-relevant environment in vitro can thus promote maturation of iPSC-derived cells. For example, artificial extracellular matrix composed of biomimetic nanofibers enhances cortical neuron morphological and functional maturation.213 Relevant paracrine signaling can also be provided by co-culture experiments, where two or more cell types interact with each other. Co-culture of cardiomyocytes with mesenchymal stem cells promotes myofibril alignment and gap junction formation in cardiomyocytes.214 Such enhanced cardiomyocyte maturation is partially mediated by mesenchymal stem cell secreted extracellular vesicles, highlighting the importance of paracrine cell-cell interactions that would be challenging to replicate using chemically defined cell culture medium alone.214 That cell-cell interactions promote maturation of iPSC-derived cells is also evident in 3D in vitro cellular assemblies, including organoids and OoCs that generally exhibit improved maturation over 2D cellular models. For example, incorporating cardiac fibroblasts into spheroids containing cardiomyocytes and epithelial cells leads to cardiomyocyte-fibroblast coupling via gap junctions as well as enhances sarcomere formation and cardiomyocyte eletrophysiological maturation.215 Similarly, a BBB-on-a-chip exhibits metabolic coupling between neurons and endothelial cells.216 Organoid maturation can be further improved by transplantation in vivo, leading to organoid vascularization, improved nutrient exchange, and exposure to physiologically-relevant systemic factors.217,218,219,220,221,222,223 For example, orthotopically transplanted lacrimal gland organoids functionally mature to produce tear-film proteins and resemble primary human tissue.217
Somatic cells are also exposed to tissue-specific mechanical and environmental conditioning, which may be partially recreated in vitro. Application of mechanical stress to iPSC-derived cardiomyocytes by stretching improves their transcriptional and functional maturation.224,225 Incremental pulsatile stretching also promotes maturation of vascular grafts composed of iPSC-derived smooth muscle cells, leading to increased mechanical strength and minimized dilation of the engineered vessels.226 Fluid shear stress enhances ciliogenesis and maturation of multiciliated airway cells, whereas cardiomyocyte maturation can be further improved by electrical field conditioning.197,200,227 Overall, paracrine signaling and mechanical cues can be readily applied to achieve advanced maturation of iPSC-derived cells.
Ultimately, iPSC-derived cells should faithfully recapitulate the cellular biology and function of their in vivo counterparts to serve as rigorous in vitro models of human development and diseases. Large omics datasets generated from primary human tissues can be used for benchmarking of iPSC-derived cells to determine their maturity and resemblance to primary cells. For example, Shin et al. performed spatial similarity mapping of single-cell transcriptomes of iPSC-derived thalamic organoids and primary human brain tissue, which revealed a strong resemblance of thalamic organoids to the primary thalamus.228 Therefore, efforts to generate multi-omics datasets of primary tissues, such as the Human Cell Atlas Project,229,230 can provide highly valuable data for iPSC-based studies and serve as a reference point for molecular profiles of functionally mature cells and tissues.
Modeling human development with iPSC-derived cells
Given that iPSCs resemble an ESC-like state after reprogramming,39 iPSC differentiation into somatic cells or organoids primarily recapitulates embryonic developmental and fetal-like cell states. Therefore, iPSCs are particularly suitable for modeling early human development. Controlled differentiation of iPSCs recapitulates key events of early embryogenesis, such as epiblast lumenogenesis, bipolar embryonic sac formation, and specification of the primitive streak and primordial germ cells.231,232,233,234 iPSC-derived primordial germ cell-like cells (PGCLCs) exhibit distinct germline-specific transcriptional programs and can be used to study germline development.232,234 Furthermore, differentiation of iPSCs towards presomitic mesoderm recapitulates human somitogenesis and the phenomenon of the segmentation clock.235 Recently, derivation of post-implantation human embryo models from ESCs has been reported.236 We anticipate that iPSCs will soon be applied to derive such sophisticated embryo models as well.237
Although human iPSCs resemble the post-implantation epiblast, they can also be reprogrammed into naïve iPSCs that resemble the pre-implantation epiblast to study human embryogenesis before blastocyst implantation.238,239,240 Derivation of naïve human iPSCs from somatic cells was first reported in 2009 and generally requires a combination of transcription factors and small-molecule compounds that modulate various signaling pathways.240,241,242 Naïve iPSCs can be used to study X chromosome inactivation, dynamics of transposable element regulation, cell fate transitions, extraembryonic lineage differentiation, and other features and events of pre-implantation embryogenesis.240,243,244 Blastoid organoids have been recently developed from naïve iPSCs to study blastocyst development and implantation.245 In addition to naïve iPSCs, trophoblast stem cells can be derived from iPSCs to model placental development.246,247,248
Differentiation of iPSCs into specific cell types reveals the principles of cell type specification and maturation. For example, profiling of dopaminergic neuron differentiation trajectories by single-cell RNA sequencing (scRNA-seq) has indicated an important role for the ASCL1 transcription factor in dopaminergic neuron specification.249 Differentiation of multiple iPSC lines can also be used to conduct population level analyses, such as the quantitative trait loci (QTL) analysis.250 In this way, gene regulatory mechanisms that play important roles in development may be uncovered. The organoid platform can be used to study the development of distinct organs. For example, temporal high-throughput profiling of brain organoid differentiation reveals transcriptional and epigenetic regulomes that orchestrate human brain development and regionalization of different brain areas.251,252 Spinal cord organoids recapitulate certain features of neural tube development by undergoing neurulation-like morphogenesis,253 whereas cardiac organoids co-cultured with epicardial-like cells mimic the envelopment of the myocardium by the epicardium that occurs during heart development.254 Finally, assembloids enable modeling of multi-tissue interactions that shape developmental programs through paracrine signaling and cell migration.255 For example, fusing anterior and posterior gut spheroids leads to the emergence of a hepato-biliary-pancreatic anlage-like structure at the interface of the two spheroids in a process that is regulated by retinoic acid signaling.256 Heterotypic brain assembloids, such as cortico-striatal assembloids, recapitulate interneuron migration that occurs during brain development as well as formation of long-range neuronal projections (Fig. 2d).187,257,258 Overall, modeling development with iPSC-derived cells can provide important insights into human-specific developmental programs and inform cell differentiation approaches for other applications as discussed next.
Modeling human diseases with iPSC-derived cells
The most common application of iPSC-derived cells is disease modeling.2,259,260 A key advantage of the iPSC technology for modeling human diseases is that iPSCs can be derived from somatic cells of patients afflicted with a particular disease and carrying causal disease mutations or genetic risk factors. Such iPSCs with a disease-relevant genetic background are subsequently differentiated into the affected cell types that can reveal disease-specific phenotypes. For example, neurons differentiated from iPSCs of patients with familial Alzheimer’s disease recapitulate amyloid β pathology, tau phosphorylation, and other phenotypes observed in Alzheimer’s disease patients.261,262,263 Alternatively, disease-relevant mutations can be introduced by CRISPR/Cas9-based gene editing, which enables derivation of isogenic disease models.264 Isogenic cell lines can be generated by correcting disease-causing mutations in patient-derived iPSCs to obtain a wild-type control iPSC line.265 The resulting pair of patient-derived iPSCs and corrected control iPSCs shares the same genetic background except for the disease-causing mutation or genetic risk variant.265,266 For example, astrocytes derived from iPSCs of patients with Alexander’s disease reveal disease-specific phenotypes caused by GFAP mutations, whereas isogenic gene-corrected controls exhibit normal cellular function (Fig. 3a). Similarly, iPSC-derived astrocytes that carry the C variant of the rs11136000 SNP of the CLU gene, a known genetic risk factor for Alzheimer’s disease, but not isogenic SNP-corrected controls, negatively affect oligodendrocyte progenitor cell (OPC) proliferation and myelination.267 Using isogenic cell lines limits confounding individual-to-individual variation and may increase the statistical power of in vitro experiments.268 On the other hand, derivation of iPSCs from large cohorts of patients enables genome-wide association studies (GWAS) combined with phenotypic analysis.269 For example, analysis of iPSC-derived cortical neurons derived from a large cohort of Alzheimer’s disease patients reveals single-nucleotide polymorphisms (SNPs) associated with amyloid β production. Similarly, liver organoids derived from multiple donors reveal pleiotropic SNP interactions associated with non-alcoholic steatohepatitis (NASH).269,270 These iPSC cohorts can also be used to perform high-content screening to rapidly detect and compare disease-relevant pathology as well as evaluate therapeutic candidates.271 Establishing iPSC biobanks that contain multiple iPSC lines representing different diseases is thus an important goal for advancing iPSC-based disease modeling.

Disease modeling with iPSC-derived cells. a Genetic diseases, such as Alexander disease (AxD), can be modeled using patient-derived iPSCs that carry disease-causing mutations.144 A tissue biopsy is first taken from a patient with AxD. Somatic cells are reprogrammed into iPSCs, and the GFAP mutations that cause AxD are corrected by gene editing. Patient-derived iPSCs and isogenic corrected controls are then differentiated into astrocytes that express GFAP at high levels. Co-culture of AxD astrocytes with oligodendrocyte progenitor cells (OPCs) reveals impaired OPC proliferation and oligodendrocyte (OL) myelination. Transcriptomic analysis indicates increased expression of the CHI3L1 gene, whereas OPC dysfunction can be partially reversed by CHI3L1 protein depletion. These observations in vitro can be further validated in primary human brain tissues as well as and experiments in vivo. b Sporadic diseases, such as Alzheimer’s disease (AD), can be modeled with patient-derived iPSCs that harbor genetic risk factors; alternatively, iPSC-derived cells can be exposed to non-genetic risk factors to induce disease-relevant pathology. For example, exposure of iPSC-derived brain organoids to human serum mimics the breakdown of the blood-brain barrier and induces AD-like pathology. Brain organoids exposed to neurotoxic serum factors have increased levels of toxic amyloid peptides and hyperphosphorylated tau as well as exhibit impaired neuronal activity. c Infectious diseases, such as COVID-19, can be modeled by exposing iPSC-derived cells and organoids to viral pathogens. iPSC-based models of viral infection can reveal human-specific tropism, mechanisms of entry, and other features of a particular virus
Given the multitude of disease modeling applications using iPSC-derived cells, the breadth of the relevant research could not be covered in a single review article. In the following sections, we consider several diseases that illustrate both the versatility of the iPSC platform as well as the different advantages and limitations of using iPSC-derived disease models. In particular, we discuss iPSC-based modeling of neurodevelopmental, psychiatric, and neurodegenerative diseases that are poorly recapitulated in animal models, require hard-to-access cell types, and can be age-related; cancer initiation that is difficult to study using primary cancer cell models that have already undergone transformation; and COVID-19 that illustrates rapid repurposing of iPSC-based cellular models to study a novel infectious disease during the height of a pandemic.
Modeling neurodevelopmental and psychiatric disorders with iPSC-derived cells
Neurodevelopmental and psychiatric disorders are unique in that their pathogenesis manifests in cognitive changes that can only be studied using animal models that exhibit cognition, whereas in vitro experiments reveal molecular and cellular disease phenotypes only.272,273 However, neurological disorders, especially those that lack clear genetic etiology, cannot be easily recapitulated in animal models due to substantial species divergence and immense complexity of the human brain.274,275,276,277 These limitations have inevitably hindered scientific discovery and therapeutic development for neurological disorders. Nonetheless, iPSC-based cellular models can provide important insights into the pathogenesis of neurological disorders, whereas state-of-the-art technologies, such as brain organoid transplantation in vivo and machine learning, pave the way for studying complex cognitive phenotypes.
Neural cells derived from iPSCs of patients with neurological disorders exhibit impaired cellular function.260,278 For example, cellular models of schizophrenia reveal aberrant proliferation and migration of neural progenitor cells, dysfunctional arborization of cortical interneurons, and impaired astrocyte glutamate uptake.279,280,281,282,283 Neural progenitor cells derived from iPSCs of patients with the autism spectrum disorder (ASD) exhibit increased proliferation and impaired migration, as well as increased DNA damage and dysregulated chromatin accessibility at the molecular level.284,285 Various assays can be used to assess neuronal network connectivity in cell culture, which is used as a proxy for cognitive dysfunction. Synaptic density can be evaluated by immunostaining, whereas electrophysiology experiments, such as multi-electrode array (MEA)-based assays, can be applied to measure neuronal activity.286,287,288 Neuronal cultures derived from iPSCs of patients with schizophrenia exhibit decreased synaptic puncta density, defective glutamatergic synaptic transmission, and molecular phenotypes related to synaptic dysfunction.289,290 On the contrary, neuronal cultures derived from iPSCs of patients with ASD exhibit increased synaptic puncta density and neuronal firing rate, indicating neuronal hyperexcitability.291 Recently, MEA has also been combined with machine learning to create simulated environments, where neural cell cultures perform complex tasks and undergo synaptic remodeling—an in vitro assay for learning.292,293 It will be interesting to determine whether neurons derived from iPSCs of patients with neurological disorders exhibit impaired synaptic remodeling in such simulated environments.
Neurological disorders can also be modeled with brain organoids that can reveal dysfunctional cell-cell interactions and complex disease phenotypes.294,295,296,297 For example, brain organoids derived from iPSCs of patients with Down syndrome or ASD exhibit dysregulated proliferation of neural progenitor cells and aberrant production of inhibitory GABAergic interneurons.298,299 An important advantage of using brain organoids for the study of neurological disorders is their complex electrophysiological phenotypes that emerge as a result of improved neuronal maturation and 3D configuration.300,301 For example, cortical-ganglionic eminence assembloids derived from iPSCs of patients with Rett syndrome exhibit neuronal hyperexcitability and epileptiform-like activity characteristic of Rett syndrome.302 Finally, transplantation of iPSC-derived cells into the rodent brain allows the evaluation of cell behavior in a complex in vivo environment as well as cognitive dysfunction associated with the disease. For example, glial progenitor cells derived from iPSCs of patients with schizophrenia exhibit impaired astrocytic and oligodendrocytic differentiation, premature cell migration into the cortex, and hypomyelination.303 The chimeric mice also exhibit behavioral deficits, such as excessive anxiety, indicating higher-order neuronal network dysfunction.303 A powerful approach of iPSC-based modeling of neurological disorders is whole brain organoid transplantation in vivo, which not only creates a complex physiological milieu for the transplanted human cells, but also preserves human cell-specific organoid environment.218,220,222,223,304,305 Although neurological disorders have successfully been modeled using brain organoids in vitro, one important limitation of the brain organoid technology is their lack of vascularization, leading to poor nutrient and oxygen exchange, cellular stress, necrosis of the organoid core, and incomplete organoid maturation.306 Remarkably, brain organoid transplantation in vivo promotes robust organoid vascularization by the host vasculature and substantially improves organoid characteristics, including neuron maturation and microglia survival.218,220,222,223,304,305 An in vivo brain organoid model of Timothy syndrome reveals abnormal neuronal morphology and increased frequency of excitatory postsynaptic potentials, whereas a model of ASD indicates microglia activation.220,222 Overall, iPSC-derived cellular models of neurological disorders reveal complex molecular, cellular, and electrophysiological disease-related phenotypes.
