
Abstract
Rare monogenetic diseases serve as natural models to dissect the molecular pathophysiology of the complex disease traits. Rheumatologic disorders by their nature are considered complex diseases with partially genetic origin, as illustrated by their heterogeneous genetic background and variable phenotypic presentation. Recent advances in genetic technologies have helped uncover multiple variants associated with disease susceptibility; however, a precise understanding of genotype–phenotype relationships is still missing. Inborn errors of immunity (IEIs), in addition to recurrent infections, may also present with autoimmune and autoinflammatory rheumatologic manifestations and have provided insights for understanding the underlying the principles of immune system homeostasis and mechanisms of immune dysregulation. This review discusses the rheumatologic manifestations in IEIs with overlapping and differentiating features in immunodeficiencies and rheumatologic disorders.
Similar content being viewed by others

Introduction
Rheumatologic disorders, including juvenile idiopathic arthritis (JIA), ankylosing spondylitis, systemic lupus erythematosus (SLE), and psoriatic arthritis, are considered as complex heterogeneous disorders where various genetic and environmental risk factors influence disease onset.1 Complex genetic inheritance patterns are major aspects of rheumatic disorders, whereby combinations of multiple variants influence disease susceptibility in addition to the course or outcome, as well as response to therapy. The heterogeneity of rheumatologic disorders in terms of presentation and outcome is a significant challenge for rheumatologists during clinical practice.1,2
Advances in genetics technologies have provided an opportunity to assess the impact of genetic aberrations on disease processes via identification of genetic risk factors using genome-wide association studies (GWAS). GWAS has been a powerful tool, extensively exploited to discover the genetic susceptibility of complex disease traits. A recent meta-analysis of collective data from GWAS on 29,800 rheumatoid arthritis (RA) patients revealed 377 candidate genes on non-major histocompatibility complex (non-MHC) loci. Ninety-eight of them were associated with two-fold increase in the risk of RA development and 15 of those genes, including AIRE, IL2RA, RAG1/2, CD40, TYK2, and CASP8, were previously identified to be causing inborn errors of immunity (IEI) particularly enriched in diseases of immune dysregulation.3 Similar attempts have been made for the pediatric cohort, for the disease groups including systemic JIA, revealing overlaps with IEI.4,5,6 Although GWAS have been useful for revealing genetic risk variants for complex disorders, linking those variants associated with disease susceptibility to disease pathogenesis has remained a challenge. Despite all efforts and recent advances, the genotype–phenotype links are still missing for many of the significant GWAS loci.7 For this reason, during the post-GWAS period, the research focus has shifted to localizing the genetic signal from GWAS to delineate which target gene may be affected, in order to validate the risk variants and refine the causative biological mechanism(s) underlying the disease.7,8
The past decade has witnessed an increase in research into the single-gene causes of the rheumatologic diseases, resulting in identification of several genetic mutations that underlie immune dysregulation and subsequently rheumatological manifestations.2 A high degree of overlap exists between autoimmune diseases and IEI in terms of genetic associations and causes (Fig. 1a, b). The 2017 analysis of French registry for IEIs revealed that there is a significant increase in the autoimmune and autoinflammatory complications in IEI patients, especially in common variable immunodeficiency (CVID) and combined immunodeficiency (CID). The overall survival time was also shorter for the patients having those complications.9

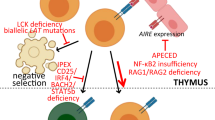
a, b The percentage of IEI having rheumatologic manifestations. Classification according to the 2017 International Union of Immunological Societies (IUIS) phenotypic classification of IEI.80 c Mechanisms involved in the pathogenesis of IEI presenting with rheumatologic manifestations. The gene list shown here displays selected examples for the respective subgroups
IEIs are genetically heterogeneous often single-gene disorders affecting development, maturation, or function of cells essential for immune system. Owing to their monogenic nature, IEIs serve as “experiments of nature.” They provide essential insights into the functioning of the immune system and contribute to the identification of the effectors of infection immunity and the checkpoints of self-reactivity. Affected individuals are prone to infections and often develop autoimmune disorders, allergies, lymphoproliferative disorders, and malignancies. IEIs are now known to be associated with a spectrum of rheumatologic diseases, including inflammatory arthritis, vasculitis, SLE, and SLE-like disorders (Fig. 1c). Investigating the consequences of genetic aberrations in patients with monogenic defects causing IEIs, allows for a mechanistic understanding of fundamental biological processes underlying these heterogenous diseases.
