
Abstract
Prolonged activation of nuclear factor (NF)-кB signaling significantly contributes to the development of colorectal cancer (CRC). New therapeutic opportunities are emerging from targeting this distorted cell signaling transduction. Here, we discovered the critical role of RING finger 138 (RNF138) in CRC tumorigenesis through regulating the NF-кB signaling, which is independent of its Ubiquitin-E3 ligase activity involved in DNA damage response. RNF138−/− mice were hyper-susceptible to the switch from colitis to aggressive malignancy, which coincided with sustained aberrant NF-кB signaling in the colonic cells. Furthermore, RNF138 suppresses the activation of NF-кB signaling pathway through preventing the translocation of NIK and IKK-Beta Binding Protein (NIBP) to the cytoplasm, which requires the ubiquitin interaction motif (UIM) domain. More importantly, we uncovered a significant correlation between poor prognosis and the downregulation of RNF138 associated with reinforced NF-кB signaling in clinical settings, raising the possibility of RNF138 dysregulation as an indicator for the therapeutic intervention targeting NF-кB signaling. Using the xenograft models built upon either RNF138-dificient CRC cells or the cells derived from the RNF138-dysregulated CRC patients, we demonstrated that the inhibition of NF-кB signaling effectively hampered tumor growth. Overall, our work defined the pathogenic role of aberrant NF-кB signaling due to RNF138 downregulation in the cascade events from the colitis switch to colonic neoplastic transformation and progression, and also highlights the possibility of targeting the NF-кB signaling in treating specific subtypes of CRC indicated by RNF138-ablation.
Similar content being viewed by others


Introduction
Colorectal carcinoma (CRC), accounting for the third leading cause of cancer-related death worldwide,1,2 often arises from sustained inflammation and presents chronic inflammation throughout their progression. Being etiologically associated with CRC progression, chronic inflammation affects all stages of CRC pathogenesis, from tumor initiation, progression, to metastasis.3,4,5,6 Recent advance in understanding the underlying mechanisms began to enable the therapeutic interventions through targeting the CRC-associated chronic inflammation.7,8
The nuclear factor-kappa B (NF-кB) signaling is central to the response to environmental stress, genotoxicity, or infections.9,10,11 NF-кB signaling is activated via two different routes, i.e., the canonical pathway (CP) and the alternative pathway.12 The latter operates predominately in the stimulated B cells as opposed to the former general activation mechanism.13 The IκB kinase (IKK) complex, constructed by three subunits, IKKα, IKKβ, and IKKγ, is key component of the CP.14 Upon the signal activation, IкB proteins become destabilized after a sequential modification process, first being phosphorylated by IKKβ and then ubiquitinated through the designated Ub E2/E3 enzymes, which ultimately releases NF-кB as its binding partner into the nucleus to switch on the transcription of targeted genes.14,15 Ubiquitination also plays important roles in the processing of NF-кB precursors as well as the assembly and regulation of the IKK complex.16,17,18 Importantly, growing evidence has linked the dysregulation of NF-кB signaling to various critical medical conditions. In CRC particularly, the aberrant signaling appears to promote the progression of a cascade events from colitis switch to colonic neoplastic transformation.12,19,20
Protein ubiquitination serves critical regulatory roles in the NF-κB signaling transduction.14,21 RING finger protein 138 (RNF138), as a Ubiquitin-E3 (Ub-E3) ligase, promotes cell survival via counteracting the apoptotic signaling as well as directly engaging in maintaining genome stability.22,23,24 With emerging evidence, RNF138 has been associated with tumorigenesis, neurotic degenerative disorders, and chronic inflammatory conditions.25,26,27,28 In particular, diverse types of cancer display differential expressions of RNF138 as documented in multiple transcriptomic and proteomic studies, although the relevant functional consequences remain largely elusive. Here, we established a close correlation between RNF138 expression and prognosis of CRC patients. Further investigation revealed that deficient RNF138 led to the dysregulation of NF-κB signaling that significantly promotes the colitis switch to malignancy, which can be effectively suppressed with specific inhibitors against individual components of the pathway.
Results
Downregulation of RNF138 in CRC tumors correlates with poor prognosis
In analyzing the data collected from The Cancer Genome Atlas (TCGA) database, we noticed that RNF138 is downregulated in different subsets of cancers, particularly in CRC, liver hepatocellular carcinoma, and thyroid carcinoma, to a less extent in cholangiocarcinoma and head and neck squamous cell carcinoma (Fig. 1a; Supplementary Table 1). This unique expression pattern was also found when using the data derived from additional independent GEO datasets (GDS4382, GDS4718, and GDS2947) (Fig. 1b). In addition, RNF138 was confirmed to be downregulated in 20 randomized CRC samples compared with the matched adjacent normal tissues at transcriptional level (Fig. 1c; Supplementary Table 2) as well as at translational level (Fig. 1d) (Supplementary Fig. S1a–c). Similar results were obtained when analyzing additional 5 CRC samples in pairs by immunofluorescence (IF) (Fig. 1e; Supplementary Fig. S1d), consistent with the results from further examining 134 pairs of CRC tissue specimens by immunohistochemistry (IHC) (Fig. 1f; Supplementary Table 3). Therefore, RNF138 downregulation is closely associated with CRC tumorigenesis.