Modeling neurodegenerative diseases with iPSC-derived cells
A distinct group of neurological disorders are age-related neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, ALS, and others.307,308,309,310 In addition to various mutations and genetic risk factors, aging is a strong risk factor for such diseases and is tightly linked to their molecular mechanisms of progression.311,312,313 However, iPSC-derived cells are fetal-like and do not naturally exhibit aging-associated phenotypes.314,315 Somatic cell reprogramming to iPSCs is associated with cellular rejuvenation, causing the loss of aging-associated phenotypes, which are not restored upon iPSC differentiation.316,317 The lack of aging-associated phenotypes is a major limitation of iPSC-derived cells for disease modeling. Nonetheless, various iPSC-based models of neurodegenerative diseases have been developed, and methods to study age-related events or induce aging-associated phenotypes are emerging (Fig. 4).306,314,318,319

Modeling aging-associated phenotypes with iPSC-derived cells. One important limitation of using iPSC-derived cells to model human diseases is their fetal-like phenotypes and the lack of aging-associated cellular features. The process of somatic cell reprogramming to iPSCs is associated with a nearly complete erasure of aging-associated epigenetic marks and phenotypes. Therefore, various strategies to induce aging-associated phenotypes in iPSC-derived cells have been developed. a Exposure of iPSC-derived cells to compounds that disrupt cellular homeostasis can be used to induce aging-associated phenotypes, such as mitochondrial stress or cellular senescence. For example, rotenone disrupts electron transfer in mitochondria, leading to an increased production of reactive oxygen species that can cause mitochondrial stress, damage other organelles, and induce cellular senescence. b Aging-associated phenotypes can also be induced by ectopic expression of progerin, a truncated variant of lamin A nuclear lamina protein. Progerin causes the Hutchinson-Gilford progeria syndrome, a disease that manifests as accelerated aging due to the disruption of the nuclear lamina. Ectopic expression of progerin is sufficient to induce senescence- and aging-associated phenotypes in iPSC-derived neurons and other cells. c Aging-associated phenotypes are preserved if target cells are derived by direct transdifferentiation without an iPSC intermediate. Primary fibroblasts can be transdifferentiated into neurons that exhibit aging-associated phenotypes and epigenetic age signatures of the fibroblast donor, and can thus be used to study age-related dysfunction of neural cells
A small proportion of cases of age-related neurodegenerative diseases are familial in nature and are driven by genetic mutations. Such causal mutations are highly penetrant and manifest in clear molecular and cellular phenotypes of iPSC-derived cells. For example, cortical neurons carrying mutations in the PSEN1 gene exhibit amyloid β pathology characteristic of Alzheimer’s disease262; dopaminergic neurons carrying mutations in the SNCA gene exhibit α-synuclein aggregation characteristic of Parkinson’s disease320; and motor neurons carrying mutations in the TDP-43 gene exhibit TDP-43 aggregation characteristic of ALS.321 However, most cases of neurodegenerative diseases are sporadic and do not have a clear etiology. Various genetic risk factors for sporadic neurodegenerative diseases have been identified through GWAS, and their subtle contributions to disease progression can be modeled with iPSC-derived cells.266,322,323,324 For example, the E4 variant of the APOE gene is the strongest genetic risk factor for Alzheimer’s disease.324,325,326 Accordingly, iPSC-derived APOE4 neurons, astrocytes, oligodendrocytes, and microglia, all exhibit dysregulated cellular homeostasis and function.266,327,328,329,330,331 Non-genetic effectors originating from outside the brain also influence progression of neurodegenerative diseases. Such effectors include the peripheral immune system that has recently been implicated in neurodegeneration as well as environmental factors, such as neurotoxins.332,333,334,335,336 For example, co-culture of iPSC-derived dopaminergic neurons with isogenic primary T cells isolated from patients with Parkinson’s disease reveals increased neuronal cell death that is mediated by T cell-secreted IL-17.336 Furthermore, exposure of iPSC-derived dopaminergic neurons to a neurotoxin 1-methyl-4-phenylpyridinium (MPP+) leads to increased expression of genes associated with Parkinson’s disease,334 whereas dopaminergic neurons carrying the A53T mutation in the SNCA gene are more susceptible to environmental pesticides than are normal controls.335 Finally, population level studies using large cohorts of iPSCs derived from patients with sporadic neurodegeneration may facilitate identification of novel biomarkers for patient stratification and reveal subtle genotype-phenotype relationships. Efforts to create disease-specific iPSC biobanks are underway; for example, hundreds of iPSC lines from patients with ALS have been established as part of the Answer ALS project.337,338 Interestingly, motor neurons derived from iPSCs of patients with sporadic ALS cluster into distinct groups based on their heterogenous phenotypes, illustrating the application of iPSC-derived cellular models to improve patient stratification.339
The models described above, however, do not incorporate aging-associated disease phenotypes that play a critical role in neurodegenerative diseases. Due to the lack of suitable models, it remains poorly defined how aging interacts with other risk factors to drive neurodegeneration. At the molecular level, aging may be associated with epigenetic erosion and DNA damage that derail homeostatic gene expression programs, resulting in suboptimal cellular phenotypes and cellular senescence.340,341,342,343,344,345 In iPSC-derived cells, aging-associated phenotypes, such as mitochondrial dysfunction, can be induced experimentally to mimic age-related cellular dysfunction (Fig. 4a). For example, iPSC-derived cells can be treated with rotenone that interferes with the mitochondrial electron transport chain, leading to increased production of reactive oxygen species (ROS), mitochondrial damage, and disruption of cellular homeostasis.346,347,348,349 However, it remains unclear whether disrupting one cellular pathway is sufficient to recapitulate aging or whether it is simply a model of cellular stress.315 An alternative strategy to induce aging-associated phenotypes is based on overexpression of progerin, a truncated variant of a nuclear lamina intermediate filament lamin A.317 Progerin is integral in the pathogenesis of Hutchinson-Gilford progeria syndrome (HGPS), a disease that causes premature aging.350 Remarkably, overexpression of progerin in iPSC-derived dopaminergic neurons induces neurite degeneration, neuromelanin accumulation, and aging-associated gene expression.317 Although progerin overexpression can induce various cellular phenotypes associated with aging, it should be noted that HGPS is a distinct disease that may not necessarily recapitulate normal human aging and may exhibit HGPS-specific phenotypes that are irrelevant to neurodegenerative diseases. Therefore, substantial efforts have been made to obtain human brain cell models without erasing aging-associated phenotypes of the somatic cells, from which the neural cells are derived. This aim can be achieved by direct transdifferentation of patient-derived fibroblasts into neurons without an iPSC intermediate (Fig. 4b).351,352,353,354,355,356 Fibroblasts can be transdifferentiated into neurons by overexpression of miRNAs or neuron fate-determining transcription factors, such as NGN2 and ASCL1, combined with a small-molecule treatment.354 Transdifferentiated neurons retain the epigenetic age and aging-associated phenotypes of the fibroblast donor and can be used to study the impact of aging on the pathogenesis of neurodegenerative diseases.316 For example, transdifferentiated neurons derived from fibroblasts of elderly patients with Alzheimer’s disease reveal aberrant neuronal phenotypes, such as Warburg-like metabolic transformation, increased post-mitotic senescence, and hypo-mature neuronal identity, that are not observed in fetal-like iPSC-derived neurons.316,357,358 Finally, iPSC-derived cellular models can also be used to study age-related events by mimicking various cell non-autonomous conditions associated with aging. For example, breakdown of the BBB may be caused by aging and is a common feature of neurodegenerative diseases, leading to leakage of potentially neurotoxic serum components into the neural tissue.359,360,361,362,363 Mimicking the BBB breakdown by exposure of iPSC-derived brain organoids to human serum induces a rapid onset of Alzheimer’s disease-like pathology, including accumulation of amyloid β and phosphorylated tau as well as impaired neuronal activity (Fig. 3b).364 We anticipate that novel approaches to induce aging-associated phenotypes and model age-related events using iPSC-derived cells will provide new insights into both neurodegenerative and other age-related diseases.
Modeling cancer initiation with iPSC-derived cells
Given their proliferative capacity, primary cancer cells derived from tumor biopsies are the most common cellular models for studying tumor cell biology and the response to therapeutic intervention.365,366,367 However, primary cancer cells have already undergone transformation, a key event that governs deregulation of cellular homeostasis and leads to cancer initiation.368 The iPSC technology offers a unique opportunity to study how various somatic mutations and other events rewire molecular and cellular programs of normal cells, so that they are transformed into cancer cells.369 For example, iPSC-derived neural stem cells carrying an H3.3K27M mutant histone H3.3 variant associated with diffuse intrinsic pontine glioma, a type of a juvenile brain tumor, exhibit aberrant gene expression programs that promote neural stem cell proliferation and stemness.370 Similarly, colonic organoids derived from iPSCs of patients carrying mutations in the APC gene associated with familial colorectal cancer exhibit elevated activity of the WNT signaling pathway and higher epithelial cell proliferation as compared to wild-type controls.371 In addition to somatic mutations, environmental factors also play a role in cell transformation. For example, chronic Helicobacter pylori infection is associated with increased incidence of gastric cancer, presumably due to persistent inflammation of the epithelial lining of the stomach.372,373 Injection of H. pylori bacteria into the lumen of iPSC-derived gastric organoids induces a rapid response of epithelial cells, including a twofold increase in cell proliferation.374 Finally, genetic manipulation of iPSCs and their subsequent differentiation into cancer-relevant cell types can be used to establish cancer evolution models that reflect successive acquisition of somatic mutations and clonal expansion of cancer cells. For example, introducing various driver mutations associated with acute myeloid leukemia into iPSCs followed by differentiation of hematopoietic progenitor cells enables modeling of leukemic transformation from premalignant cell states to transplantable leukemia.375 High-throughput profiling of gene expression across the continuum of leukemogenesis reveals distinct molecular pathways, such as dysregulated inflammatory signaling, that promote tumorigenesis.375 Overall, iPSC-based cellular models can provide important insights into molecular and cellular events governing cancer initiation, which may facilitate patient stratification for early screening and cancer prevention.
Modeling COVID-19 with iPSC-derived cells
Modeling viral infection with iPSC-derived cellular models can reveal unique interactions between viruses and human cells (Fig. 3c).288,376,377,378,379 The COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has prompted the scientific community to rapidly repurpose experimental platforms, so that SARS-CoV-2 cellular tropism, molecular mechanisms of entry, life cycle, and SARS-CoV-2 targeting therapeutics could be investigated.380,381,382 Although animal cell lines and models permissive to SARS-CoV-2 have been identified and developed, human iPSC-derived cellular models have the advantage of revealing human-specific SARS-CoV-2 tropism and vulnerabilities.383,384,385,386,387 Therefore, iPSC-based cellular models of SARS-CoV-2 infection have been swiftly applied to study COVID-19, revealing disease-specific phenotypes.388 For example, SARS-CoV-2 infection of iPSC-derived alveolar epithelial type 2 (AT2) cells cultured at air-liquid interface, a model for respiratory tract infection, induces cytotoxicity and a pro-inflammatory phenotype of AT2 cells.389 Co-culture of iPSC-derived macrophages and lung epithelial cells reveals a protective role of macrophages against the SARS-CoV-2 infection of epithelial cells; however, M1 and M2 polarized macrophages exhibit different inflammatory responses.390 Given widespread extrapulmonary manifestations of COVID-19,391,392,393 permissiveness of different tissues and organs to SARS-CoV-2 can be evaluated using tissue-specific organoids.388,394 For example, SARS-CoV-2 infects and productively replicates in salivary gland organoids, indicating the potential role for salivary glands as a reservoir of SARS-CoV-2.221 Similarly, SARS-CoV-2 actively replicates in capillary organoids, which may explain SARS-CoV-2-associated viremia.395,396 SARS-CoV-2 infection also induces cytotoxicity in iPSC-derived cardiomyocytes and cardiospheres, causing myofibrillar disruption, impaired cardiomyocyte beating, and cell death.397,398,399 Neurological manifestations of COVID-19 have also been documented.400,401,402 SARS-CoV-2 infects iPSC-derived neural progenitor cells, neurons, astrocytes, and brain organoids.403,404,405,406 SARS-CoV-2 infection of neural tissues leads to increased tau hyperphosphorylation, a hallmark of Alzheimer’s disease, suggesting that SARS-CoV-2 infection may have long-term neurological effects that could contribute to the onset of neurodegeneration.403,407 Interestingly, the susceptibility of iPSC-derived neurons and astrocytes to SARS-CoV-2 infection is dependent on the APOE variant; APOE4 cells exhibit increased susceptibility to SARS-CoV-2 infection as compared to APOE3 cells.406 Overall, the iPSC technology has been rapidly adapted to investigate human-specific disease phenotypes of COVID-19, providing vital insights into this life-threatening disease.