We herein review selected IEIs presenting within a spectrum of rheumatologic manifestations and how detailed studies on the their molecular pathophysiology has contributed to our understanding of immune homeostasis.
Defects of lymphocyte development and tolerance
The adaptive immune system is orchestrated by the crosstalk of T and B lymphocytes, which express immune receptors, immunoglobulins (Igs), and T cell receptors (TCRs), all showing an immense diversity of antigen repertoire to discriminate between self and non-self. Genetic aberrations impairing lymphocyte development, selection, activation, and differentiation can lead to immune signaling disturbances, resulting in IEIs presenting with autoimmunity and autoinflammation.10,11
Severe combined immunodeficiencies (SCIDs) are a prototypic group of disorders of the adaptive immune system in which both components, T and B cells, are lacking or functionally impaired. Mutations in genes involved in T-cell development can result in SCID. SCID patients have absent or very low numbers of T cells and absent or non-functional B cells, predisposing them to life-threatening bacterial, fungal, and viral infections.12 Hypomorphic mutations in SCID-causing genes may lead to residual or less functional protein production, which gives rise to a spectrum of phenotypes, including susceptibility to infections and dysregulated immunity. Consequently, all partial T-cell disorders may present with autoimmune or autoinflammatory manifestations.11
Receptor rearrangement with VDJ-recombination is crucial for the development of lymphocytes. Together with other enzymes and recombinases, RAG1 and RAG2 form a complex that initiates DNA cleavage and then repairs DNA breaks during VDJ-recombination. Classical RAG deficiency leads to SCID with absent T and B cells. Hypomorphic mutations of RAG have recently been shown to underlie a broader spectrum of clinical phenotypes including CID with granulomatous reactions or CD4-lymphopenia, CVID, or even hyper-IgM syndromes.13,14 A recent review of patient databases showed that 67% of the patients with RAG deficiency have documented autoimmune/autoinflammatory complications, such as autoimmune cytopenia, granuloma, vasculitis and neuropathies. Notably, in these patients, vasculitis occurred early in the disease course and was often complicated by significant end-organ involvement and, in most cases, was not responsive to first- or second-line therapy but required hematopoietic stem cell transplantation (HSCT) for definitive management.15
RAG1/2 mutations are the most common cause of Omenn syndrome—a defect in the activation and/or regulation of T-cell proliferation presenting with erythroderma, eosinophilia, hepatosplenomegaly, lymphadenopathy, and elevated serum IgE levels. Mutations in DCLRE1C, encoding Artemis, another protein involved in VDJ-recombination, can also cause Omenn syndrome. Similarly, hypomorphic mutations of DCLRE1C have been reported to be presenting as patients having CVID with autoimmune manifestations, including Hashimoto thyroiditis and JIA.16
Adenosine deaminase deficiency (ADA) is one of the common forms of SCID that accounts for 15–20% of all cases.17 Metabolic deficiency of the ADA enzyme results in the accumulation of toxic substrates, such as adenosine, 2′-deoxyadenosine, and deoxyribonucleotides, subsequently leading to severe lymphopenia (affecting T, B, and natural killer (NK) cells). Immune dysregulation in ADA deficiency may disrupt adaptive and innate immune pathways that normally lead to the development of tolerance.17 A wide spectrum of autoimmune manifestations have been documented (i.e., hemolytic anemia and immune thrombocytopenia) particularly in delayed and late-onset forms of ADA deficiency.17
TCR maturation and integrity is essential for the development and regulatory functions of T lymphocytes. The strength of TCR signaling plays a critical role in governing positive and negative selection in the thymus as well as responses of effector and regulatory T (Treg) cells in the periphery. In contrast to other TCR components where the mutations result in T−B+NK+ SCID, CD3γ deficiency causes a milder phenotype mainly presenting with autoimmunity. Clinical manifestations may extend from autoimmune thyroiditis and autoimmune hepatitis to ANA+ arthritis and nephrotic syndrome.18,19