Downregulation of RNF138 in CRC tumors correlates with poor prognosis. RNF138 mRNA levels in 10 cancer types in the TCGA database (a) and three independent GEO datasets (b). T tumor tissues, N normal tissues. n.s. not significant; *P < 0.05; **P < 0.01; ****P < 0.0001. c Heatmap depiction of RNF138 mRNA levels assessed by qPCR in 20 CRC samples. d Immunoblots and quantification of RNF138 in 10 CRC samples. e H&E and IF staining for RNF138 and DAPI in a representative CRC sample. Dashed white line represents the boundary between normal and neoplastic tissues. Histogram (right) shows the quantification of RNF138-staining in CRC or adjacent normal tissues (n = 20). Scale bar, 20 μm. f H&E and RNF138-staining in 134 CRC tissues (top, colon tissues, n = 67; bottom, rectum tissues, n = 67). Histograms show quantification of RNF138-staining in CRC and adjacent normal tissues (right). Scale bar, 100 μm. g Representative H&E and RNF138-staining images in high and low RNF138 expression in 420 CRC TMAs. The inset box shows an overview of the specimen staining. Scale bar, 25 μm. Kaplan–Meier survival curves showing the correlation between RNF138 expression and DFS (h) or OS (i) in 420 CRC samples
We probed the expression of RNF138 in tissue microarrays (TMAs) of 420 CRC samples (Supplementary Table 4) with the clinicopathological parameter setup established (Supplementary Fig. S2a). The reduction in RNF138 expression was found specifically associated with more aggressive tumor phenotypes (P = 0.027), reflected by both the increase in the size and extent of primary tumors (P = 0.003) and lymph node invasion (P = 0.003), while no significant correlation was observed with CRC tumor metastasis (P = 0.246) (Supplementary Fig. S2b–d; Supplementary Table 4). Importantly, the Kaplan–Meier survival analysis revealed that the increase in RNF138 expression closely correlates with enhanced disease-free survival (DFS) and improved overall survival (OS) prognoses of patients (Fig. 1g–i), suggesting RNF138 expression level as a prognosis indicator of CRC.
Ablation of RNF138 expression exacerbates the progression from colitis to aggressive malignancy
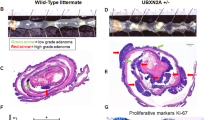
Chronic inflammation, often in the form of colitis in colorectal tissue, plays important roles in the colonic neoplastic transformation and progression.6,29 We next interrogated the possible role of RNF138 in the colitis-associated colorectal tumorigenesis. The chronic colitis and the colitis-associated colorectal cancer (CAC) animal models were developed as illustrated in Fig. 2a. Upon the colitis occurrence, RNF138−/− mice deteriorated more rapidly than RNF138fl/fl mice under co-housing condition, which was reflected in both the change in clinical scores and weight loss (Fig. 2b; Supplementary Fig. S3a), and ultimately suffered a higher mortality rate (Fig. 2c). The colorectum in RNF138−/− mice were significantly reduced in length and exhibited splenomegaly as opposed to the control (Fig. 2d; Supplementary Fig. S3b–d). In RNF138−/− mice, extensive inflammation was manifested throughout the mucosa along with reinforced lymphocytic infiltration as revealed by IHC, which coincided with altered epithelial structures and loss of crypts (Fig. 2e). In contrast, only inflammatory lesions were found in the control mice. Furthermore, precancerous lesions in the colorectal tissue were more evident in the RNF138−/− (57.14%) (Fig. 2f). Two out of seven RNF138−/− mice developed substantial CRC tumors with high-grade dysplasia (Supplementary Fig. S3e). Meanwhile, excessive cell proliferation indicated by Ki67 occurred in the colonic tissues of RNF138−/− mice (Fig. 2g).

Ablation of RNF138 expression exacerbates the progression from colitis to aggressive malignancy. a Schematic overview of chronic colitis and CAC models. b Clinical scores of RNF138fl/fl and RNF138−/− mice (n = 7) on the 9th day after DSS administration. c Kaplan–Meier survival analysis of RNF138fl/fl (n = 15) and RNF138−/− (n = 23) mice during DSS treatment. d Macroscopic appearance of colons of RNF138fl/fl and RNF138−/− mice (n = 7) cohorts at the end of DSS treatment. e Representative H&E images (left) with histogram of histology scores (right) of colon tissues for chronic colitis model. Data are mean ± SEM (n = 7). Scale bar, 100 μm. f Representative H&E staining of precancerous lesions in colitis models. Histogram of precancerous percentages from RNF138fl/fl and RNF138−/− mice after DSS administration (n = 7). Scale bar, 100 μm. g Representative Ki67-staining (left) and quantification positive cells per crypt (right) in RNF138fl/fl and RNF138−/− colitis models (n = 7). Scale bar, 25 μm. h Clinical scores of RNF138fl/fl and RNF138−/− mice (n = 7) on the 12th day after AOM injection. i Kaplan–Meier survival analysis of RNF138fl/fl (n = 17) and RNF138−/− (n = 23) mice during the course of AOM/DSS treatment. (j) Representative longitudinal luminal views of colons from RNF138fl/fl and RNF138−/− mice (n = 7) at the end of the AOM/DSS treatment. Tumor numbers (k), tumor load (l), and tumor size distribution (m) of RNF138fl/fl and RNF138−/− mice (n = 7) after AOM/DSS treatment. n Representative H&E images (left) and histological analysis (right) of colon tissues from RNF138fl/fl and RNF138−/− mice (n = 7) after AOM/DSS administration. Scale bar, 100 μm. Percentages of tumor types (o) and differentiation grade (p) in RNF138fl/fl vs. RNF138−/− mice. q Representative Ki67-staining (left) and quantification of positive cells per crypt (right) in RNF138fl/fl and RNF138−/− CAC models (n = 7). Scale bar, 25 μm
Similarly, RNF138−/− mice exhibited higher clinical scores and suffered more significant weight loss and higher mortality rate during the induction of CAC with azoxymethane (AOM) and dextran sodium sulfate (DSS) (Fig. 2h, i; Supplementary Fig. S3f). In these mice, both colon and spleen tissues were also significantly shortened in length and displayed more severe damage (Fig. 2j; Supplementary Fig. S3g, h). Particularly, all RNF138−/− mice (23/23) developed CRC tumors as opposed to 86.7% in the control mice (15/17) (Supplementary Fig. S3i). Furthermore, we found that both the tumor number and tumor load were nearly doubled in the RNF138−/− mice (Fig. 2k–l) along with significant increase in size (Fig. 2m). The tumors in these mice appeared to be more advanced (Fig. 2n), developing higher levels of intramucosal carcinoma and adenocarcinoma that displayed lower degree of differentiation (Fig. 2o, p; Supplementary Fig. S3j). Meanwhile, augmented cell proliferation, highlighted by the Ki67-staining, was also identified in the RNF138−/− mice tissues (Fig. 2q). Thus, we propose that RNF138 suppresses the chronic colitis switch to colonic neoplastic transformation.