Drug development using iPSC-derived cells
Various advantages of iPSC-derived cellular models discussed throughout this review are also applicable to drug development applications.408 Given their human origin, iPSC-derived cells can be used as a preclinical platform to test drug efficacy and toxicity as well as uncover human-specific molecular mechanisms of drug action. Various somatic cell types, including those that are inaccessible from primary sources, can be derived from disease-specific iPSCs that harbor relevant causal mutations or genetic risk variants to assess drug efficacy in the context of a specific genetic background. iPSC-based experiments can also be scaled to perform high-throughput drug screening with thousands of small-molecule candidates. For example, Gu et al. performed a survival screen of 4500 compounds based on the caspase 3/7 activity to identify anti-apoptotic compounds that limited death of endothelial cells derived from iPSCs of patients with pulmonary arterial hypertension.409 When combined with high-content imaging technologies, drug screening assays can be used to evaluate complex phenotypes, such as changes to cellular morphology or accumulation of disease-associated protein aggregates.271,410,411 Park et al. developed a high-throughput drug screening pipeline to evaluate amyloid β and tau pathology in brain organoids derived from iPSCs of patients with Alzheimer’s disease.271 In particular, the authors used tissue-clearing techniques and high-content imaging to visualize and quantify the burden of amyloid β and phosphorylated tau upon drug treatment.271 Combining iPSC-based drug screening with computational analyses and machine learning can reveal targetable regulatory nodes associated with a specific disease as well as therapeutic candidates for drug repurposing.412 Taubes et al. performed an in silico drug repurposing analysis to identify candidates that could reverse APOE4-associated gene expression signatures in Alzheimer’s disease.413 Having identified bumetanide as a potential candidate, the authors validated its efficacy in iPSC-derived APOE4 neurons.413 Furthermore, Theodoris et al. used machine learning to identify small-molecule compounds that could reverse aberrant gene expression associated with haploinsufficiency for the NOTCH1 gene in calcific aortic disease.414 The authors screened over 1500 predicted candidates using iPSC-derived endothelial cells and identified an inverse agonist of the estrogen-related receptor α (ERRα) as a potent hit.414
iPSC-derived cellular models can also be used to evaluate drug toxicity, which is a major cause of drug attrition in therapeutic development.415,416 Although preclinical toxicology is based on animal studies, human-specific drug toxicity may not necessarily manifest in animal models, leading to costly drug withdrawals late in the drug development pipeline. Therefore, the iPSC technology can be used as a complementary platform to assess drug toxicity and its human-specific molecular mechanisms.417,418,419 For example, drug nephrotoxicity may be evaluated using iPSC-derived podocytes that form the epithelial lining of the kidney glomerulus.420 A microfluidics-based glomerulus-on-a-chip recapitulates adriamycin-induced podocyte injury and albuminuria.421 Similarly, iPSC-derived 3D cardiac tissues recapitulate doxorubicin-induced cardiotoxicity, leading to disruption of sarcomeres and cessation of beating.422 Evaluating drug toxicity using patient-specific iPSCs may also facilitate precision medicine-driven patient stratification based on individual patient susceptibility to particular therapeutics. For example, transcriptomic analysis of a panel of iPSC-derived cardiomyocytes reveals patient-specific cardiomyocyte susceptibility to oxidative stress associated with decreased expression of the NFE2L2 gene.423 Cardiomyocytes with low NFE2L2 expression are more susceptible to tacrolimus- and rosiglitazone-mediated cardiotoxicity as compared to cardiomyocytes with high NFE2L2 expression.423 Uncovering the mechanisms of drug toxicity can facilitate the development of novel therapeutic strategies to mitigate such toxicity. Sharma et al. found that exposure of iPSC-derived cardiomyocytes to cardiotoxic tyrosine kinase inhibitors leads to compensatory insulin signaling that may be cardioprotective.424 Indeed, adding exogenous insulin or IGF1 improves cardiomyocyte viability in the presence of tyrosine kinase inhibitors.424 Finally, drug toxicity can be elicited by unexpected drug distribution or accumulation in certain human tissues. Drug pharmacokinetics can be assessed in barrier-forming organoids, such as choroid plexus organoids that form fluid-filled cysts and exhibit selective permeability to various drugs.425 Drug absorption and metabolism by the cytochrome P450 (CYP) family enzymes can be evaluated using iPSC-derived intestinal epithelial cells.426 Humanized animal models can also reveal human tissue-specific drug pharmacokinetics and accumulation; for example, transplantation of iPSC-derived kidney organoids into athymic rats has been used to evaluate organoid exposure to systemically administered drugs.427 Overall, the iPSC technology enables complementary evaluation of drug efficacy and toxicity using human-specific models.
iPSC-based cell therapy
Cell therapy has recently emerged as a promising approach to repair or replace damaged tissue as well as engineer immune responses to a disease, such as cancer.428,429,430,431,432,433 The success of adoptive chimeric antigen receptor (CAR) T cell therapy to treat acute lymphoblastic leukemia and large B cell lymphoma has paved the way for developing novel cell therapies, including those based on the iPSC technology.11,434,435,436 Although primary cells, such as T cells, natural killer (NK) cells, and mesenchymal stem cells, can be isolated from a patient and later used as autologous cell therapy, other cell types, such as neurons, cannot be harvested for transplantation. Furthermore, the quality of primary cells may be compromised by a disease or by germline mutations as well as exhibit unwanted heterogeneity. The iPSC technology can be used to overcome these limitations, given that iPSCs can be genetically engineered, clonally expanded, and differentiated into most somatic cell types.11 Furthermore, iPSC-based cell therapy has fewer ethical constraints as compared to ESC-based cell therapy because iPSCs are derived from somatic cells.437,438 Xenotransplantation experiments serve as a proof of principle that transplanted iPSC-derived cells can mitigate disease-associated tissue dysfunction and restore homeostasis. For example, transplanted human iPSC-derived pancreatic islets secrete insulin and control glycemia in diabetic mice439 and macaques.440 Similarly, human iPSC-derived OPCs rescue myelination in myelin-deficient mice upon transplantation, indicating the potential application of OPC-based cell therapy for treating demyelinating white matter disorders.123,145,441 These examples indicate that the iPSC technology can be used to derive hard-to-access cell types and restore normal tissue physiology upon transplantation. As a result, various clinical trials using iPSC-derived cellular products to treat human diseases have been initiated (Table 1).
Autologous iPSC-based cell therapy
iPSC-based cell therapy can be divided into two categories—autologous and allogeneic (Fig. 5). In autologous cell therapy, iPSCs are derived from the same patient who will receive the cell transplant.442,443,444 Autologous cell therapy is meant to prevent immune rejection of the transplant by the recipient because the immune system recognizes the transplanted cells as “self” tissue. A tissue biopsy is first collected from the patient who will undergo autologous cell therapy, and the isolated somatic cells are reprogrammed into iPSCs. These iPSCs can then be genetically modified to correct undesired mutations or introduce new gene expression cassettes. For example, if a patient has a monogenic disease that is caused by a germline mutation, gene correction can be performed. After genetic modification, iPSCs are differentiated into the desired cellular product that will be used for transplantation. Extensive quality control of iPSCs and iPSC-derived cells is required to ensure that the cellular product is functional and does not contain any deleterious or tumorigenic mutations. The feasibility of gene correction-based autologous cell therapy has been demonstrated in preclinical animal models. For example, transplantation of hepatocytes derived from gene-corrected iPSCs of a patient with hereditary antithrombin deficiency leads to normalization of antithrombin levels in the plasma of antithrombin-lacking mice, thus mitigating the thrombophilic state.445 Similarly, transplantation of pancreatic beta cells derived from gene-corrected iPSCs of a patient with monogenic Wolfram syndrome restores normal glucose homeostasis in diabetic mice.446 A detailed example of preclinical development of iPSC-based autologous cell therapy for Canavan disease, a monogenic neurodevelopmental disorder, is shown in Fig. 6.

Autologous and allogeneic iPSC-based cell therapy. In autologous cell therapy, somatic cells are collected from the patient who will receive the cell transplant. The isolated somatic cells are reprogrammed into iPSCs, which can then be genetically engineered to correct disease-associated mutations or introduce new gene expression vectors. Modified iPSCs are differentiated into the cellular product that will be transplanted into the patient and rigorously evaluated for quality. In allogeneic cell therapy, iPSCs are taken from a biobank and genetically engineered for immune cloaking. The resulting hypoimmunogenic universal donor iPSCs can be further genetically modified to introduce cell therapy-specific gene expression vectors, such as a chimeric antigen receptor (CAR) expression cassette, and then differentiated into the desired cell type. After rigorous quality assessment, cellular products can be stocked and distributed as off-the-shelf therapeutics for transplantation into multiple recipients. KO, knockout; KI, knockin

Development of iPSC-based autologous cell therapy. Despite the success of adoptive immune cell therapy, multiple other diseases affect cell types that cannot be easily isolated from patients for genetic engineering and transplantation back into the patient. For example, Canavan disease (CD) is a monogenic autosomal recessive neurological disorder caused by mutations in the aspartoacylase (ASPA) gene. These mutations disrupt ASPA enzymatic activity, leading to the accumulation of N-acetylaspartate (NAA) in the brain and causing spongy degeneration. ASPA enzymatic activity can be restored by transplantation of autologous neural progenitor cells (NPCs) that harbor CRISPR/Cas9-corrected ASPA or ectopically express wild-type ASPA delivered by lentiviral (LV) transduction. a A skin biopsy is obtained from a CD patient, and patient-specific iPSCs are derived from the isolated skin fibroblasts. b iPSCs are genetically engineered to restore wild-type ASPA expression and differentiated into NPCs that will be used for transplantation. c To demonstrate the efficacy of iPSC-derived NPC therapy for CD, preclinical experiments using a CD mouse model (Nur7) can be performed. CD mice exhibit characteristic spongy degeneration with vacuolation, myelin defects, and motor dysfunction. In our studies,122,123,441 we transplanted WT-ASPA-NPCs into the corpus callosum (CC), the subcortical region (SC), and the brainstem (BS) by stereotactic injection. We found that WT-ASPA-NPC-transplanted CD mice exhibited increased ASPA activity and reduced NAA levels, increased myelination and reduced vacuolation, and improved motor function. GMP, good manufacturing practice
Allogeneic iPSC-based cell therapy
In allogeneic cell therapy, iPSCs derived from a universal donor are used for transplantation, circumventing the lengthy and costly process of iPSC production from each patient who will receive the cell transplant (Fig. 5).447,448 The desired cells can be differentiated, characterized, and stocked in advance, so that the cellular product is available on demand or “off-the-shelf” without the need for in-house manufacturing. However, allogeneic cell therapy poses a risk of immune rejection and graft-versus-host disease, requiring additional “immune cloaking” strategies to evade the host immune system (Fig. 7a).449 Commonly used genetic modifications include knockout of the B2M gene, which encodes a component of human leukocyte antigen (HLA, also known as major histocompatibility complex, MHC) class I molecules, to disrupt foreign antigen presentation to cytotoxic CD8+ T cells; knockout of the CIITA gene to disrupt foreign antigen presentation to CD4+ helper T cells; overexpression of the B2M-HLA-E fusion construct to inhibit the “missing-self” response of NK cells; and overexpression of CD47 to provide the “don’t-eat-me” signal to macrophages.449 A combination of such modifications is often used to evade different immune cell types. For example, Wang et al. engineered hypoimmunogenic universal donor iPSCs by knocking out B2M, CIITA, and PVR (encoding a ligand for NK cell activation) as well as overexpressing B2M-HLA-E.450 Hu et al. also knocked out B2M and CIITA but instead overexpressed CD47, having observed that not only macrophages but also most IL-2 stimulated NK cells present the SIRPα receptor of CD47.451 It should be noted that extensive genetic engineering required for immune cloaking can introduce off-target mutations, whereas prolonged iPSC culture and clonal expansion can lead to accumulation of spontaneous genetic aberrations. In our recent study, we knocked out B2M and CIITA and took advantage of endogenously expressed CD47 in OPCs, our cell type of interest, to evade the NK response.441 Therefore, our approach requires two steps of genetic engineering only, reducing the likelihood of undesired mutational events. Having engineered the universal donor cells, their immune evasive properties can be validated in preclinical models. Universal donor cells and primary immune cells from an unrelated donor can be co-cultured together in vitro or co-injected in vivo to evaluate their survival and persistence (Fig. 7b).

Engineering universal donor cells for allogeneic cell therapy. a Universal donor cells are genetically engineered to prevent the host immune response despite their foreign origin. CD8+ cytotoxic T cells recognize foreign cells via their T cell receptor (TCR) that interacts with human leukocyte antigen (HLA) class I molecules presenting unique antigens. If a foreign antigen is presented by the HLA class I molecules, CD8+ cytotoxic T cells initiate destruction of the encountered cell. Knockout of the β2 microglobulin (B2M) gene is sufficient to disrupt the universal donor cell interaction with CD8+ cytotoxic T cells. However, ablation of HLA class I molecules elicits a “missing-self” response by natural killer (NK) cells, leading to cell lysis. Therefore, B2M knockout is often combined with ectopic expression of HLA-E, which interacts with the inhibitory NK cell receptor NKG2A/CD94 to suppress the missing-self response. Knockout of CIITA disrupts foreign antigen presentation to CD4+ T cells via HLA class II molecules. To prevent macrophage-mediated cell killing, CD47 surface protein can be ectopically expressed in universal donor cells. CD47 interacts with the signal-regulatory protein α (SIRPα) and acts as the “don’t-eat-me” signal to suppress macrophage-mediated phagocytosis. b Hypoimmunogenicity of universal donor cells can be tested by performing in vitro and in vivo cytotoxicity assays, in which universal donor cells are mixed with primary immune cells, such as T cells, derived from an unrelated donor. Universal donor cells exhibit increased survival and stable persistence in the presence of primary immune cells of a mismatched donor, indicating successful immune evasion
An alternative approach to prevent immune rejection of allogeneic cell therapy is to establish HLA-homozygous iPSC haplobanks to match the donor-patient genotypes of the main HLA molecules involved in immune rejection.452,453,454,455 Several dozens of iPSC lines are sufficient to cover a large proportion of the population by HLA matching. For example, Yoshida et al. established a clinical-grade HLA haplobank of 27 iPSC lines derived from 7 donors, theoretically covering 40% of the Japanese population for HLA-matched iPSCs.455 Overall, allogeneic cell therapy holds great promise to streamline the production pipeline, but the safety concerns, especially those related to immune rejection, remain to be fully addressed.