ZAP70 is a protein found on the surface membrane of lymphocytes and essential for TCR signaling. ZAP70 deficiency leads to a SCID form in which CD8 T cells are absent and CD4 T cells are present but defective in function. Although disruption in thymic stromal cells of ZAP70-deficient individuals implicated a defect in T-cell tolerance, clinical evidence was not available for autoimmunity in these patients.20 More recently, a combination of hypomorphic and hyperactivating compound heterozygous mutations in ZAP70 have been reported to be causing a CID having autoimmune phenotype with therapy-resistant bullous pemphigoid, nephrotic syndrome, and inflammatory colitis documenting evidence for systemic autoimmunity.21
Diseases of immune dysregulation
Immunodeficiencies with immune dysregulation arise from abrogation of signaling pathways involved in development and maintenance of immune tolerance mechanisms, conventionally categorized under central and peripheral tolerance. Central tolerance occurs in the thymus and involves maturation and selection of T cells for developing antigenic repertoire to differentiate between self and non-self. Loss of central tolerance due to mutations in AIRE leads to autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED). AIRE mediates the promiscuous gene expression of a large number of tissue-specific antigens and is involved in the negative selection of autoreactive T cells.22 APECED patients present with a characteristic combination of chronic mucocutaneous candidiasis and autoimmunity that affects several endocrine and non-endocrine tissues. Non-endocrine autoimmune complications have recently been recognized as common disease manifestations, such as urticarial eruptions, Sjögren-like syndrome, autoimmune gastritis, hepatitis, pneumonitis, and tubulointerstitial nephritis.23 These manifestations develop in patients depending on the combinatorial disruption of tissue-specific antigenic repertoire.
A small percentage of lymphocytes that emigrate from the thymus develop into CD4+CD25+ Treg cells and express the transcription factor forkhead box protein P3 (FOXP3). These cells become the key regulators of peripheral immune tolerance mechanisms with the suppressive effect that is exerted at the level of the tissues and peripheral lymphoid organs. Mutations in the FOXP3 gene cause immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, an inherited disorder of systemic autoimmunity that is characterized by defective peripheral immune tolerance. The most common initial presentation of patients is a classical triad of enteropathy, eczematous dermatitis, and endocrinopathy (type-I diabetes or thyroiditis). Over the course of the disease, additional symptoms of systemic autoimmunity including cytopenias, nephropathy, hepatitis, arthritis, and alopecia have been documented.24,25
CD25 is a surface receptor constitutively expressed at high levels by Treg cells, enables them to be the first responders to interleukin (IL)-2 during an immune response, and promotes the transcription of FOXP3.26 CD25 deficiency is an autosomal-recessive disorder manifesting with severe autoimmune enteritis, viral infections, and lymphoproliferation. Similar to IPEX, over the disease course, additional autoimmune symptoms such as arthritis and alopecia can be observed.27 The signaling through CD25 has also been exploited for therapeutic approach to restore Treg function in autoimmune diseases including SLE and steroid refractory chronic graft versus host disease.28,29