RNF138 deletion reinforces tumorigenesis
We constructed the tumor organoids system to understand RNF138’s role in the CRC tumorigenesis and progression (Fig. 3a), in which the growth of organoids was monitored for 9 days from day 0.5 post transplantation. From day 7, larger in diameter became apparent in the organoids derived from the RNF138−/− tumors (Fig. 3b; Supplementary Fig. S4a) along with alternated morphology and more rapid increase in number (Fig. 3c; Supplementary Fig. S4b). The xenografts from the RNF138−/− organoids developed larger in volume, on average two times of the ones from the control (Fig. 3d; Supplementary Fig. S4c, d), and exhibited a higher level of excessive growth indicated by Ki67-staining (Fig. 3e). Altogether, these results confirmed that RNF138 suppresses the tumor growth independent of the orthotopic microenvironment.

RNF138 inhibits chronic colitis and CRC tumorigenesis via NF-кB signaling. a Schematic overview of in vitro organoid culture of primary mouse colorectal tumors (top) and xenografts (bottom). b Representative images of mouse tumor organoids at different time-points. Scale bar, 200 μm. c Quantification of mouse intestinal tumor organoid numbers per field from the 5th to 9th day. d Representative images of xenograft tumor organoids in RNF138fl/fl and RNF138−/− mice (n = 5) on the 60th day after implantation (left). Quantification of the tumor volume at endpoint (right). e Representative images for Ki67-staining and DAPI of xenograft tumor organoids (left). Quantification of number of Ki67-positive cells per crypt (n = 20, right). Scale bar, 20 μm. f Heatmap depiction of differentially expressed genes in RN138fl/fl and RNF138−/− colorectal cells. g GSEA of NF-kappa B signaling relative to RNF138−/− against RNF138fl/fl mice in colitis, CAC, and organoid models. Enrichment plots are shown along with the normalized enrichment score and P value. h Protein-protein interaction analysis of significant GO terms in colitis, CAC, and organoid models. Circles at nodes represent GO terms in one model (orange, colitis; blue, CAC; red, organoid model; yellow, I-κB kinase/NF-κB, and NIK/NF-κB signaling). i Representative p65-staining of colorectal sections from RNF138fl/fl and RNF138−/− mouse colitis (top) and CAC (bottom) models. Quantification of nuclear p65-positive cells per crypt (n = 20) (right). Scale bar, 25 μm. j Representative images of p65-immunostaining and DAPI from xenograft tumor organoids (left). Quantification of nuclear p65-positive cells per crypt (n = 20) (right). Scale bar, 20 μm. k Immunoblots of phosphorylation and total levels of p65 in RNF138WT and RNF138KO CRC cells. l Representative images of p65-immunostaining in RNF138WT and RNF138KO HCT116 (left) and RKO (right) cells. Histogram shows the quantification of nuclear p65 intensity (n = 20). Scale bar, 5 μm. m qPCR analysis of NF-кB target genes expression in RNF138WT and RNF138KO HCT116 (top) and RKO (bottom) cells (n = 3). **P < 0.01 and ***P < 0.001
RNF138 dysregulation gives rise to aberrant NF-кB signaling underlying chronic inflammation, colonic neoplastic transformation, and progression
We systematically examined the global changes imposed by RNF138 deficiency in either the colon tissues dissected from the colitis and CAC models or the organoids derived from the CAC model. In all three sets of samples, substantial differences were identified at transcriptomic level between the RNF138fl/fl and RNF138−/− genetic backgrounds (Fig. 3f; Supplementary Fig. S5a). As revealed in the gene ontology (GO) term analysis, these differences primarily attributed to changes in the intestinal epithelial cell differentiation, proliferation, apoptosis, and cellular senescence (Supplementary Fig. S5b). Further analysis of signaling networks underlying these functional changes illuminated the central role of the NF-κB signaling interconnecting with the diverse functional elements (Fig. 3g, h; Supplementary Fig. S5c, d). Nuclear staining of p65 as an indicator downstream of NF-κB signaling were significantly enhanced upon RNF138 deletion (Fig. 3i, j). Thus, RNF138 is functionally linked to the NF-κB signaling that is critical for a cascade of events from the onset of colitis to the colitis-to-tumor transition and further CAC tumor progression.
We also recapitulated this phenomenon in human CRC cell lines where the RNF138 expression was suppressed via specific small interfering RNA (siRNA). In both HCT116 and RKO cells, upon RNF138 RNAi, p65 levels were moderately increased though with significant increase in the phosphorylation of p65 (pp65) (Supplementary Fig. S5e), consistent with results observed when disrupting RNF138 by CRISPR-Cas9 (Fig. 3k). Furthermore, RNF138 deficiency enhanced the nuclear translocation of p65 (Fig. 3l) that led to the increase in the downstream gene expression (NFκB1, IκBα, CXCL1, and IL8), overall highlighting enhanced activation of the NF-κB signaling pathway (Fig. 3m; Supplementary Fig. S5f). In brief, RNF138 negatively regulates the NF-κB signaling during colorectal tumorigenesis.