Challenges associated with iPSC-based cell therapy
Compared to pharmacological therapy, cell therapy is extremely complex and poses major safety, quality assurance, and logistical challenges, including those specific to iPSC-based therapeutics.456,457 A major concern is the propensity of iPSCs for teratoma formation; it is critical to ensure that undifferentiated iPSCs and stem cell-like intermediates are completely removed from the cellular product that will be transplanted into the patient to prevent tumor formation.458 Residual iPSCs can be removed from differentiated cell cultures by selective elimination of highly proliferative cells using chemotherapeutic drugs, such as doxorubicin,459 or by selective elimination of alkaline phosphatase-positive cells using toxic substrates of alkaline phosphatase.460 Introducing a gene encoding a self-destruction switch can provide an additional safety mechanism to selectively remove transplanted cells if they acquire tumorigenic properties.461 Such self-destruction systems include inducible activation of apoptosis, expression of enzymes that can convert non-toxic substrates into toxic compounds, and expression of surface receptors that can be targeted by infusion of monoclonal antibodies.461 As discussed earlier, iPSCs can also exhibit higher intrinsic genetic heterogeneity as compared to ESCs, and acquire mutations during reprogramming, prolonged culture, and gene editing.111,462,463 Such mutations may confer tumorigenic potential or lead to the emergence of novel immunogenic epitopes. Therefore, genetic analysis may be required at different stages of iPSC preparation to ensure that the cellular product is free of deleterious mutations.
Incomplete maturation of iPSC-derived cells remains a major hurdle in developing efficacious cell therapies. For example, iPSC-derived CAR T cells are often not as functional as CAR T cells derived from primary T cells, which may limit their tumor cell killing ability and persistence.431,464 Various approaches to improve iPSC differentiation and maturation protocols for cell therapy applications are under active investigation. For example, T cells can be differentiated using hematopoietic or thymic organoids that mimic the in vivo environment of the developing T cells.465,466,467 Challenges associated with efficacy of iPSC-based cell therapy for solid tissues include poor transplant engraftment and limited therapeutic response. Systemic infusion of cellular therapeutics may not be sufficient to establish a solid organ graft or may result in off-target engraftment.456 For example, intrasplenic infusion of iPSC-derived hepatocytes leads to their engraftment into various organs, including the liver, stomach, spleen, and large intestine.468 Engraftment can be controlled by using biomimetic scaffolds to differentiate cells as structured assemblies, followed by their direct transplantation into the recipient organ. Transplantation of iPSC-derived hepatocytes as a cell sheet generated using a supportive membrane promotes successful liver engraftment with no cells detected in other organs.468 Biodegradable scaffolds also promote integration and improve functionality of iPSC-derived retinal pigment epithelium patches as compared to epithelial cells cultured and transplanted without a scaffold.469 Similarly, bio-ink polymers with favorable rheological properties support osteogenic differentiation of iPSC-derived mesenchymal stromal cells and promote repair of cranial defects upon transplantation into a mouse model of cranial injury.470 Combination therapy can also improve the efficacy of iPSC-based cell therapy via synergistic mechanisms. For example, a combination therapy of iPSC-derived NK cells and anti-PD-1 immunotherapy synergize to kill tumor cells.471 Similarly, a combination therapy of the neurotrophic factor GDNF and iPSC-derived dopaminergic neurons to treat Parkinson’s disease results in brain-wide dopaminergic neuron innervation in a rat model, whereas transplantation of dopaminergic neurons alone is associated with poor long-distance innervation.472
Logistics, reproducibility, and the overall cost of iPSC-based cell therapies should also be considered. Logistical challenges include manufacturing and quality assurance of iPSC-based cell therapies.457 Off-the-shelf iPSC-derived cellular products for allogeneic cell therapy can be generated and distributed in a centralized manner, whereas autologous cell therapies might require hospital-affiliated personnel and facilities to routinely generate cellular products compliant with good manufacturing practices (GMP).473 Reproducibility and consistency of iPSC-derived cellular products can be improved by automating cell culture with liquid-handling robots, whereas large-scale differentiation of iPSCs can be achieved by using bioreactors. Stirred-tank bioreactors enable the scaling of suspension culture as well as monitoring of cell growth and various biophysical parameters, such as pH.474,475 Automation as well as optimization of iPSC derivation, maintenance, and differentiation protocols can also reduce the overall costs of iPSC-based cell therapies. For example, developing growth factor-free media formulations that do not require costly recombinant proteins could make iPSC maintenance more cost-effective.476 Although various challenges remain to be overcome, iPSC-based cell therapy holds great promise to restore tissue homeostasis and function in a way that cannot be achieved with pharmacological therapy.
Concluding remarks and future perspectives
Since its development less than two decades ago, the iPSC platform has opened new frontiers for scientific discovery and therapeutic development. The study of somatic cell reprogramming has revealed immense complexity of cellular transformation that occurs during the induction of the pluripotent stem cell state and encompasses both deterministic and stochastic elements.6 These mechanisms have shed light on the central role of transcription factors in orchestrating gene expression programs, the importance of epigenetic regulation of cell fate, and the cooperative nature of different effectors of reprogramming. With increasing understanding of reprogramming mechanisms, novel methods for efficient and cost-effective derivation of iPSCs continue to emerge. For example, recent reports of fully chemical iPSC derivation methods hold promise for the development of fully defined, scalable, and rapid somatic cell reprogramming protocols.128,129,130
As in vitro models of human development, iPSCs and iPSC-derived cells have been used to investigate the principles and mechanisms of cell fate transitions, self-organization, and developmental disorders. Furthermore, iPSC-based cellular models for numerous other diseases, ranging from genetic to sporadic and age-related disorders, enable the study of human-specific disease mechanisms and the testing of potential therapeutic candidates in vitro.2 Sophisticated cellular models, including organs-on-a-chip, organoids, assembloids, and others, can be used to study higher-order tissue architecture, compartmentalization, and long-range interactions in human development and diseases.10,160,163,188,477 These advanced models of human tissues can also be used to evaluate drug efficacy, toxicity, and pharmacokinetics, thus serving as an additional preclinical platform for drug screening.408 We anticipate that the complexity and functional maturation of iPSC-derived cells and tissues will continue to improve and will reveal yet unappreciated mechanisms and phenotypes of human biology. For example, emerging methods for brain organoid transplantation and vascularization pave the way for obtaining highly functional and mature human cell-based neural tissues that can integrate into the host circuitry and influence animal behavior.218,220,222 Such models enable the study of neuronal network connectivity and its dysfunction in human-specific neurodevelopmental disorders that are challenging to reproduce in preclinical models.
Finally, the promise of the iPSC-based cell therapy has substantially materialized in the past decade, with numerous preclinical studies and early-stage clinical trials being conducted across the spectrum of human diseases (Table 1).11 These efforts are focused on various cancers, for which autologous and allogeneic iPSC-based immune cell therapies are being developed, genetic developmental disorders that require cell transplantation to restore tissue homeostasis, and even sporadic age-related diseases to replace degenerating tissues. Of notable interest are allogeneic cell therapies that utilize universal donor cells engineered to evade immune rejection.448 Universal donor cells can be prepared, characterized, and stocked in advance, considerably simplifying the manufacturing pipeline and reducing the turnaround time. Although important challenges associated with iPSC-based cell therapy remain to be resolved, the technology holds great promise to alleviate human diseases.
The technological advances that evolve alongside the iPSC technology offer new opportunities to define molecular mechanisms of iPSC induction, optimize protocols of iPSC differentiation into somatic cells, develop sophisticated drug screening platforms, and create efficacious cell therapies. We anticipate that improving technologies, such as microscopy tools,478,479 multiomics,480 CRISPR/Cas9-based studies of gene and protein function,481,482,483,484 epigenetic engineering,485,486,487,488 machine learning algorithms,489,490,491,492 and others, will provide new insights into the molecular events that govern somatic cell reprogramming to pluripotency and iPSC differentiation into terminal somatic cell types. The study of human development and diseases using iPSC-based models will benefit from enhanced collaboration, including the development of deeply characterized benchmark iPSC lines493 as well as ethnically diverse iPSC biobanks.494 Automation of iPSC differentiation into somatic cells and organoids will increase reproducibility of in vitro studies required for rigorous high-throughput applications, including drug screening.495 Finally, improving iPSC differentiation and maturation protocols will enable derivation of efficacious cellular products for therapeutic development, whereas production of entire iPSC-derived organs may be possible by chimeric organogenesis.496,497,498 Overall, the iPSC technology will continue to propel fundamental research and therapeutic development to accelerate scientific discovery and relieve human diseases.
References
Rowe, R. G. & Daley, G. Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 20, 377–388 (2019).
Shi, Y., Inoue, H., Wu, J. C. & Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130 (2017).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Yu, J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007).
Takahashi, K. & Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 17, 183–193 (2016).
Breschi, A., Gingeras, T. R. & Guigo, R. Comparative transcriptomics in human and mouse. Nat. Rev. Genet. 18, 425–440 (2017).
Gharib, W. H. & Robinson-Rechavi, M. When orthologs diverge between human and mouse. Brief. Bioinform. 12, 436–441 (2011).
Lynch, V. J. Use with caution: developmental systems divergence and potential pitfalls of animal models. Yale J. Biol. Med. 82, 53–66 (2009).
Takebe, T. & Wells, J. M. Organoids by design. Science 364, 956–959 (2019).
Yamanaka, S. Pluripotent Stem Cell-based Cell Therapy- Promise And Challenges. Cell Stem Cell 27, 523–531 (2020).
Gurdon, J. B. The generation of diversity and pattern in animal development. Cell 68, 185–199 (1992).
Kiefer, J. C. Epigenetics in development. Dev. Dyn. 236, 1144–1156 (2007).
Tompkins, J. D. Discovering DNA methylation, the history and future of the writing on DNA. J. Hist. Biol. 55, 865–887 (2022).
Roe, S. A. Matter, life, and generation: eighteen-century embryology and the Haller-Wolff Debate, (Cambridge University Press, 1981).
Kilgour, F. G. William Harvey and his contributions. Circulation 23, 286–296 (1961).
Aulie, R. P. Caspar Friedrich Wolff and his ‘Theoria generationis’, 1759. J. Hist. Med. Allied Sci. 16, 124–144 (1961).
Weismann, A. Das Keimplasma; eine Theorie der Vererbung, (Jena, Fischer, 1892).
Waddington, C. H. The Strategy of the Genes; A Discussion of Some Aspects of Theoretical Biology, (Cambridge: Cambridge University Press, 1957).
Nanney, D. L. Epigenetic control systems. Proc. Natl. Acad. Sci. USA 44, 712–717 (1958).
Gurdon, J. B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 10, 622–640 (1962).
Gurdon, J. B. The transplantation of nuclei between two species of Xenopus. Dev. Biol. 5, 68–83 (1962).
Gurdon, J. B. Adult frogs derived from the nuclei of single somatic cells. Dev. Biol. 4, 256–273 (1962).
Gurdon, J. B. Multiple genetically identical frogs. J. Hered. 53, 5–9 (1962).
Gurdon, J. B., Elsdale, T. R. & Fischberg, M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature 182, 64–65 (1958).
Jeltsch, A. & Jurkowska, R. Z. New concepts in DNA methylation. Trends Biochem. Sci. 39, 310–318 (2014).
Riggs, A. D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 14, 9–25 (1975).
Robertson, K. D. & Wolffe, A. P. DNA methylation in health and disease. Nat. Rev. Genet. 1, 11–19 (2000).
Schubeler, D. Function and information content of DNA methylation. Nature 517, 321–326 (2015).
Evans, M. J. & Kaufman, M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981).
Martin, G. R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 78, 7634–7638 (1981).
Thomson, J. A. et al. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998).
Tada, M., Takahama, Y., Abe, K., Nakatsuji, N. & Tada, T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 11, 1553–1558 (2001).
Cowan, C. A., Atienza, J., Melton, D. A. & Eggan, K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 309, 1369–1373 (2005).
Davis, R. L., Weintraub, H. & Lassar, A. B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 (1987).
Halder, G., Callaerts, P. & Gehring, W. J. Induction of ectopic eyes by targeted expression of the eyeless gene in drosophila. Science 267, 1788–1792 (1995).
Kulessa, H., Frampton, J. & Graf, T. Gata-1 reprograms Avian Myelomonocytic cell-lines into Eosinophils, Thromboblasts, and Erythroblasts. Gene Dev. 9, 1250–1262 (1995).
Xie, H., Ye, M., Feng, R. & Graf, T. Stepwise reprogramming of B cells into macrophages. Cell 117, 663–676 (2004).
Wernig, M. et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324 (2007).
Huangfu, D. W. et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 26, 795–797 (2008).
Huangfu, D. et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 26, 1269–1275 (2008).
Hou, P. et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341, 651–654 (2013).
Zhu, J. et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654 (2013).
Apostolou, E. & Hochedlinger, K. Chromatin dynamics during cellular reprogramming. Nature 502, 462–471 (2013).
Apostolou, E. & Stadtfeld, M. Cellular trajectories and molecular mechanisms of iPSC reprogramming. Curr. Opin. Genet. Dev. 52, 77–85 (2018).
Cacchiarelli, D. et al. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell 162, 412–424 (2015).
Nefzger, C. M. et al. Cell type of origin dictates the route to pluripotency. Cell Rep. 21, 2649–2660 (2017).
Borkent, M. et al. A serial shRNA screen for roadblocks to reprogramming identifies the protein modifier SUMO2. Stem Cell Rep. 6, 704–716 (2016).
Buckley, S. M. et al. Regulation of Pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell 11, 783–798 (2012).
Qin, H. et al. Systematic identification of barriers to human iPSC generation. Cell 158, 449–461 (2014).
Simic, M. S. et al. Transient activation of the UPR(ER) is an essential step in the acquisition of pluripotency during reprogramming. Sci. Adv. 5, eaaw0025 (2019).
Wu, Y. et al. Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv. 5, eaax7525 (2019).
Pei, D. Q., Shu, X. D., Gassama-Diagne, A. & Thiery, J. P. Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 21, 44–53 (2019).