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) is a checkpoint inhibitor molecule constitutively expressed in Treg cells, essential for their suppressive functions. CTLA-4, normally located in intracellular vesicles, is transferred to membrane upon T-cell activation. Competing with co-stimulatory molecule of CD28, CTLA-4 captures the CD80/86 receptors expressed on antigen-presenting cells and by means of transendocytosis and inhibits the T-cell activation through restriction of activatory costimulation. Patients with defects in CTLA-4 further show autoantibody-mediated cytopenias, lymphoproliferation, hypogammaglobulinemia, organ-specific autoimmunity, and lymphocytic infiltration of non-lymphoid organs. A recent publication on a cohort of patients with CTLA4 haploinsufficiency described hypogammaglobulinemia, lymphoproliferation, respiratory involvement, gastrointestinal symptoms, and autoimmune cytopenia as main features of the disease.30,31,32
Although homozygous mutations of CTLA4 have not been reported in humans, homozygous mutations in LRBA phenocopy CTLA4 haploinsufficiency. LRBA mutations have been linked to defective vesicle trafficking including transendocytosis and turnover of CTLA-4.33 A decrease in LRBA protein function leads to the diminished expression of CTLA-4 on the surface of Tregs.33 Although features of CTLA-4 and LRBA deficiencies are similar, a notable difference is the typically earlier age of onset of LRBA deficiency. Furthermore, additional autoimmune features, including type-I diabetes, autoimmune thyroiditis, arthritis, psoriasis or other skin disorders, uveitis, and vitiligo, appear to be more common in LRBA deficiency.34,35
In order to restore the loss of function of CTLA-4 in patients with LRBA and CTLA4 mutations, abatacept and belatacept CTLA-4-fusion proteins have been used as a therapeutic approach. To date, both molecules are under clinical trial for use in LRBA and CTLA-4 deficiency with individual reports stating beneficial effects on autoimmune and lymphoproliferative symptoms.31,33
Wiskott-Aldrich syndrome (WAS) is a primary immune deficiency with an X-linked inheritance pattern, classically characterized by thrombocytopenia with small platelets, eczema, and recurrent infections. WAS is caused by the mutations in the WAS gene coding WAS protein (WASp). WASp is involved in cell signaling and cytoskeleton reorganization through actin regulation. Autoimmune manifestations are observed in 70% of WAS patients and serve as a major complicating factor for HSCT outcome. Autoimmune cytopenias are most commonly reported complications, followed by arthritis, vasculitis, inflammatory bowel disease (IBD), and immune-mediated renal disease, including IgA-nephropathy and IgA-vasculitis. Dysfunctional Tregs and expansion in autoreactive B-cell population are the suggested mechanisms for autoimmunity in WAS patients.36
More recently, mutations in another actin regulator, ARPC1B, have been identified in a group of patients presenting with severe recurrent infections, inflammation, and allergy, similar to WAS. Although low platelets and recurrent infections were the initial manifestations, cutaneous vasculitis was the characteristic symptom. Leucocytoclastic vasculitis was documented in all cases undergoing a skin biopsy. Defective Treg cell function has been shown to be delineating the pathology seen in these patients.37
Signal transducer and activator of transcription (STAT) molecules are evolutionarily conserved transcription factors involved in proliferation, differentiation, migration, apoptosis, and cell survival. Autosomal-dominant STAT3-GOF mutations can cause early-onset lymphoproliferation and autoimmunity.38 The most frequent clinical manifestations are autoimmune cytopenias and lymphoproliferation followed by enteropathy, interstitial lung disease, thyroiditis, diabetes, and arthritis. Although there is no standard of care therapy, the use of anti-IL-6 receptor and Janus-activated kinase (JAK) inhibitors are promising strategies.39
Similar to STAT3, STAT1-GOF also present with multiple autoimmunities. Impaired TH17 cell differentiation and exaggerated responsiveness to type-I and -II interferons (IFNs) manifest in patients with chronic mucocutaneous candidiasis and autoimmunity, including autoimmune cytopenias, enteropathy, type-I diabetes, and SLE-like disease. Owing to the hyperinflammatory state, allogeneic HSCT outcomes have been very poor. Treatment of patients with ruxolitinib, JAK1/2 inhibitor, appear to be beneficial in controlling autoimmunity in these patients.40,41