RNF138 restrains the activation of NF-κB signaling by retaining NIBP in the nucleus
RNF138 seems not engaged in the canonical NF-κB signaling pathway directly (Supplementary Table 5; Supplementary Fig. S6a) but through the association with NIK- and IKK-β-binding protein (NIBP), a NF-κB signaling transduction regulator identified previously.30,31 The interaction between RNF138 and NIBP was confirmed by immunoprecipitation, and most likely occurs in the nucleus (Fig. 4a) as NIBP distributes in both the nucleus and cytoplasm whereas RNF138 resides primarily in the nucleus (Fig. 4b). As revealed in the proximity ligation assay, NIBP interacts with RNF138 directly in the nucleus (Fig. 4c) whereas it is associated with IKKβ (Fig. 4d), in the cytoplasm.30 Upon the RNAi against NIBP, the activation of NF-κB was significantly compromised, which was reflected in the reduction both in the phosphorylation of p65, (Fig. 4e) and in the transcription of downstream genes featured by NFкB1, IκBα, CXCL1, and IL8 (Fig. 4f), suggesting that NIBP likely mediates the regulation of NF-κB by RNF138. Furthermore, the loss of NIBP restored the NF-кB signaling that was distorted due to the defective RNF138 (Fig. 4g). In summary, RNF138 regulates the NF-κB signaling at least partially via NIBP.

Nuclear-cytoplasmic partitioning of NIBP is regulated by RNF138 and contributes to the activation of NF-κB signaling. a Co-immunoprecipitation analysis of endogenous RNF138 and NIBP interactions in HCT116 total lysates (left) and nuclear extracts (right). IgG was used as control. b Immunostaining analysis of RNF138-NIBP co-localization in HCT116 and RKO cells. Nuclei were stained with DAPI. Insets show stained areas in more detail. Scale bar, 20 μm. c Representative confocal images (left) and quantification (right, n = 20) of endogenous RNF138 and NIBP interactions in HCT116 and RKO cells using proximity ligation assay. IgG were used as controls. Scale bar, 5 μm. d Immunostaining analysis of the co-localization of endogenous IKKβ and NIBP in HCT116 and RKO cells. Scale bar, 5 μm. e Immunoblots of phosphorylation, total levels of p65 in HCT116 and RKO cells transfected with si-NIBP or control siRNA. f qPCR analysis of NF-κB target gene expression in HCT116 and RKO cells (n = 3) with NIBP inhibition. *P < 0.05; **P < 0.01 and ****P < 0.0001. g Immunoblots of phosphorylation and total levels of p65 and IKKβ in RNF138WT and RNF138KO HCT116 and RKO cells with NIBP inhibition. h Representative immunostaining images of NIBP localization in RNF138WT and RNF138KO HCT116 and RKO cells, and the distribution of relative fluorescence intensities of lines scanned across the nucleus (right). Quantification of cytoplasmic NIBP intensity in HCT116 and RKO cells (n = 20). Scale bar, 5 μm. i Immunostaining analysis of IKKβ-NIBP co-localization in RNF138WT and RNF138KO HCT116 and RKO cells and relative pixel intensity of lines scanned across the nucleus. Scale bar, 20 μm
RNF138 serves as a Ub-E3 ligase in DNA damage response.22,23,32,33 In contrast, the level of NIBP remained unchanged upon the disruption of RNF138 by either RNAi or knockout (Supplementary Fig. S6b), suggesting the functional association between two is likely independent of ubiquitination and protein-degradation. Interestingly, the paradigm of NIBP subcellular distribution was shifted upon RNF138 depletion, with significant increase in the nuclear partitioning coincided with the decreased cytoplasmic presence (Fig. 4h), which consequently enhanced the co-localization of NIBP and IKKβ as observed in the RNF138-deficient HCT116 and RKO cells (Fig. 4i).
Thus, we propose that the nuclear-cytoplasmic partitioning of NIBP contributes significantly to the key mechanism by which RNF138 regulates the NF-κB signaling transduction.
The Ub-E3 ligase activity is dispensable for the regulation of NIBP by RNF138
The ubiquitin interacting motif (UIM) located in RNF138 extreme C-terminus (AA225-243) is essential for its interaction with NIBP as opposed to the RING domain (AA18-58) and triple Zinc finger (ZNF) (ZNF1 AA86-105, ZNF2 AA159-180, and ZNF3 AA189-215) (Fig. 5a, c). However, little impact on the interaction between RNF138 and NIBP was observed when RNF138, as a Ub-E3 ligase, became inactive with specific mutations introduced to the catalytic cysteines (C18A/C54A) (Fig. 5c). We also identified the second (AA360-669) and the third Trs120 domain (AA882-1100) in NIBP were primarily responsible for its interaction with RNF138 (Fig. 5b, d).

The regulation of NIBP by RNF138 is independent of the Ub-E3 ligase activity. a Schematic representation of RNF138 mutant constructs. Asterisk indicates amino acid residue mutant location (C/A) in RING domain. b Schematic representation of NIBP truncation constructs map. c Co-IP analysis of the interaction between HA-NIBP and full-length or truncated Flag-RNF138 mutants in 293T cells. d Co-IP analysis of the interaction between RNF138 and Flag-NIBP truncations in 293T cells. e Representative images of NIBP-immunostaining in HCT116 and RKO RNF138KO cell lines transfected with empty vector or RNF138 truncations (flag-red), with co-localization as shown by the pixel intensity profile. Quantification of NIBP nucleus intensity in HCT116 and RKO cells transfected with different RNF138 mutants (right). Scale bar, 5 μm. f Immunoblots of phosphorylation and total p65 levels in HCT116 and RKO-knockout cells transfected with EV or different RNF138 truncations. g qPCR analysis of NF-κB target gene expression in HCT116 and RKO-knockout cells transfected with EV or RNF138 truncations. h Cell viability of HCT116- and RKO-knockout cells transfected with EV or RNF138 truncations
This molecular connection was also reflected at functional level. The distorted nuclear partitioning of NIBP due to RNF138 deficiency could be restored by ectopically expressing either the full-length protein (FL), RING domain protein-truncated protein (ΔRING) or the catalytic cysteines C/A mutant (Mutant), but much less effective with the UIM-truncated protein (Fig. 5e). Furthermore, in contrast to the UIM-truncated protein, either the full length, the RING domain-truncated, or the catalytic cystine C/A mutant could restore the NF-κB signaling distorted by the loss of RNF138, which was manifested first by changes in the phosphorylation of p65 (Fig. 5f), then the expression of downstream genes including NF-кB1, IκBα, CXCL1, and IL8 (Fig. 5g), and further in cell proliferation (Fig. 5h). Importantly, the UIM-truncated RNF138 failed to exert impact on the proliferation as opposed to the full-length protein, the RING domain-truncated, and the catalytic cystine C/A mutant RNF138. Therefore, RNF138 regulates NF-κB signaling through its physical and functional interaction with NIBP independent of its Ub-E3 ligase activity.