Soufi, A., Donahue, G. & Zaret, K. S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012).
Soufi, A. et al. Pioneer transcription factors target partial DNA Motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568 (2015).
Chen, J. et al. Hierarchical Oct4 binding in concert with primed epigenetic rearrangements during somatic cell reprogramming. Cell Rep. 14, 1540–1554 (2016).
Chronis, C. et al. Cooperative binding of transcription factors orchestrates reprogramming. Cell 168, 442–459.e420 (2017).
Zaret, K. S. & Carroll, J. S. Pioneer transcription factors: establishing competence for gene expression. Gene Dev. 25, 2227–2241 (2011).
Vanzan, L. et al. High throughput screening identifies SOX2 as a super pioneer factor that inhibits DNA methylation maintenance at its binding sites. Nat. Commun. 12, 3337 (2021).
Roberts, G. A. et al. Dissecting OCT4 defines the role of nucleosome binding in pluripotency. Nat. Cell Biol. 23, 834–845 (2021).
Di Giammartino, D. C. et al. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat. Cell Biol. 21, 1179–1190 (2019).
Rahl, P. B. et al. c-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010).
Garcia-Gutierrez, L., Delgado, M. D. & Leon, J. MYC oncogene contributions to release of cell cycle brakes. Genes 10, 244 (2019).
Smith, Z. D., Sindhu, C. & Meissner, A. Molecular features of cellular reprogramming and development. Nat. Rev. Mol. Cell Biol. 17, 139–154 (2016).
Deng, W., Jacobson, E. C., Collier, A. J. & Plath, K. The transcription factor code in iPSC reprogramming. Curr. Opin. Genet. Dev. 70, 89–96 (2021).
Hernandez, C. et al. Dppa2/4 facilitate epigenetic remodeling during reprogramming to pluripotency. Cell Stem Cell 23, 396–411.e398 (2018).
Liu, J. et al. The oncogene c-Jun impedes somatic cell reprogramming. Nat. Cell Biol. 17, 856–867 (2015).
Markov, G. J. et al. AP-1 is a temporally regulated dual gatekeeper of reprogramming to pluripotency. Proc. Natl. Acad. Sci. USA 118, e2104841118 (2021).
Silva, J. et al. Nanog is the gateway to the pluripotent ground state. Cell 138, 722–737 (2009).
Nakagawa, M. et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106 (2008).
Mai, T. et al. NKX3-1 is required for induced pluripotent stem cell reprogramming and can replace OCT4 in mouse and human iPSC induction. Nat. Cell Biol. 20, 900–908 (2018).
Kim, J. B. et al. Direct reprogramming of human neural stem cells by OCT4. Nature 461, 649–653 (2009).
Kim, J. B. et al. Oct4-induced pluripotency in adult neural stem cells. Cell 136, 411–419 (2009).
Radzisheuskaya, A. & Silva, J. C. Do all roads lead to Oct4? the emerging concepts of induced pluripotency. Trends Cell Biol. 24, 275–284 (2014).
Li, D. et al. Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell 21, 819–833.e816 (2017).
Xing, Q. R. et al. Diversification of reprogramming trajectories revealed by parallel single-cell transcriptome and chromatin accessibility sequencing. Sci. Adv. 6, eaba1190 (2020).
Stadhouders, R. et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 50, 238–249 (2018).
Knaupp, A. S. et al. Transient and permanent reconfiguration of chromatin and transcription factor occupancy drive reprogramming. Cell Stem Cell 21, 834–845.e836 (2017).
Cheloufi, S. et al. The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218–224 (2015).
dos Santos, R. L. et al. MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 15, 102–110 (2014).
Onder, T. T. et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 483, 598–602 (2012).
Chen, J. et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 45, 34–42 (2013).
Sridharan, R. et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nat. Cell Biol. 15, 872–882 (2013).
Li, L. P. et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade (vol 2, pg 882, 2020). Nat. Metab. 2, 1179–1179 (2020).
Tran, K. A. et al. Defining reprogramming checkpoints from single-cell analyses of induced pluripotency. Cell Rep. 27, 1726–1741.e1725 (2019).
Sun, G., Fu, C., Shen, C. & Shi, Y. Histone deacetylases in neural stem cells and induced pluripotent stem cells. J. Biomed. Biotechnol. 2011, 835968 (2011).
Yin, Y. et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239 (2017).
Lyko, F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92 (2018).
Pastor, W. A., Aravind, L. & Rao, A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 14, 341–356 (2013).
Piccolo, F. M. & Fisher, A. G. Getting rid of DNA methylation. Trends Cell Biol. 24, 136–143 (2014).
Rasmussen, K. D. & Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750 (2016).
Caldwell, B. A. et al. Functionally distinct roles for TET-oxidized 5-methylcytosine bases in somatic reprogramming to pluripotency. Mol. Cell 81, 859–869.e858 (2021).
Doege, C. A. et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 488, 652–655 (2012).
Sardina, J. L. et al. Transcription factors drive Tet2-mediated enhancer demethylation to reprogram cell fate. Cell Stem Cell 23, 727–741.e729 (2018).
Zviran, A. et al. Deterministic somatic cell reprogramming involves continuous transcriptional changes governed by Myc and epigenetic-driven modules. Cell Stem Cell 24, 328–341.e329 (2019).
Hu, X. et al. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 14, 512–522 (2014).
Costa, Y. et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374 (2013).
Gao, Y. W. et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA Methylation and Hydroxymethylation in reprogramming. Cell Stem Cell 12, 453–469 (2013).
Chen, J. et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 45, 1504–1509 (2013).
Guo, L. et al. Resolving cell fate decisions during somatic cell reprogramming by single-cell RNA-Seq. Mol. Cell 73, 815–829.e817 (2019).
Shakiba, N. et al. Cell competition during reprogramming gives rise to dominant clones. Science 364, eaan0925 (2019).
Francesconi, M. et al. Single cell RNA-seq identifies the origins of heterogeneity in efficient cell transdifferentiation and reprogramming. Elife 8, e41627 (2019).
Schwarz, B. A. et al. Prospective Isolation of Poised iPSC intermediates reveals principles of cellular reprogramming. Cell Stem Cell 23, 289–305.e285 (2018).
Bar-Nur, O., Russ, H. A., Efrat, S. & Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 9, 17–23 (2011).
Kim, K. et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 29, 1117–1119 (2011).
Marchetto, M. C. et al. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS One 4, e7076 (2009).
Ohi, Y. et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat. Cell Biol. 13, 541–549 (2011).
Kim, K. et al. Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290 (2010).
Rouhani, F. J. et al. Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat. Genet. 54, 1406–1416 (2022).
Wei, W., Gaffney, D. J. & Chinnery, P. F. Cell reprogramming shapes the mitochondrial DNA landscape. Nat. Commun. 12, 5241 (2021).
Deuse, T. et al. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat. Biotechnol. 37, 1137–1144 (2019).
Narsinh, K. H. et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J. Clin. Investig. 121, 1217–1221 (2011).
Malik, N. & Rao, M. S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 997, 23–33 (2013).
Manzini, S., Viiri, L. E., Marttila, S. & Aalto-Setala, K. A comparative view on easy to deploy non-integrating methods for patient-specific iPSC production. Stem Cell Rev. Rep. 11, 900–908 (2015).
Scesa, G., Adami, R. & Bottai, D. iPSC preparation and epigenetic memory: does the tissue origin matter? Cells 10, 1470 (2021).
Macarthur, C. C. et al. Generation of human-induced pluripotent stem cells by a nonintegrating RNA Sendai virus vector in feeder-free or xeno-free conditions. Stem Cells Int. 2012, 564612 (2012).
Seki, T. et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 7, 11–14 (2010).
Zhou, W. B. & Freed, C. R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27, 2667–2674 (2009).
Haridhasapavalan, K. K. et al. An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 686, 146–159 (2019).
Kaji, K. et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458, 771–775 (2009).
Woltjen, K. et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770 (2009).
Chao, J. et al. Therapeutic development for Canavan disease using patient iPSCs introduced with the wild-type ASPA gene. iScience 25, 104391 (2022).
Feng, L. et al. Cell-based therapy for canavan disease using human iPSC-Derived NPCs and OPCs. Adv. Sci. 7, 2002155 (2020).
Warren, L. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630 (2010).
Wen, W. et al. Enhanced generation of integration-free iPSCs from human adult peripheral blood mononuclear cells with an optimal combination of episomal vectors. Stem Cell Rep. 6, 873–884 (2016).
Anokye-Danso, F. et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388 (2011).
Miyoshi, N. et al. Reprogramming of mouse and human cells to pluripotency using mature MicroRNAs. Cell Stem Cell 8, 633–638 (2011).
Kim, Y., Jeong, J. & Choi, D. Small-molecule-mediated reprogramming: a silver lining for regenerative medicine. Exp. Mol. Med. 52, 213–226 (2020).
Liuyang, S. et al. Highly efficient and rapid generation of human pluripotent stem cells by chemical reprogramming. Cell Stem Cell 30, 450–459.e459 (2023).
Guan, J. et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 605, 325–331 (2022).
Li, W. et al. Identification of Oct4-activating compounds that enhance reprogramming efficiency. Proc. Natl. Acad. Sci. USA 109, 20853–20858 (2012).
Zhu, S. et al. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 7, 651–655 (2010).
Lin, T. et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 6, 805–808 (2009).
Cao, S. et al. Chromatin accessibility dynamics during chemical induction of pluripotency. Cell Stem Cell 22, 529–542.e525 (2018).
Zhao, Y. et al. A XEN-like state bridges somatic cells to pluripotency during chemical reprogramming. Cell 163, 1678–1691 (2015).
Velychko, S. et al. Excluding Oct4 from Yamanaka cocktail unleashes the developmental potential of iPSCs. Cell Stem Cell 25, 737–753.e734 (2019).
Shi, Y. Induced pluripotent stem cells, new tools for drug discovery and new hope for stem cell therapies. Curr. Mol. Pharm. 2, 15–18 (2009).
Lo, B. & Parham, L. Ethical issues in stem cell research. Endocr. Rev. 30, 204–213 (2009).
Robertson, J. A. Human embryonic stem cell research: ethical and legal issues. Nat. Rev. Genet. 2, 74–78 (2001).
Fernandopulle, M. S. et al. Transcription factor–mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell Biol. 79, e51 (2018).
Lin, Y. & Zou, J. Differentiation of cardiomyocytes from human pluripotent stem cells in fully chemically defined conditions. STAR Protoc. 1, 100015 (2020).
Iriguchi, S. et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 12, 430 (2021).
Douvaras, P. & Fossati, V. Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat. Protoc. 10, 1143–1154 (2015).
Li, L. et al. GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and Myelination in an hiPSC model of alexander disease. Cell Stem Cell 23, 239–251.e236 (2018).
Wang, S. et al. Human iPSC-derived oligodendrocyte progenitor cells can Myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–264 (2013).
Hurley, K. et al. Reconstructed single-cell fate trajectories define lineage plasticity windows during differentiation of human PSC-derived distal lung progenitors. Cell Stem Cell 26, 593–608.e598 (2020).
Joung, J. et al. A transcription factor atlas of directed differentiation. Cell 186, 209–229.e226 (2023).
Li, Q. V. et al. Genome-scale screens identify JNK-JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat. Genet. 51, 999–1010 (2019).
Washer, S. J. et al. Single-cell transcriptomics defines an improved, validated monoculture protocol for differentiation of human iPSC to microglia. Sci. Rep. 12, 19454 (2022).
Zheng, H. et al. Generating hematopoietic cells from human pluripotent stem cells: approaches, progress and challenges. Cell Regen. 12, 31 (2023).
Pratumkaew, P., Issaragrisil, S. & Luanpitpong, S. Induced pluripotent stem cells as a tool for modeling hematologic disorders and as a potential source for cell-based therapies. Cells 10, 3250 (2021).
Chambers, S. M. et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280 (2009).
Qi, Y. et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 35, 154–163 (2017).
Drager, N. M. et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nat. Neurosci. 25, 1149–1162 (2022).
Leng, K. et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat. Neurosci. 25, 1528–1542 (2022).
Tian, R. et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 24, 1020–1034 (2021).
Tian, R. et al. CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 104, 239–255.e212 (2019).
Guttikonda, S. R. et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 24, 343–354 (2021).
Park, J. et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951 (2018).
Kim, J., Koo, B. K. & Knoblich, J. A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584 (2020).
Schutgens, F. & Clevers, H. Human organoids: tools for understanding biology and treating diseases. Annu Rev. Pathol. 15, 211–234 (2020).
Hofer, M. & Lutolf, M. P. Engineering organoids. Nat. Rev. Mater. 6, 402–420 (2021).
Corsini, N. S. & Knoblich, J. A. Human organoids: new strategies and methods for analyzing human development and disease. Cell 185, 2756–2769 (2022).
Rossi, G., Manfrin, A. & Lutolf, M. P. Progress and potential in organoid research. Nat. Rev. Genet. 19, 671–687 (2018).
Cederquist, G. Y. et al. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 37, 436–444 (2019).
Morizane, R. et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 33, 1193–1200 (2015).
Bershteyn, M. et al. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial Glia. Cell Stem Cell 20, 435–449.e434 (2017).
Qian, X. et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 26, 766–781.e769 (2020).
Abbott, J. et al. Generation and characterization of NGLY1 patient-derived midbrain organoids. Front Cell Dev. Biol. 11, 1039182 (2023).
Sabate-Soler, S. et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 70, 1267–1288 (2022).
Jacob, F. et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal SARS-CoV-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell 27, 937–950.e939 (2020).
Ballabio, C. et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 11, 583 (2020).
van Essen, M. J. et al. PTCH1-mutant human cerebellar organoids exhibit altered neural development and recapitulate early medulloblastoma tumorigenesis. Dis. Model Mech. 17, dmm050323 (2024).
Gabriel, E. et al. Human brain organoids assemble functionally integrated bilateral optic vesicles. Cell Stem Cell 28, 1740–1757.e1748 (2021).
Gagliardi, G. et al. Characterization and transplantation of CD73-positive photoreceptors isolated from human iPSC-derived retinal organoids. Stem Cell Rep. 11, 665–680 (2018).