Predominantly antibody disorders
CVID is the most common symptomatic group of PIDs with an estimated prevalence of 1/25,000. Patients manifest with recurrent sinopulmonary infections: autoimmune, inflammatory, or neoplastic diseases. There is no single disease classification for CVID due to the heterogeneous manifestations, therefore the diagnostic approach is based on exclusion. The clinical diagnostic criteria for CVID in ESID (European Society of Immune Deficiencies) registry are (1) increased susceptibility to infections, autoimmunity, lymphoproliferation, and granulomatous disease; (2) marked reduction of serum IgG, IgA, and/or IgM levels two standard deviations below the mean; (3) impaired vaccine responses or absent isohemagglutinins or low switched memory B cells; (4) exclusion of other causes of hypogammaglobulinemia; (5) exclusion of T-cell defects and (6) establishment of diagnosis after the fourth year of life.42 The prevalence of autoimmune disorders in CVID patients have been reported to be 23% in the ESID registry.43 The most prevalent autoimmune disorder in patients with CVID is autoimmune cytopenia followed by systemic and organ-specific autoimmunities.43 More recently, the analysis of the CVID cohort in USIDNET registry revealed that 1/3 of the patients having systemic autoimmunity presented with rheumatologic complications. CVID-associated rheumatologic diseases were reported to be overlapping with other inflammatory CVID complications in about one-third of the patients.44 The tendency to develop autoimmune manifestations can potentially be explained by multiple defects, including the dysregulation of B cells, impaired class-switch recombination and B cell receptor editing, increased survival of autoreactive B cells, expansion of CD21low B cells, impaired activation-induced cell death, decreased clearance of apoptotic cells and immune complexes, and persistent infections.45
During the past 10 years with the next-generation sequencing, several novel genes have been identified to be causing CVID or CVID-like diseases. Currently, approximately 10–15% of CVID patients receive genetic diagnosis.43
Identification of single-gene mutations in only a small group of the CVID population led to the idea that CVID is rather a complex disease not only inherited with classical Mendelian pattern. The proposed complex picture of CVID immunopathogenesis involves an interplay of genes, environmental factors, and dysregulation of immune cells, where gut microbiota and gastrointestinal inflammation can both be important contributors to the systemic immune activation seen in CVID and epigenetic mechanisms may be the undiscovered link between these contributors.45
XLA (X-linked agammaglobulinemia) is one of the earliest recognized primary immunodeficiencies, presenting with B-cell maturation deficiency due to the mutations in the BTK gene. In a recent multicentric study, IBD and arthritis were reported in 3.4% and 7.9% of the patients, respectively.46 BTK has been identified as a direct regulator of a key innate inflammatory pathway, the nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing 3 (NLRP3) inflammasome and NK-cell activation. Normal BTK function is essential for normal TLR-induced IL-10 production in macrophages. The impact of BTK deficiency is not fully defined, although patients exhibit higher than expected rates of inflammatory diseases suggesting dysregulation of the innate immune response.46
Hyper-IgM syndrome denotes to a group of IEIs characterized by defective CD40 signaling in B cells, which subsequently affects the class-switch recombination and somatic hypermutation. The most common hyper-IgM syndrome is caused by mutations in CD40L, which is expressed by activated CD4+ T cells.47 Four other genes (CD40, AICDA, UNG, NEMO) expressed by B cells have been associated with the hyper-IgM phenotype. In addition to the susceptibility to recurrent and opportunistic infections, these patients are also prone to autoimmune complications, especially hematologic abnormalities, autoimmune thyroid disease, nephritis, IBD, and RA. Moreover, organ-specific autoantibodies are commonly found in hyper-IgM patients.48