RNF138 downregulation coincides with the aberrant NF-кB signaling in CRC associated with unfavorable clinical outcomes
A significant positive correlation was established between RNF138 expression and p65 total level using human primary CRC TMAs (n = 420) (Supplementary Fig. S7a). Similar association was also established between RNF138 expression with the cytoplasmic accumulation of p65 as opposed to the nuclear translocation of p65 as an indicator of NF-кB activation in CRC tissue specimens (Supplementary Fig. S7b, c), suggesting that RNF138 negatively regulates the NF-кB signaling transduction. We further observed significant downregulation of RNF138 in the CRC tissue specimens that coincided with the hyperactivation of NF-кB signaling pathway indicated by the changes in the p65 nucleic/cytoplasmic (N/C) ratio (Fig. 6a, b). Specifically, the CRC patients were stratified into four groups based on the level of RNF138 and the pp65 N/C ratio, i.e., RNF138high-pp65 N/C ratiolow, RNF138low-pp65 N/C ratiohigh, RNF138high-pp65 N/C ratiohigh, and RNF138low-pp65 N/C ratiolow. The Kaplan–Meier survival analysis shown that CRC patients with RNF138low-pp65 N/C ratiohigh had shorter disease-free survival and overall survival compared with the other three CRC subtypes (Fig. 6c, d). Importantly, RNF138low-pp65 N/C ratiohigh tumors exhibited much more aggressive clinical features (Fig. 6e, f; Supplementary Fig. S7d, e). Moreover, RNF138, the pp65 ratio (N/C), and the progression and prognosis of CRC patients were inversely correlated (Fig. 6g, h; Supplementary Fig. S7f). In particular, the sustained activation of NF-кB signaling occurred in two RNF138-ablated CRC subtypes associated with much more aggressive tumors and adverse outcomes, indicating the inverse correlation between RNF138 expression and the pp65 N/C ratio in tumors (n = 284) (Fig. 6i–l; Supplementary Fig. S7g, h), consistent with the changes in downstream gene expression highlighted in the cases of ICAM1 and PTGS2 (Fig. 6m, n).

RNF138 reduction coincided with aberrant activation of NF-кB signaling and correlated with unfavorable clinical outcomes in the CRC patients. a Correlation between log(pp65 N/C) and RNF138-staining according to Pearson’s test. The formula, coefficient of correlation (r), and the P value are indicated (n = 420). b Assessment of log(pp65 N/C) expression levels in low (n = 207) and high (n = 213) RNF138 CRC tissues. Data are mean ± SEM. Kaplan–Meier survival curves of DFS (c) and OS (d) stratified by RNF138 and pp65 N/C expression in CRC TMAs (n = 420). Log-rank test was used. e Percentages of RNF138high-pp65 N/C ratiolow (red) and RNF138low-pp65 N/C ratiohigh (blue) according to pTNM stage (1, 2, 3, 4) (n = 284). Correlation among RNF138, log(pp65 N/C), and pTNM stage (f), DFS (g), and OS (h) in CRC patient TMAs (n = 420). Log(N/C) indicates log(pp65 N/C). i Percentages of pTNM stage in RNF138high-pp65 N/C ratiolow (n = 138) and RNF138low-pp65 N/C ratiohigh (n = 146) in CRC patient TMAs (n = 284). Correlation among RNF138, log(pp65 N/C), and pTNM stage (j), DFS (k), and OS (l) in CRC patient TMAs (n = 284). Log(N/C), log(pp65 N/C). m IHC analysis of pp65, ICAM1, and PTGS2 in low- and high-RNF138-expressing CRC samples. Scale bar, 100 μm. n Correlation between RNF138 mRNA levels and NF-κB target genes according to qPCR analysis. The formula, coefficient of correlation (r), and the P value were provided via Pearson’s test (n = 30)
In brief, poor prognosis in CRC patients significantly correlates with the elevated NF-кB signaling coincided with RNF138 downregulation in the affiliated tumors. Therefore, we highlighted the possibility of using RNF138 downregulation as indicator for both prognosis and the therapeutic interventions targeting the NF-κB signaling.
Targeting the NF-кB signaling suppresses CRC growth associated with RNF138-ablation
We then further explored the therapeutic potential in targeting the distorted NF-κB signaling associated with RNF138 downregulation. SC75741 appeared effective in suppressing CRC tumor cell growth by the NF-κB pathway (Supplementary Fig. S8a–d), and could restore the aberrant NF-κB signaling with deficient RNF138-NIBP axis (Supplementary Fig. S8e). We therefore selected SC75741 as a specific chemical probe to address the effect of targeting the NF-κB signaling on the CRC progression. The RNF138WT and RNF138KO xenografts (CDX) were generated in parallel by injecting the corresponding HCT116 cells into the nude mice prior to the SC75741 treatment (Fig. 7a). Upon the treatment, the tumor growth was remarkably repressed in the RNF138KO xenografts, reflected in the reduction in both tumor volume and weights, whereas no significant difference was observed in the RNF138WT counterparts (Fig. 7b, c; Supplementary Fig. S9a, b). This inhibition was also marked at molecular level by the significant reduction in the Ki67 and PCNA staining of the RNF138KO xenografts compared with the WT, indicating an impaired proliferation as the result of the inhibition of NF-κB signaling (Fig. 7d; Supplementary Fig. S9c).