Lane, A. et al. Modeling and rescue of RP2 Retinitis Pigmentosa using iPSC-derived retinal organoids. Stem Cell Rep. 15, 67–79 (2020).
Del Dosso, A., Urenda, J. P., Nguyen, T. & Quadrato, G. Upgrading the physiological relevance of human brain organoids. Neuron 107, 1014–1028 (2020).
Di Lullo, E. & Kriegstein, A. R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 18, 573–584 (2017).
Cerneckis, J. & Shi, Y. Myelin organoids for the study of Alzheimer’s disease. Front. Neurosci. 17, 1283742 (2023).
Feng, L. et al. Developing a human iPSC-derived three-dimensional myelin spheroid platform for modeling myelin diseases. iScience 26, 108037 (2023).
Broda, T. R., McCracken, K. W. & Wells, J. M. Generation of human antral and fundic gastric organoids from pluripotent stem cells. Nat. Protoc. 14, 28–50 (2019).
McCracken, K. W. et al. Wnt/beta-catenin promotes gastric fundus specification in mice and humans. Nature 541, 182–187 (2017).
Kanton, S. & Pasca, S. P. Human assembloids. Development 149, dev201120 (2022).
Pasca, S. P. Assembling human brain organoids. Science 363, 126–127 (2019).
Pasca, S. P. et al. A nomenclature consensus for nervous system organoids and assembloids. Nature 609, 907–910 (2022).
Martins, J. M. F. et al. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell 26, 172–186.e176 (2020).
Andersen, J. et al. Generation of functional Human 3D Cortico-Motor Assembloids. Cell 183, 1913–1929.e1926 (2020).
Leung, C. M. et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Prim. 2, 33 (2022).
Ma, C., Peng, Y., Li, H. & Chen, W. Organ-on-a-Chip: a new paradigm for drug development. Trends Pharm. Sci. 42, 119–133 (2021).
Wu, Q. et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomed. Eng. Online 19, 1–9 (2020).
Zhang, B. Y., Korolj, A., Lai, B. F. L. & Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 3, 257–278 (2018).
Low, L. A., Mummery, C., Berridge, B. R., Austin, C. P. & Tagle, D. A. Organs-on-chips: into the next decade. Nat. Rev. Drug Discov. 20, 345–361 (2021).
Tavakol, D. N., Fleischer, S. & Vunjak-Novakovic, G. Harnessing organs-on-a-chip to model tissue regeneration. Cell Stem Cell 28, 993–1015 (2021).
Vunjak-Novakovic, G., Ronaldson-Bouchard, K. & Radisic, M. Organs-on-a-chip models for biological research. Cell 184, 4597–4611 (2021).
van der Helm, M. W., van der Meer, A. D., Eijkel, J. C., van den Berg, A. & Segerink, L. I. Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 4, e1142493 (2016).
Zakharova, M. et al. Multiplexed blood-brain barrier organ-on-chip. Lab Chip 20, 3132–3143 (2020).
Sone, N. et al. Multicellular modeling of ciliopathy by combining iPS cells and microfluidic airway-on-a-chip technology. Sci. Transl. Med 13, eabb1298 (2021).
Vatine, G. D. et al. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell 24, 995–1005.e1006 (2019).
Michas, C. et al. Engineering a living cardiac pump on a chip using high-precision fabrication. Sci. Adv. 8, eabm3791 (2022).
Zhao, Y. et al. A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell 176, 913–927.e918 (2019).
Shultz, L. D. et al. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 30, 123–142 (2019).
Flahou, C., Morishima, T., Takizawa, H. & Sugimoto, N. Fit-for-all iPSC-derived cell therapies and their evaluation in humanized mice with NK cell immunity. Front. Immunol. 12, 662360 (2021).
Moquin-Beaudry, G. et al. Autologous humanized mouse models of iPSC-derived tumors enable characterization and modulation of cancer-immune cell interactions. Cell Rep. Methods 2, 100153 (2022).
Zeleniak, A. et al. De novo construction of T cell compartment in humanized mice engrafted with iPSC-derived thymus organoids. Nat. Methods 19, 1306–1319 (2022).
Sharma, A., Sances, S., Workman, M. J. & Svendsen, C. N. Multi-lineage human iPSC-derived platforms for disease modeling and drug discovery. Cell Stem Cell 26, 309–329 (2020).
Abud, E. M. et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e279 (2017).
Fattorelli, N. et al. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat. Protoc. 16, 1013–1033 (2021).
Hasselmann, J. et al. Development of a chimeric model to study and manipulate human microglia in vivo. Neuron 103, 1016–1033.e1010 (2019).
Svoboda, D. S. et al. Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc. Natl. Acad. Sci. USA 116, 25293–25303 (2019).
Xu, R. et al. Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat. Commun. 11, 1577 (2020).
Wimmer, R. A. et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565, 505–510 (2019).
Ho, R. et al. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 19, 1256–1267 (2016).
Alvarez, Z. et al. Artificial extracellular matrix scaffolds of mobile molecules enhance maturation of human stem cell-derived neurons. Cell Stem Cell 30, 219–238.e214 (2023).
Yoshida, S. et al. Maturation of human induced pluripotent stem cell-derived cardiomyocytes by soluble factors from human mesenchymal stem cells. Mol. Ther. 26, 2681–2695 (2018).
Giacomelli, E. et al. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell 26, 862–879.e811 (2020).
Maoz, B. M. et al. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat. Biotechnol. 36, 865–874 (2018).
Hayashi, R. et al. Generation of 3D lacrimal gland organoids from human pluripotent stem cells. Nature 605, 126–131 (2022).
Mansour, A. A. et al. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441 (2018).
Munera, J. O. et al. Differentiation of human pluripotent stem cells into colonic organoids via transient activation of BMP signaling. Cell Stem Cell 21, 51–64.e56 (2017).
Revah, O. et al. Maturation and circuit integration of transplanted human cortical organoids. Nature 610, 319–326 (2022).
Tanaka, J. et al. Human induced pluripotent stem cell-derived salivary gland organoids model SARS-CoV-2 infection and replication. Nat. Cell Biol. 24, 1595–1605 (2022).
Schafer, S. T. et al. An in vivo neuroimmune organoid model to study human microglia phenotypes. Cell 186, 2111–2126.e2120 (2023).
Cerneckis, J. & Shi, Y. Context matters: hPSC-derived microglia thrive in a humanized brain environment in vivo. Cell Stem Cell 30, 909–910 (2023).
Ronaldson-Bouchard, K. et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243 (2018).
Tu, C. Y., Chao, B. S. & Wu, J. C. Strategies for improving the maturity of human induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 123, 512–514 (2018).
Luo, J. et al. Tissue-engineered vascular grafts with advanced mechanical strength from human iPSCs. Cell Stem Cell 26, 251–261.e258 (2020).
Ronaldson-Bouchard, K. et al. Engineering of human cardiac muscle electromechanically matured to an adult-like phenotype. Nat. Protoc. 14, 2781–2817 (2019).
Shin, D. et al. Thalamocortical organoids enable in vitro modeling of 22q11.2 microdeletion associated with neuropsychiatric disorders. Cell Stem Cell 31, 421–432.e428 (2024).
Regev, A. et al. The Human Cell Atlas. Elife 6, e27041 (2017).
Rozenblatt-Rosen, O., Stubbington, M. J. T., Regev, A. & Teichmann, S. A. The human cell atlas: from vision to reality. Nature 550, 451–453 (2017).
Zheng, Y. et al. Controlled modelling of human epiblast and amnion development using stem cells. Nature 573, 421–425 (2019).
Sasaki, K. et al. Robust In vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell 17, 178–194 (2015).
Hayashi, M., Kawaguchi, T., Durcova-Hills, G. & Imai, H. Generation of germ cells from pluripotent stem cells in mammals. Reprod. Med. Biol. 17, 107–114 (2018).
Esfahani, S. N. et al. Derivation of human primordial germ cell-like cells in an embryonic-like culture. Nat. Commun. 15, 167 (2024).
Matsuda, M. et al. Recapitulating the human segmentation clock with pluripotent stem cells. Nature 580, 124–129 (2020).
Weatherbee, B. A. T. et al. Pluripotent stem cell-derived model of the post-implantation human embryo. Nature 622, 584–593 (2023).
Zernicka-Goetz, M. The evolution of embryo models. Nat. Methods 20, 1844–1848 (2023).
Manor, Y. S., Massarwa, R. & Hanna, J. H. Establishing the human naive pluripotent state. Curr. Opin. Genet. Dev. 34, 35–45 (2015).
Weinberger, L., Ayyash, M., Novershtern, N. & Hanna, J. H. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17, 155–169 (2016).
Zhou, J., Hu, J., Wang, Y. & Gao, S. Induction and application of human naive pluripotency. Cell Rep. 42, 112379 (2023).
Giulitti, S. et al. Direct generation of human naive induced pluripotent stem cells from somatic cells in microfluidics. Nat. Cell Biol. 21, 275–286 (2019).
Li, W. et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell 4, 16–19 (2009).
Sahakyan, A. et al. Human naive pluripotent stem cells Model X chromosome dampening and X inactivation. Cell Stem Cell 20, 87–101 (2017).
Theunissen, T. W. et al. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell 19, 502–515 (2016).
Kagawa, H. et al. Human blastoids model blastocyst development and implantation. Nature 601, 600–605 (2022).
Wei, Y. et al. Efficient derivation of human trophoblast stem cells from primed pluripotent stem cells. Sci. Adv. 7, eabf4416 (2021).
Castel, G. et al. Induction of human trophoblast stem cells from somatic cells and pluripotent stem cells. Cell Rep. 33, 108419 (2020).
Jang, Y. J., Kim, M., Lee, B. K. & Kim, J. Induction of human trophoblast stem-like cells from primed pluripotent stem cells. Proc. Natl. Acad. Sci. USA 119, e2115709119 (2022).
Earley, A. M., Burbulla, L. F., Krainc, D. & Awatramani, R. Identification of ASCL1 as a determinant for human iPSC-derived dopaminergic neurons. Sci. Rep. 11, 22257 (2021).
Jerber, J. et al. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet. 53, 304–312 (2021).
Camp, J. G. et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 112, 15672–15677 (2015).
Fleck, J. S. et al. Inferring and perturbing cell fate regulomes in human brain organoids. Nature 621, 365–372 (2023).
Lee, J. H. et al. Production of human spinal-cord organoids recapitulating neural-tube morphogenesis. Nat. Biomed. Eng. 6, 435–448 (2022).
Hofbauer, P. et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell 184, 3299–3317.e3222 (2021).
Marton, R. M. & Pasca, S. P. Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. 30, 133–143 (2020).
Koike, H. et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature 574, 112–116 (2019).
Miura, Y. et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 38, 1421–1430 (2020).
Xiang, Y. et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383–398.e387 (2017).
Soldner, F. & Jaenisch, R. iPSC disease modeling. Science 338, 1155–1156 (2012).
Li, L., Chao, J. & Shi, Y. Modeling neurological diseases using iPSC-derived neural cells : iPSC modeling of neurological diseases. Cell Tissue Res. 371, 143–151 (2018).
Israel, M. A. et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220 (2012).
Kwart, D. et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP beta-CTFs, Not Abeta. Neuron 104, 256–270.e255 (2019).
Liu, Q. et al. Effect of potent gamma-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 71, 1481–1489 (2014).
Hendriks, D., Clevers, H. & Artegiani, B. CRISPR-Cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell 27, 705–731 (2020).
Firth, A. L. et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 12, 1385–1390 (2015).
Lin, Y. T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1294–1294 (2018).
Liu, Z. et al. Astrocytic response mediated by the CLU risk allele inhibits OPC proliferation and myelination in a human iPSC model. Cell Rep. 42, 112841 (2023).
Brunner, J. W. et al. Power and optimal study design in iPSC-based brain disease modelling. Mol. Psychiatry 28, 1545–1556 (2023).
Kondo, T. et al. Dissection of the polygenic architecture of neuronal Aβ production using a large sample of individual iPSC lines derived from Alzheimer’s disease patients. Nat. Aging 2, 125–139 (2022).
Kimura, M. et al. En masse organoid phenotyping informs metabolic-associated genetic susceptibility to NASH. Cell 185, 4216–4232.e4216 (2022).
Park, J. C. et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 12, 280 (2021).
Parenti, I., Rabaneda, L. G., Schoen, H. & Novarino, G. Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci. 43, 608–621 (2020).
Thapar, A., Cooper, M. & Rutter, M. Neurodevelopmental disorders. Lancet Psychiatry 4, 339–346 (2017).
Fang, R. et al. Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science 377, 56–62 (2022).
Hodge, R. D. et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019).
Pembroke, W. G., Hartl, C. L. & Geschwind, D. H. Evolutionary conservation and divergence of the human brain transcriptome. Genome Biol. 22, 1–33 (2021).
Zhu, Y. et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362, eaat8077 (2018).
Li, L. & Shi, Y. When glia meet induced pluripotent stem cells (iPSCs). Mol. Cell Neurosci. 109, 103565 (2020).
Shao, Z. et al. Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat. Neurosci. 22, 229–242 (2019).
Szabo, A. et al. A human iPSC-astroglia neurodevelopmental model reveals divergent transcriptomic patterns in schizophrenia. Transl. Psychiatry 11, 554 (2021).
Topol, A. et al. Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Rep. 15, 1024–1036 (2016).
Yoon, K. J. et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15, 79–91 (2014).
Murai, K. et al. The TLX-miR-219 cascade regulates neural stem cell proliferation in neurodevelopment and schizophrenia iPSC model. Nat. Commun. 7, 10965 (2016).
Schafer, S. T. et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 22, 243–255 (2019).
Wang, M. et al. Increased neural progenitor proliferation in a hiPSC model of autism induces replication stress-associated genome instability. Cell Stem Cell 26, 221–233.e226 (2020).
Kathuria, A. et al. Synaptic deficits in iPSC-derived cortical interneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Transl. Psychiatry 9, 321 (2019).
Kizner, V., Fischer, S. & Naujock, M. Multielectrode Array (MEA)-based detection of spontaneous network activity in human iPSC-derived cortical neurons. Methods Mol. Biol. 1994, 209–216 (2019).