Phagocytic disorders
Chronic granulomatous disease (CGD) is an IEI caused by mutations in one of the five subunits of NADPH oxidase complex, leading to a profound defect in a burst of oxygen consumption that normally accompanies phagocytosis in all myeloid cells. Patients suffer from recurrent infections along with autoinflammatory and autoimmune disorders.49 Rheumatic manifestations include antiphospholipid syndrome, recurrent pericardial effusion, JIA, IgA nephropathy, SLE, and autoimmune pulmonary disease. Analysis of a French cohort of patients with CGD revealed that 17% of the patients develop autoimmune complications, the most frequent being discoid lupus erythematosus.50
Several studies delineating the role of NADPH oxidase in CGD illustrate that its effects extend beyond the predisposition to infections. NADPH oxidase has been shown to be modulating MHC-II presentation in B cells. Defects in oxidase activity may alter the peptide repertoire displayed by MHC-II, which might potentially be a mechanism contributing to the development of autoimmunity in patients with CGD.51
Complement deficiencies
The complement pathway is a complex auto-regulated system comprising of plasma proteins and cell surface receptors that react with each other initiating a cascade to opsonize the pathogens and generate small-molecule mediators to regulate both the innate and adaptive immune system.52 As part of innate immune system, complement components destroy pathogens and prevent autologous tissue damage by removing immunocomplexes and apoptotic and tumoral cells. The complement pathway contributes to adaptive immunity as well by enhancing antibody generation through priming of B cells.53
Complement deficiencies manifest with recurrent infections with encapsulated bacteria and SLE. Despite being rare, deficiencies in the early components of the complement pathway are strongly associated with SLE. In all, 93% of C1r- and C1s-deficient patients, 75% of C4-deficient patients, and 57% of C1q-deficient patients have SLE.54
Autoinflammatory disorders
Autoinflammatory diseases represent a group of immunedysregulatory disorders typically presenting in early childhood with fever and features of clinical and laboratory inflammation. Identification of disease-specific gene defects has contributed substantially to elucidate the key mechanisms of pathogenesis, including excessive IL-1 signaling, constitutive nuclear factor kappa B (NF-κB) activation, and chronic type-I IFN signaling.55
Mutations in MEFV, a gene encoding the intracellular sensor pyrin/marenostrin result in Familial Mediterranean Fever (FMF). FMF is the most prevalent monogenic autoinflammatory disease, particularly affecting the eastern Mediterranean population. FMF classically presents with episodic flares, including fever, generalized peritonitis, and non-erosive oligoarthritis. Pleuritis, pericarditis, scrotal pain might also develop. Although symptom-free intervals are characteristic for FMF, the subclinical inflammation leads to secondary amyloidosis and other sequelae in untreated patients. Pyrin is an essential protein in the NLRP3 inflammasome complex, where the mutations cause excess inflammation via caspase-1 activation and IL-1b production.55,56 A critical role of pyrin has been implicated in several important cellular functions, such as the assembly of inflammasome, intracellular danger signal sensing and activation of inflammation through inflammasome, resolution of inflammation by the autophagy of granulocytes, and regulation of inflammation through cytoskeletal rearrangements.57,58 More recently, pyrin has also been shown to modulate inactivation of Rho-GTPases involved in actin regulation in response to pathogen modifications.59 Elucidating pathways where pyrin is involved has helped us understand the efficacy of colchicine treatment, which has been the standard of care for these patients.