Targeting NF-кB signaling suppresses the RNF138-ablated CRC tumor growth. a Schematic overview of HCT116 CDX model establishment and SC75741 treatment. b The tumor growth curve of CDX model in RNF138WT and RNF138KO HCT116 cells treated with vehicle or SC75741 for 18 days n = 7. c Quantification of tumor weights from CDX model 18 days post-implantation (n = 7). d Representative H&E and IHC-staining of RNF138, pp65, and Ki67 from nude mice tumors. Scale bar, 25 μm. e Schematic overview of PDX model establishment and SC75741 treatment. f Tumor growth curve of PDX model of RNF138high-pp65 N/C ratiolow and RNF138low-pp65 N/C ratiohigh treated with vehicle or SC75741 for 18 days (n = 9). g Quantification of tumor weights from PDX models on the 18th day post-injection SC75741 (n = 9). h Representative H&E and IHC-staining with anti-RNF138, -pp65, and -Ki67 antibodies in tumor tissue from PDX models. Scale bar, 25 μm. i Proposed model for the role of NF-κB signaling regulated by RNF138 in colonic neoplastic transformation, progression, and SC75741 treatment
We further evaluate the results in a more clinical-relevant context with the CRC patient-derived xenograft (PDX) models (Fig. 7e). Specifically, the corresponding fragments derived from either RNF138high-pp65 N/C ratiolow (patient-1 and patient-2) or RNF138low-pp65 N/C ratiohigh (patient-3 and patient-4) CRC tumors were transplanted into NOD/SCID mice for PDX models (Supplementary Fig. S9f; Supplementary Table 6). SC75741 effectively blocked the activation of NF-κB signaling in all these models and specifically inhibited the growth of the RNF138low-pp65 N/C ratiohigh PDX model (Fig. 7f, g; Supplementary Fig. S9d, e). In contrast, no significant change was observed in the RNF138high-pp65 N/C ratiolow PDX models (Fig. 7f, g; Supplementary Fig. S9e). The Ki67- and PCNA-staining also indicated specific reduction in cell proliferation, accompanied by the diminished NF-κB signaling, in the RNF138low-pp65 N/C ratiohigh model (Fig. 7h; Supplementary Fig. S9g).
Collectively, these results strongly support the role of aberrant activation of NF-κB pathway in the CRC progression that is specially associated with RNF138 dysregulation. We propose NF-κB signaling-targeted therapy as the potential effective clinical intervention for the CRC with poor prognosis specifically associated with RNF138 downregulation (Fig. 7i).
Discussion
RNF138 functions as a Ub-E3 ligase to protect genome stability.22,23,34 However, mounting evidence suggests more complex functionality around RNF138, particularly between its RNF138 expression with carcinogenesis and chemoresistance.35,36 Multiple studies confirmed differential expressions of RNF138 in different physiological and pathological settings.36,37,38 Therefore, it is conceivable that RNF138 may reveal different aspects of its functionality under different circumstances. For example, RNF138 drives NF-κB activation and lymphomagenesis through destabilizing MYD88 L265P by ubiquitination,25 which differs from the mechanism proposed by this work. We revealed here a close association between RNF138 expression and NF-κB-mediated inflammation that is central to CRC tumorigenesis and prognosis. Using the ex vivo colitis and CAC models, we confirmed that RNF138 functions as a suppressor of CRC tumorigenesis through preventing excessive activation of NF-κB signaling. These findings together raise a possible interplay between DNA damage and repair process and inflammation in the context of tumorigenesis, particularly when considering the increasing amount of evidence showing that disrupting DNA damage response and repair accelerates inflammation.39,40 On the other hand, loss of cell/tissue integrity as well as other pathological alterations due to chronic inflammation leave higher vulnerability of DNA damages and potential mutations as the base of tumorigenesis.41,42 Hence investigating the role of RNF138 in these medical contexts could provide crucial insights into the interplay between these two processes. However, no significant correlation was observed between RNF138 and DNA damage response, indicated by γH2AX, in our colitis, CAC, or CAC-derived organoids models (Supplementary Fig. S10a, b), therefore in line with the notion of complex functionality of RNF138 in different physiological and pathological settings. RNA-seq analysis of RNF138-deficient models and in-depth data analysis revealed NF-кB signaling as the key target of RNF138 whereas other CRC-correlated genetic pathways, such as Wnt signaling and STAT3 signaling, were not remarkably altered with RNF138 depletion (Supplementary Fig. S10c–f). In the same line, the enhanced activation of NF-кB signaling occurred in CRC cells with RNF138 deficiency, implying a new regulatory mechanism for NF-кB signaling by RNF138. Although we appreciate the fact that RNF138’s E3 ligase activity may play important roles in different tumorigenesis processes,25 our work nevertheless highlights its functionality aspects independent of the E3 ligase activity by showing its control of the nucleocytoplasmic partition of NIBP as a key regulator of NF-κB signaling. Specifically, the UIM domain of RNF138 mediates its interaction with NIBP, critical for balancing the activation of NF-κB.
In diverse cancer types, the regulation of NF-кB signaling frequently become distorted, resulting in constitutive and unwarranted activation of the pathway associated with chronic inflammation.12,15 Therefore, reversing this process by targeting the NF-кB signaling have been an attractive direction in therapeutic development.43 Significant advance in the field is manifested in a collection of promising NF-кB inhibitors both in the development pipeline and in clinic.44,45,46 However, much left behind is our knowledge on the prerequisites for determining the timing and potential clinical outcome of such interventions. Particularly, effective early clinical indicators of aberrant NF-кB pathways are lacking, greatly hindering the applications of corresponding chemotherapeutic innovations. We demonstrated that downregulation of RNF138 significantly promoted the transition from colitis to tumor and sensitized the CRC cells to SC75741, a highly potent and specific NF-κB signaling inhibitor. More importantly, this notion was further supported by the observations in the CDX and PDX models, highlighting the potential of SC75741 in specific NF-κB-targeting therapy for treating the CRC patients with low RNF138 expression and hyperactivation of NF-κB (high pp65 N/C ratio). Critically, this clinical index was associated with a significant proportion (~34.8%, [146/420]) of CRC patients in our cohort, and with poorer prognoses compared with the rest of the cohort. This prompts us to wonder the possibility that low RNF138 and high pp65 N/C ratio in combination can be utilized as a useful indicator in clinic for the choice of NF-κB chemotherapeutic strategies.