Sun, G. et al. Modeling human cytomegalovirus-induced microcephaly in human iPSC-derived brain organoids. Cell Rep. Med. 1, 100002 (2020).
Brennand, K. J. et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225 (2011).
Wen, Z. et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014).
Zaslavsky, K. et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 22, 556–564 (2019).
Cai, H. W. et al. Brain organoid reservoir computing for artificial intelligence. Nat. Electron 6, 1032–1039 (2023).
Kagan, B. J. et al. In vitro neurons learn and exhibit sentience when embodied in a simulated game-world. Neuron 110, 3952–3969.e3958 (2022).
Chiaradia, I. & Lancaster, M. A. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat. Neurosci. 23, 1496–1508 (2020).
Wang, H. Modeling neurological diseases with human brain organoids. Front. Synaptic Neurosci. 10, 15 (2018).
Velasco, S., Paulsen, B. & Arlotta, P. 3D brain organoids: studying brain development and disease outside the embryo. Annu Rev. Neurosci. 43, 375–389 (2020).
Cerneckis, J. & Shi, Y. Modeling brain macrophage biology and neurodegenerative diseases using human iPSC-derived neuroimmune organoids. Front. Cell Neurosci. 17, 1198715 (2023).
Mariani, J. et al. FOXG1-dependent dysregulation of GABA/Glutamate neuron differentiation in autism spectrum disorders. Cell 162, 375–390 (2015).
Xu, R. et al. OLIG2 drives abnormal neurodevelopmental phenotypes in human iPSC-based organoid and chimeric mouse models of down syndrome. Cell Stem Cell 24, 908–926.e908 (2019).
Trujillo, C. A. et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell Stem Cell 25, 558–569.e557 (2019).
Passaro, A. P. & Stice, S. L. Electrophysiological analysis of brain organoids: current approaches and advancements. Front. Neurosci. 14, 622137 (2020).
Samarasinghe, R. A. et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 24, 1488–1500 (2021).
Windrem, M. S. et al. Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208.e196 (2017).
Dong, X. et al. Human cerebral organoids establish subcortical projections in the mouse brain after transplantation. Mol. Psychiatry 26, 2964–2976 (2021).
Wilson, M. N. et al. Multimodal monitoring of human cortical organoids implanted in mice reveal functional connection with visual cortex. Nat. Commun. 13, 7945 (2022).
Cerneckis, J., Bu, G. & Shi, Y. Pushing the boundaries of brain organoids to study Alzheimer’s disease. Trends Mol. Med. 29, 659-672 (2023).
Dugger, B. N. & Dickson, D. W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9, a028035 (2017).
Hardiman, O. et al. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 3, 1–19 (2017).
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet 386, 896–912 (2015).
Knopman, D. S. et al. Alzheimer disease. Nat. Rev. Dis. Prim. 7, 33 (2021).
Gonzales, M. M. et al. Biological aging processes underlying cognitive decline and neurodegenerative disease. J. Clin. Investig. 132, e158453 (2022).
Camandola, S. & Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. Embo J. 36, 1474–1492 (2017).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Cornacchia, D. & Studer, L. Back and forth in time: directing age in iPSC-derived lineages. Brain Res. 1656, 14–26 (2017).
Studer, L., Vera, E. & Cornacchia, D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell 16, 591–600 (2015).
Mertens, J. et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell 28, 1533–1548.e1536 (2021).
Miller, J. D. et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705 (2013).
Giacomelli, E. et al. Human stem cell models of neurodegeneration: from basic science of amyotrophic lateral sclerosis to clinical translation. Cell Stem Cell 29, 11–35 (2022).
Okano, H. & Morimoto, S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell 29, 189–208 (2022).
Virdi, G. S. et al. Protein aggregation and calcium dysregulation are hallmarks of familial Parkinson’s disease in midbrain dopaminergic neurons. Npj Parkinsons Dis. 8, 162 (2022).
Egawa, N. et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4, 145ra104 (2012).
Young, J. E. et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16, 373–385 (2015).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. J. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Belloy, M. E., Napolioni, V. & Greicius, M. D. A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101, 820–838 (2019).
Serrano-Pozo, A., Das, S. & Hyman, B. T. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 20, 68–80 (2021).
Sienski, G. et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 13, eaaz4564 (2021).
Tcw, J. et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185, 2213–2233.e2225 (2022).
Blanchard, J. W. et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779 (2022).
Murdock, M. H. & Tsai, L. H. Insights into Alzheimer’s disease from single-cell genomic approaches. Nat. Neurosci. 26, 181–195 (2023).
Victor, M. B. et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 29, 1197–1212.e1198 (2022).
Chen, X. et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615, 668–677 (2023).
Gate, D. et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020).
Krauskopf, J. et al. Transcriptomics analysis of human iPSC-derived dopaminergic neurons reveals a novel model for sporadic Parkinson’s disease. Mol. Psychiatry 27, 4355–4367 (2022).
Ryan, S. D. et al. Isogenic human iPSC parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1 alpha transcription. Cell 155, 1351–1364 (2013).
Sommer, A. et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23, 123–131.e126 (2018).
Baxi, E. G. et al. Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat. Neurosci. 25, 226–237 (2022).
Workman, M. J. et al. Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron 111, 1191–1204.e1195 (2023).
Fujimori, K. et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589 (2018).
Chakrabarti, S. & Mohanakumar, K. P. Aging and neurodegeneration: a tangle of models and mechanisms. Aging Dis. 7, 111–113 (2016).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Franceschi, C., Garagnani, P., Vitale, G., Capri, M. & Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 28, 199–212 (2017).
Grimm, A. & Eckert, A. Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 143, 418–431 (2017).
Bertucci, E. M. & Parrott, B. B. Is CpG density the link between epigenetic aging and lifespan? Trends Genet. 36, 725–727 (2020).
Kosan, C., Heidel, F. H., Godmann, M. & Bierhoff, H. Epigenetic erosion in adult stem cells: drivers and passengers of aging. Cells 7, 237 (2018).
Little, D. et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 8, 9033 (2018).
Du, F., Yu, Q., Chen, A., Chen, D. & Yan, S. S. Astrocytes attenuate mitochondrial dysfunctions in human dopaminergic neurons derived from iPSC. Stem Cell Rep. 10, 366–374 (2018).
Cheng, X. Y. et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 9, 1–14 (2020).
Zagoura, D., Canovas-Jorda, D., Pistollato, F., Bremer-Hoffmann, S. & Bal-Price, A. Evaluation of the rotenone-induced activation of the Nrf2 pathway in a neuronal model derived from human induced pluripotent stem cells. Neurochem. Int. 106, 62–73 (2017).
Benson, E. K., Lee, S. W. & Aaronson, S. A. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci. 123, 2605–2612 (2010).
Ambasudhan, R. et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9, 113–118 (2011).
Carter, J. L., Halmai, J. & Fink, K. D. The iNs and outs of direct reprogramming to induced neurons. Front. Genome Ed. 2, 7 (2020).
Drouin-Ouellet, J., Pircs, K., Barker, R. A., Jakobsson, J. & Parmar, M. Direct neuronal reprogramming for disease modeling studies using patient-derived neurons: what have we learned? Front. Neurosci. 11, 530 (2017).
Mertens, J., Marchetto, M. C., Bardy, C. & Gage, F. H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 17, 424–437 (2016).
Wang, H., Yang, Y., Liu, J. & Qian, L. Direct cell reprogramming: approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 22, 410–424 (2021).
Wapinski, O. L. et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635 (2013).
Herdy, J. R. et al. Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer’s disease. Cell Stem Cell 29, 1637–1652.e1636 (2022).
Traxler, L. et al. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metab. 34, 1248–1263.e1246 (2022).
Barisano, G. et al. Blood–brain barrier link to human cognitive impairment and Alzheimer’s disease. Nat. Cardiovasc. Res. 1, 108–115 (2022).
Knox, E. G., Aburto, M. R., Clarke, G., Cryan, J. F. & O’Driscoll, C. M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatr. 27, 2659–2673 (2022).
Montagne, A. et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302 (2015).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Zlokovic, B. V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738 (2011).
Chen, X. et al. Modeling sporadic Alzheimer’s disease in human brain organoids under serum exposure. Adv. Sci. 8, e2101462 (2021).
Mirabelli, P., Coppola, L. & Salvatore, M. Cancer cell lines are useful model systems for medical research. Cancers 11, 1098 (2019).
Gillet, J. P., Varma, S. & Gottesman, M. M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 105, 452–458 (2013).
Wilding, J. L. & Bodmer, W. F. Cancer cell lines for drug discovery and development. Cancer Res. 74, 2377–2384 (2014).
Wijewardhane, N., Dressler, L. & Ciccarelli, F. D. Normal somatic mutations in cancer transformation. Cancer Cell 39, 125–129 (2021).
Smith, R. C. & Tabar, V. Constructing and deconstructing cancers using human pluripotent stem cells and organoids. Cell Stem Cell 24, 12–24 (2019).
Haag, D. et al. H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell 39, 407–422.e413 (2021).
Crespo, M. et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 23, 878–884 (2017).
Ford, A. C., Yuan, Y. & Moayyedi, P. Long-term impact of helicobacter pylori eradication therapy on gastric cancer incidence and mortality in healthy infected individuals: a meta-analysis beyond 10 years of follow-up. Gastroenterology 163, 754–756.e751 (2022).
Polk, D. B. & Peek, R. M. Jr. Helicobacter pylori: gastric cancer and beyond. Nat. Rev. Cancer 10, 403–414 (2010).
McCracken, K. W. et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014).
Wang, T. et al. Sequential CRISPR gene editing in human iPSCs charts the clonal evolution of myeloid leukemia and identifies early disease targets. Cell Stem Cell 28, 1074–1089.e1077 (2021).
Garcez, P. P. et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818 (2016).
Scoon, W. A. et al. Ebola virus infection induces a delayed type I IFN response in bystander cells and the shutdown of key liver genes in human iPSC-derived hepatocytes. Stem Cell Rep. 17, 2286–2302 (2022).
Luo, Y., Zhang, M., Chen, Y., Chen, Y. & Zhu, D. Application of human induced pluripotent stem cell-derived cellular and organoid models for COVID-19 research. Front. Cell Dev. Biol. 9, 720099 (2021).
Harschnitz, O. & Studer, L. Human stem cell models to study host-virus interactions in the central nervous system. Nat. Rev. Immunol. 21, 441–453 (2021).
Lamers, M. M. & Haagmans, B. L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol 20, 270–284 (2022).
Cevik, M., Kuppalli, K., Kindrachuk, J. & Peiris, M. Virology, transmission, and pathogenesis of SARS-CoV-2. BMJ 371, m3862 (2020).
Harrison, A. G., Lin, T. & Wang, P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 41, 1100–1115 (2020).
Bestion, E., Halfon, P., Mezouar, S. & Mege, J. L. Cell and animal models for SARS-CoV-2 research. Viruses 14, 1507 (2022).
Chu, H., Chan, J. F. & Yuen, K. Y. Animal models in SARS-CoV-2 research. Nat. Methods 19, 392–394 (2022).
Cleary, S. J. et al. Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. Br. J. Pharm. 177, 4851–4865 (2020).
Lee, C. Y. & Lowen, A. C. Animal models for SARS-CoV-2. Curr. Opin. Virol. 48, 73–81 (2021).
Takayama, K. In vitro and animal models for SARS-CoV-2 research. Trends Pharm. Sci. 41, 513–517 (2020).
Simoneau, C. R. & Ott, M. Modeling multi-organ infection by SARS-CoV-2 using stem cell technology. Cell Stem Cell 27, 859–868 (2020).
Huang, J. et al. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar Type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell 27, 962–973.e967 (2020).
Lian, Q. et al. Differential effects of macrophage subtypes on SARS-CoV-2 infection in a human pluripotent stem cell-derived model. Nat. Commun. 13, 2028 (2022).
Elrobaa, I. H. & New, K. J. COVID-19: pulmonary and extra pulmonary manifestations. Front. Public Health 9, 711616 (2021).
Gupta, A. et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 26, 1017–1032 (2020).
Ning, Q. et al. The mechanism underlying extrapulmonary complications of the coronavirus disease 2019 and its therapeutic implication. Signal. Transduct. Target Ther. 7, 57 (2022).
Chen, K. G., Park, K. & Spence, J. R. Studying SARS-CoV-2 infectivity and therapeutic responses with complex organoids. Nat. Cell Biol. 23, 822–833 (2021).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913.e907 (2020).
Wang, W. L. et al. Detection of SARS-CoV-2 in different types of clinical specimens. Jama 323, 1843–1844 (2020).
Bojkova, D. et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc. Res. 116, 2207–2215 (2020).
Perez-Bermejo, J. A. et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci. Transl. Med. 13, eabf7872 (2021).
Sharma, A. et al. Human iPSC-derived cardiomyocytes are susceptible to SARS-CoV-2 infection. Cell Rep. Med. 1, 100052 (2020).
Ahmad, I. & Rathore, F. A. Neurological manifestations and complications of COVID-19: a literature review. J. Clin. Neurosci. 77, 8–12 (2020).
Niazkar, H. R., Zibaee, B., Nasimi, A. & Bahri, N. The neurological manifestations of COVID-19: a review article. Neurol. Sci. 41, 1667–1671 (2020).
Yassin, A. et al. Neurological manifestations and complications of coronavirus disease 2019 (COVID-19): a systematic review and meta-analysis. BMC Neurol. 21, 1–17 (2021).
Ramani, A. et al. SARS-CoV-2 targets neurons of 3D human brain organoids. Embo J. 39, e106230 (2020).
Zhang, B. Z. et al. SARS-CoV-2 infects human neural progenitor cells and brain organoids. Cell Res. 30, 928–931 (2020).
Cui, Q. et al. Compound screen identifies the small molecule Q34 as an inhibitor of SARS-CoV-2 infection. iScience 25, 103684 (2022).
Wang, C. et al. ApoE-isoform-dependent SARS-CoV-2 neurotropism and cellular response. Cell Stem Cell 28, 331–342.e335 (2021).