In conclusion, our work promotes the potential of targeting NF-κB signaling in chemotherapy for CRC by dissecting the molecular mechanism underlines the aberrant activation of NF-κB signaling coincided with the loss of RNF138 and addresses the importance of RNF138 as a prognosis indicator for such treatments (Supplementary Fig. S11).
Materials and methods
Human specimens
The primary CRC tissues and 420 CRC tissue microarrays (TMAs) with follow-up were obtained from the Cancer Hospital, Chinese Academy of Medical Sciences (Beijing, China). Patient consent and approval were obtained from the Institutional Research Ethics Committee for research purpose (NCC2019C-016). The 134 paired CRC specimens were obtained from Shanghai Biochip. The detailed clinicopathological characteristics of the patients are summarized in Supplementary Tables 3, 4 and 6.
Mice
RNF138-deficient mice were generated as previously described.24 Female BALB/c nude mice and NOD/SCID mice were obtained from Beijing Vital River Laboratory Animal Technology. All protocols were approved by the Institutional Animal Care and Use Committee at the Institution of Basic Medical Sciences (005-2016).
Cell culture
HCT116, RKO, and HEK293T cell lines were obtained from the Cell Resource Center of Peking Union Medical College (Beijing, China) and cultured as described elsewhere.47 All cell lines were maintained at 37 °C, in a 5% CO2 incubator and used before reaching 20 passages.
Quantification and statistical analysis
A comparison of two groups was analyzed by Student’s t-test. Kaplan–Meier curves for survival were generated and analyzed by a log-rank test. The clinicopathological parameters in CRC cases were analyzed using Pearson’s chi-square test or Fisher’s exact chi-square test. Correlation studies were analyzed with Pearson’s or Spearman’s rank correlation tests. P value less than 0.05 were considered statistically significant. GraphPad Prism 8.0, SPSS 20.0, and Origin2019b were used to generate graphs and statistical analyses. The results are repeated at least three times and are presented as the means ± SEM for all quantified data, unless stated otherwise.
Data availability
All data supporting the findings of this study are available from the corresponding authors on reasonable request.
References
Biragyn, A. & Ferrucci, L. Gut dysbiosis: a potential link between increased cancer risk in ageing and inflammaging. Lancet Oncol. 19, e295–e304 (2018).
Lasry, A., Zinger, A. & Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 17, 230–240 (2016).
Dewson, G. & Silke, J. The Walrus and the Carpenter: complex regulation of tumor immunity in colorectal cancer. Cell 174, 14–16 (2018).
Song, M., Chan, A. T. & Sun, J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology 158, 322–340 (2020).
Garner, H. & de Visser, K. E. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat. Rev. Immunol. 20, 483–497 (2020).
Burke, K. E. et al. Interval colorectal cancer in inflammatory bowel disease: the role of guideline adherence. Dig. Dis. Sci. 65, 111–118 (2020).
Gee, M. H. et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell 172, 549–563 (2018).
Kesselring, R. et al. IRAK-M expression in tumor cells supports colorectal cancer progression through reduction of antimicrobial defense and stabilization of STAT3. Cancer Cell 29, 684–696 (2016).
Yu, Z., Quan, Y. N., Huang, Z. Q., Wang, H. H. & Wu, L. F. Monitoring oxidative stress, immune response, Nrf2/NF-κB signaling molecules of Rhynchocypris lagowski living in BFT system and exposed to waterborne ammonia. Ecotoxicol. Environ. Saf. 205, 111161 (2020).
Püschel, F. et al. Starvation and antimetabolic therapy promote cytokine release and recruitment of immune cells. Proc. Natl Acad. Sci. USA 117, 9932–9941 (2020).
Kusnadi, A. et al. Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8(+) T cells. Sci. Immunol. 6, eabe4782 (2021).
Taniguchi, K. & Karin, M. NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 18, 309–324 (2018).
Ben-Neriah, Y. Regulatory functions of ubiquitination in the immune system. Nat. Immunol. 3, 20–26 (2002).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362 (2008).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Chen, Z. J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 7, 758–765 (2005).
Mitxitorena, I. et al. The deubiquitinase USP7 uses a distinct ubiquitin-like domain to deubiquitinate NF-ĸB subunits. J. Biol. Chem. 295, 11754–11763 (2020).
Liu, J. et al. Targeting the ubiquitination/deubiquitination process to regulate immune checkpoint pathways. Signal Transduct. Target Ther. 6, 28 (2021).
Ben-Neriah, Y. & Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 (2011).
Zhu, G. et al. MyD88 mediates colorectal cancer cell proliferation, migration and invasion via NF‑κB/AP‑1 signaling pathway. Int. J. Mol. Med. 45, 131–140 (2020).
Hayden, M. S. & Ghosh, S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 (2012).
Ismail, I. H. et al. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol 17, 1446–1457 (2015).
Schmidt, C. K. et al. Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting DNA repair. Nat. Cell Biol. 17, 1458–1470 (2015).
Xu, L. et al. Rnf138 deficiency promotes apoptosis of spermatogonia in juvenile male mice. Cell Death Dis. 8, e2795 (2017).
Yu, X. et al. MYD88 L265P elicits mutation-specific ubiquitination to drive NF-κB activation and lymphomagenesis. Blood 137, 1615–1627 (2021).
Lee, K. et al. Proteome-wide discovery of mislocated proteins in cancer. Genome Res. 23, 1283–1294 (2013).
Long, P., Samnakay, P., Jenner, P. & Rose, S. A yeast two-hybrid screen reveals that osteopontin associates with MAP1A and MAP1B in addition to other proteins linked to microtubule stability, apoptosis and protein degradation in the human brain. Eur. J. Neurosci. 36, 2733–2742 (2012).
Nielsen, O. H., Bjerrum, J. T., Csillag, C., Nielsen, F. C. & Olsen, J. Influence of smoking on colonic gene expression profile in Crohn’s disease. PLoS ONE 4, e6210 (2009).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Hu, W. H. et al. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J. Biol. Chem. 280, 29233–29241 (2005).
Fu, Z. H. et al. NIK‑ and IKKβ‑binding protein contributes to gastric cancer chemoresistance by promoting epithelial‑mesenchymal transition through the NF‑κB signaling pathway. Oncol. Rep. 39, 2721–2730 (2018).
Han, D. et al. Ubiquitylation of Rad51d mediated by E3 ligase Rnf138 promotes the homologous recombination repair pathway. PLoS ONE 11, e0155476 (2016).
Yard, B. D., Reilly, N. M., Bedenbaugh, M. K. & Pittman, D. L. RNF138 interacts with RAD51D and is required for DNA interstrand crosslink repair and maintaining chromosome integrity. DNA Repair 42, 82–93 (2016).
Bekker-Jensen, S. & Mailand, N. RNF138 joins the HR team. Nat. Cell Biol. 17, 1375–1377 (2015).
Lu, Y. et al. RNF138 confers cisplatin resistance in gastric cancer cells via activating Chk1 signaling pathway. Cancer Biol. Ther. 19, 1128–1138 (2018).
Wu, H. et al. Downregulation of RNF138 inhibits cellular proliferation, migration, invasion and EMT in glioma cells via suppression of the Erk signaling pathway. Oncol. Rep. 40, 3285–3296 (2018).
Zhou, Y. X. et al. A novel gene RNF138 expressed in human gliomas and its function in the glioma cell line U251. Anal. Cell. Pathol. 35, 167–178 (2012).
Kim, W. et al. RNF138-mediated ubiquitination of rpS3 is required for resistance of glioblastoma cells to radiation-induced apoptosis. Exp. Mol. Med. 50, e434 (2018).
Paludan, S. R., Reinert, L. S. & Hornung, V. DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat. Rev. Immunol. 19, 141–153 (2019).
Nastasi, C., Mannarino, L. & D’Incalci, M. DNA damage response and immune defense. Int. J. Mol. Sci. 21, 7504 (2020).
Fishbein, A., Hammock, B. D., Serhan, C. N. & Panigrahy, D. Carcinogenesis: failure of resolution of inflammation? Pharmcol. Ther. 218, 107670 (2021).
de Bono, J. S. et al. Prostate carcinogenesis: inflammatory storms. Nat. Rev. Cancer 20, 455–469 (2020).
Tornatore, L. et al. Clinical proof of concept for a safe and effective NF-κB-targeting strategy in multiple myeloma. Br. J. Haematol 185, 588–592 (2019).
Rasmi, R. R., Sakthivel, K. M. & Guruvayoorappan, C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed. Pharmacother. 130, 110569 (2020).
Wang, W., Nag, S. A. & Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 22, 264–289 (2015).
Prescott, J. A. & Cook, S. J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 7, 115 (2018).
Song, W. et al. Rhomboid domain containing 1 promotes colorectal cancer growth through activation of the EGFR signalling pathway. Nat. Commun. 6, 8022 (2015).
Acknowledgements
We deeply thank Dr. Xiaodong Zhang (Wuhan University) for kindly providing plasmids to construct RNF138 and NIBP expressing vectors. We thank Dr. Dong Wang (Southern Medical University) for assisting the bioinformatics analysis. This work was supported by CAMS Innovation Fund for Medical Sciences (CIFMS, 2021-I2M-1-066, 2017-I2M-4-002 to H.Z., 2021-I2M-1-019 to W.S., 2021-1-I2M-014 to C.Z.L.), the National Natural Science Foundation of China (81972311, 82141127 and 81672461 to H.Z., 81672472, and 31970794 to W.S., 81570780 to C.Z.L., 32000586 to K.L.), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT310026 to H.Z.), and Sanming Project of Medicine in Shenzhen (SZSM202011010 to H.Z.), the National Key Research and Development Program of China (2018YFC1003500 to W.S.), the State Key Laboratory Special fund from the Ministry of Science (2060204 to W.S.), the State Key Project on Infection Diseases of China (2017ZX10201021-007-003 to H.Z.).
Author information
Authors and Affiliations
Contributions
Conceptualization, W.S., H.Z. and C.Z.L.; Methodology, W.S., H.Z., C.Z.L., Y.L.L. and R.H.; Investigation and Validation, Y.L.L., R.H., J.M,Y., X.C.L., T.J., L.G., A.T., J.M.L., Q.C.C., Y.H.W., L.Y.S., C.Y.G., F.X., Z.Y.W., Y.L., K.L., J.B.L., X.C., J.J.Y., H.J.H., X.W.C. Y.F.W. and X.Y.C.; Formal Analysis, Y.L.L., R.H., J.M.Y., X.C.L., H.T.Z., H.W. and Z.H.; Data Curation, W.S., C.Z.L. and H.Z.; Visualization, Y.L.L., C.Z.L., W.S. and R.H.; Resources, W.S., H.Z., J.M.Y. and J.Q.C.; Writing-Orginal Draft, C.Z.L., Y.L.L. and R.H.; Writing-Review & Edition, W.S., C.Z.L., H.Z., N.Z. and Y.L.L.; Supervision, H.Z., W. S., C.Z.L., J.Q.C., S.Y.M. and L.F.W.; Funding Acquisition, H.Z., W.S., C.Z.L. and K.L. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, Y., Huang, R., Ying, J. et al. RING finger 138 deregulation distorts NF-кB signaling and facilities colitis switch to aggressive malignancy. Sig Transduct Target Ther 7, 185 (2022). https://doi.org/10.1038/s41392-022-00985-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-00985-1
This article is cited by
-
Gliclazide Reduces Colitis-Associated Colorectal Cancer Formation by Deceasing Colonic Inflammation and Regulating AMPK-NF-κB Signaling Pathway
Digestive Diseases and Sciences (2024)