Shen, W. B. et al. SARS-CoV-2 invades cognitive centers of the brain and induces Alzheimer’s-like neuropathology. Preprint at BioRxiv (2022).
Kleiman, R. J. & Engle, S. J. Human inducible pluripotent stem cells: Realization of initial promise in drug discovery. Cell Stem Cell 28, 1507–1515 (2021).
Gu, M. et al. iPSC-endothelial cell phenotypic drug screening and in silico analyses identify tyrphostin-AG1296 for pulmonary arterial hypertension. Sci. Transl. Med. 13, eaba6480 (2021).
Bray, M. A. et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 11, 1757–1774 (2016).
Chin, M. Y., Espinosa, J. A., Pohan, G., Markossian, S. & Arkin, M. R. Reimagining dots and dashes: visualizing structure and function of organelles for high-content imaging analysis. Cell Chem. Biol. 28, 320–337 (2021).
Vamathevan, J. et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18, 463–477 (2019).
Taubes, A. et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease. Nat. Aging 1, 932–947 (2021).
Theodoris, C. V. et al. Network-based screen in iPSC-derived cells reveals therapeutic candidate for heart valve disease. Science 371, eabd0724 (2021).
Pangalos, M. N., Schechter, L. E. & Hurko, O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat. Rev. Drug Discov. 6, 521–532 (2007).
Waring, M. J. et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475–486 (2015).
Inoue, H. & Yamanaka, S. The use of induced pluripotent stem cells in drug development. Clin. Pharm. Ther. 89, 655–661 (2011).
Liu, W., Deng, Y., Liu, Y., Gong, W. & Deng, W. Stem cell models for drug discovery and toxicology studies. J. Biochem. Mol. Toxicol. 27, 17–27 (2013).
Pasteuning-Vuhman, S., de Jongh, R., Timmers, A. & Pasterkamp, R. J. Towards advanced iPSC-based drug development for neurodegenerative disease. Trends Mol. Med. 27, 263–279 (2021).
Reiser, J. & Sever, S. Podocyte biology and pathogenesis of kidney disease. Annu Rev. Med. 64, 357–366 (2013).
Musah, S. et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat. Biomed. Eng. 1, 0069 (2017).
Richards, D. J. et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 4, 446–462 (2020).
Matsa, E. et al. Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 19, 311–325 (2016).
Sharma, A. et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 9, eaaf2584 (2017).
Pellegrini, L. et al. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 369, eaaz5626 (2020).
Kwon, O. et al. The development of a functional human small intestinal epithelium model for drug absorption. Sci. Adv. 7, eabh1586 (2021).
Westerling-Bui, A. D. et al. Transplanted organoids empower human preclinical assessment of drug candidate for the clinic. Sci. Adv. 8, eabj5633 (2022).
Brown, C. et al. Mesenchymal stem cells: cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 13, 1738–1755 (2019).
Chien, K. R. et al. Regenerating the field of cardiovascular cell therapy. Nat. Biotechnol. 37, 232–237 (2019).
Huang, K., Hu, S. & Cheng, K. A new era of cardiac cell therapy: opportunities and challenges. Adv. Health. Mater. 8, e1801011 (2019).
Sterner, R. C. & Sterner, R. M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11, 69 (2021).
Brown, C. E. & Mackall, C. L. CAR T cell therapy: inroads to response and resistance. Nat. Rev. Immunol. 19, 73–74 (2019).
Finck, A. V., Blanchard, T., Roselle, C. P., Golinelli, G. & June, C. H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 28, 678–689 (2022).
Bashor, C. J., Hilton, I. B., Bandukwala, H., Smith, D. M. & Veiseh, O. Engineering the next generation of cell-based therapeutics. Nat. Rev. Drug Discov. 21, 655–675 (2022).
Desgres, M. & Menasche, P. Clinical translation of pluripotent stem cell therapies: challenges and considerations. Cell Stem Cell 25, 594–606 (2019).
Stevens, K. R. & Murry, C. E. Human pluripotent stem cell-derived engineered tissues: clinical considerations. Cell Stem Cell 22, 294–297 (2018).
Doss, M. X. & Sachinidis, A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 8, 403 (2019).
Lovell-Badge, R. et al. ISSCR guidelines for stem cell research and clinical translation: the 2021 update. Stem Cell Rep. 16, 1398–1408 (2021).
Balboa, D. et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 40, 1042–1055 (2022).
Du, Y. et al. Human pluripotent stem-cell-derived islets ameliorate diabetes in non-human primates. Nat. Med. 28, 272–282 (2022).
Feng, L. et al. Developing hypoimmunogenic human iPSC-derived oligodendrocyte progenitor cells as an off-the-shelf cell therapy for myelin disorders. Adv. Sci. 10, e2206910 (2023).
Madrid, M., Sumen, C., Aivio, S. & Saklayen, N. Autologous induced pluripotent stem cell-based cell therapies: promise, progress, and challenges. Curr. Protoc. 1, e88 (2021).
Schweitzer, J. S. et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med. 382, 1926–1932 (2020).
Schweitzer, J. S., Song, B. & Kim, K. S. A step closer to autologous cell therapy for Parkinson’s disease. Cell Stem Cell 28, 595–597 (2021).
Tang, L. V. et al. Gene editing of human iPSCs rescues thrombophilia in hereditary antithrombin deficiency in mice. Sci. Transl. Med. 14, eabq3202 (2022).
Maxwell, K. G. et al. Gene-edited human stem cell-derived beta cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci. Transl. Med. 12, eaax9106 (2020).
Depil, S., Duchateau, P., Grupp, S. A., Mufti, G. & Poirot, L. Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 19, 185–199 (2020).
Crow, D. Could iPSCs enable “off-the-shelf” cell therapy? Cell 177, 1667–1669 (2019).
Lanza, R., Russell, D. W. & Nagy, A. Engineering universal cells that evade immune detection. Nat. Rev. Immunol. 19, 723–733 (2019).
Wang, B. et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 5, 429–440 (2021).
Hu, X. et al. Hypoimmune induced pluripotent stem cells survive long term in fully immunocompetent, allogeneic rhesus macaques. Nat. Biotechnol. 42, 413–423 (2023).
Alvarez-Palomo, B. et al. Evaluation of the Spanish population coverage of a prospective HLA haplobank of induced pluripotent stem cells. Stem Cell Res Ther. 12, 233 (2021).
Lee, S. et al. Repurposing the cord blood bank for haplobanking of HLA-Homozygous iPSCs and their usefulness to multiple populations. Stem Cells 36, 1552–1566 (2018).
Sullivan, S. et al. Haplobanking induced pluripotent stem cells for clinical use. Stem Cell Res. 49, 102035 (2020).
Yoshida, S. et al. A clinical-grade HLA haplobank of human induced pluripotent stem cells matching approximately 40% of the Japanese population. Med 4, 51–66.e10 (2023).
Nguyen, P. K., Neofytou, E., Rhee, J.-W. & Wu, J. C. Potential strategies to address the major clinical barriers facing stem cell regenerative therapy for cardiovascular disease: a review. JAMA Cardiol. 1, 953–962 (2016).
Aijaz, A. et al. Biomanufacturing for clinically advanced cell therapies. Nat. Biomed. Eng. 2, 362–376 (2018).
Lee, A. S., Tang, C., Rao, M. S., Weissman, I. L. & Wu, J. C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 19, 998–1004 (2013).
Chour, T. et al. Method for selective ablation of undifferentiated human pluripotent stem cell populations for cell-based therapies. JCI Insight 6, e142000 (2021).
Kuang, Y. et al. Efficient, selective removal of human pluripotent stem cells via ecto-alkaline phosphatase-mediated aggregation of synthetic peptides. Cell Chem. Biol. 24, 685–694.e684 (2017).
Jones, B. S., Lamb, L. S., Goldman, F. & Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharm. 5, 254 (2014).
Lund, R. J., Narva, E. & Lahesmaa, R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 13, 732–744 (2012).
Ma, H. et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 511, 177–183 (2014).
Guo, R. et al. Generation and clinical potential of functional T lymphocytes from gene-edited pluripotent stem cells. Exp. Hematol. Oncol. 11, 1–17 (2022).
Motazedian, A. et al. Multipotent RAG1+ progenitors emerge directly from haemogenic endothelium in human pluripotent stem cell-derived haematopoietic organoids. Nat. Cell Biol. 22, 60–73 (2020).
Seet, C. S. et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 14, 521–530 (2017).
Wang, Z. et al. 3D-organoid culture supports differentiation of human CAR(+) iPSCs into highly functional CAR T cells. Cell Stem Cell 29, 651–653 (2022).
Nagamoto, Y. et al. Transplantation of a human iPSC-derived hepatocyte sheet increases survival in mice with acute liver failure. J. Hepatol. 64, 1068–1075 (2016).
Sharma, R. et al. Clinical-grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Sci. Transl. Med. 11, eaat5580 (2019).
Glaeser, J. D. et al. iPSC-neural crest derived cells embedded in 3D printable bio-ink promote cranial bone defect repair. Sci. Rep. 12, 18701 (2022).
Cichocki, F. et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy. Sci. Transl. Med. 12, eaaz5618 (2020).
Moriarty, N. et al. A combined cell and gene therapy approach for homotopic reconstruction of midbrain dopamine pathways using human pluripotent stem cells. Cell Stem Cell 29, 434–448.e435 (2022).
Iancu, E. M. & Kandalaft, L. E. Challenges and advantages of cell therapy manufacturing under good manufacturing practices within the hospital setting. Curr. Opin. Biotechnol. 65, 233–241 (2020).
Ackermann, M. et al. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat. Protoc. 17, 513–539 (2022).
Ackermann, M. et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 9, 5088 (2018).
Yasuda, S. Y. et al. Chemically defined and growth-factor-free culture system for the expansion and derivation of human pluripotent stem cells. Nat. Biomed. Eng. 2, 173–182 (2018).
Zhao, Z. et al. Organoids. Nat. Rev. Methods Prim. 2, 94 (2022).
Basu, S. et al. Live-cell three-dimensional single-molecule tracking reveals modulation of enhancer dynamics by NuRD. Nat. Struct. Mol. Biol. 30, 1628–1639 (2023).
Dodonova, S. O., Zhu, F., Dienemann, C., Taipale, J. & Cramer, P. Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672 (2020).
Wang, J. et al. Phase separation of OCT4 controls TAD reorganization to promote cell fate transitions. Cell Stem Cell 28, 1868–1883.e1811 (2021).
He, W. et al. De novo identification of essential protein domains from CRISPR-Cas9 tiling-sgRNA knockout screens. Nat. Commun. 10, 4541 (2019).
Hsu, J. Y. et al. CRISPR-SURF: discovering regulatory elements by deconvolution of CRISPR tiling screen data. Nat. Methods 15, 992–993 (2018).
Yang, L. et al. High-resolution characterization of gene function using single-cell CRISPR tiling screen. Nat. Commun. 12, 4063 (2021).
Liu, P., Chen, M., Liu, Y., Qi, L. S. & Ding, S. CRISPR-based chromatin remodeling of the endogenous Oct4 or Sox2 Locus enables reprogramming to pluripotency. Cell Stem Cell 22, 252–261.e254 (2018).
Baumann, V. et al. Targeted removal of epigenetic barriers during transcriptional reprogramming. Nat. Commun. 10, 2119 (2019).
Takahashi, Y. et al. Transgenerational inheritance of acquired epigenetic signatures at CpG islands in mice. Cell 186, 715–731.e719 (2023).
Tompkins, J. et al. Engineering CpG island DNA methylation in pluripotent cells through synthetic CpG-free ssDNA insertion. Cell Rep. Methods 3, 100465 (2023).
Cerneckis, J., Ming, G. L., Song, H., He, C. & Shi, Y. The rise of epitranscriptomics: recent developments and future directions. Trends Pharm. Sci. 45, 24–38 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Stahl, K., Graziadei, A., Dau, T., Brock, O. & Rappsilber, J. Protein structure prediction with in-cell photo-crosslinking mass spectrometry and deep learning. Nat. Biotechnol. 1−10 (2023).
Greener, J. G., Kandathil, S. M., Moffat, L. & Jones, D. T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 23, 40–55 (2022).
Coronnello, C. & Francipane, M. G. Moving towards induced pluripotent stem cell-based therapies with artificial intelligence and machine learning. Stem Cell Rev. Rep. 18, 559–569 (2022).
Pantazis, C. B. et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell Stem Cell 29, 1685–1702.e1622 (2022).
Bisogno, L. S. et al. Ancestry-dependent gene expression correlates with reprogramming to pluripotency and multiple dynamic biological processes. Sci. Adv. 6, eabc3851 (2020).
Czerniecki, S. M. et al. High-throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell 22, 929–940.e924 (2018).
Lu, Y., Zhou, Y., Ju, R. & Chen, J. Human-animal chimeras for autologous organ transplantation: technological advances and future perspectives. Ann. Transl. Med. 7, 576 (2019).
Takebe, T. et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484 (2013).
Suchy, F., Yamaguchi, T. & Nakauchi, H. iPSC-derived organs in vivo: challenges and promise. Cell Stem Cell 22, 21–24 (2018).
Acknowledgements
The authors would like to thank Louise and Herbert Horvitz, the Christopher Family, the Judy and Bernard Briskin Fund, and the Sidell Kagan Foundation for their generosity and forethought. This work was supported by the National Institute on Aging of the National Institutes of Health R01 AG072291 and RF1 AG079307 and the National Institute of Neurological Disorders and Stroke of the National Institutes of Health U01 NS122101 to Y.S. J.C. is a predoctoral scholar in the Stem Cell Biology and Regenerative Medicine Research Training Program of the California Institute for Regenerative Medicine (CIRM). Figures 1–7 were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
J.C. and Y.S. conceptualized the review article. J.C. drafted the manuscript and prepared the figures. H.C. drafted the table. J.C. revised the manuscript with inputs from H.C. and Y.S. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Y.S. is the editorial board member of Signal Transduction and Targeted Therapy, but was not involved in the handling of this manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cerneckis, J., Cai, H. & Shi, Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Sig Transduct Target Ther 9, 112 (2024). https://doi.org/10.1038/s41392-024-01809-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01809-0