Cryopyrin-associated periodic syndromes (CAPS) are another classical example for disorders associated with excessive IL-1 signaling and thus respond to IL-1-inhibiting therapy. Autosomal-dominant mutations in NLRP3, encoding cryopyrin have been linked to CAPS. NLRP3 is a key protein of the NLRP3 inflammasome, activated by several stimuli and ultimately triggering conversion of pro-IL-1 to mature IL-1 by caspase-1 activation. Common clinical features of patients with CAPS are episodes of fever, neutrophilic urticaria, conjunctivitis, arthralgia, and elevated acute-phase reactants.55 Although in mild patients symptoms are limited to cold-induced episodes of inflammation with mentioned disease manifestations, most patients can develop permanent hearing loss, and severe ones also develop joint and central nervous system damage early in life. Despite this clinical heterogeneity, IL-1 inhibition is the standard therapy recommended for CAPS patients.60
Mevalonate kinase deficiency (also called hyper-IgD syndrome) is caused by homozygous mutations in the MVK gene. In addition to elevated IgD, patients present with non-erosive arthritis of large joints, cervical lymphadenopathy, abdominal pain, vomiting, diarrhea, and variable skin lesions.61,62 Mutations in MVK cause abrogation of geranylgeranyl pyrophosphate synthesis, which is an intermediate for isoprenoid and substrate used for protein prenylation. Attacks are likely caused by uncontrolled release of IL-1β as a consequence of insufficient geranylgeranyl pyrophosphate generation.63
Autosomal-dominant mutations in tumor necrosis factor receptor superfamily member 1A (TNFRSF1A), encoding the TNF receptor TNFR1, have been linked to tumor necrosis factor receptor-associated periodic fever syndrome (TRAPS). The impaired clearance of TNFR1 aggregates leads to accumulation in the cytoplasm and induction of NF-κB and triggers innate immunity and excessive IL-1b secretion.60 Patients often present with recurrent fever, abdominal pain, pleuritis, myalgias, arthralgias, periorbital edema, and conjunctivitis. Myalgias caused by a monocytic fasciitis can also be present. Neurological manifestations include headaches and, rarely, aseptic meningitis, optic neuritis, and behavioral alterations.64
Activating heterozygous mutations in NLRC4 cause a rare disorder with recurrent fever and systemic inflammation reminiscent of macrophage activation syndrome. NLRC4 encodes a nucleotide-binding oligomerization domain-like protein, which oligomerizes upon activation of caspase 1. Gain-of-function mutations cause hyperactivation of caspase 1, which induces the excess production of IL-1 and IL-18, in due course leading to an inflammatory cell death termed pyroptosis.65,66
Autoinflammatory diseases were conventionally thought of as disorders of IL-1 and related cytokines, without the presence of autoantibodies. However, recent years have revealed a group of autoinflammatory diseases due to an intrinsic IFN-alpha excess, termed the “interferonopathies.”67 The main diseases in these group are the Aicardi Goutières Syndrome (AGS),67 chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome,68 and STING-associated vasculopathy with onset in infancy (SAVI).68
Abnormal nucleic acid generation is one of the consequences of the viral replication and a major trigger for the antiviral response of the host. Therefore, the accumulation of free DNA and RNA nucleotides initiate an innate immune response activating the IFN pathway. AGS has been the prototype for type-I interferonopathies.67 There is ample evidence of pathogenesis due to mutations in TREX1; however, the underlying mechanisms of other gene defects are still unclear. Concisely, TREX1 encodes for a DNA exonuclease whose loss-of-function results in accumulation of single-stranded DNA (ssDNA). The accumulated ssDNA in turn activate DNA sensors to induce type-I IFN. Patients often present with subacute encephalomyelitis resembling a viral infection in very early infancy that causes demyelination and neurological decline.55
De novo gain-of-function mutations in TMEM173 have been reported in SAVI patients. TMEM173 encodes STING, an adaptor protein located in endoplasmic reticulum (ER). Cytosolic cGAP detects double-stranded DNA and activates to produce cGAMP, a mediator inducing STING translocation from ER to the ER–Golgi intermediate compartment.69 This translocation results in recruitment of TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3, leading to induction of type-I IFN.68 Severe vasculopathy affecting particularly distal extremities and often requiring amputation is a characteristic disease manifestation.55
Another group of autoinflammatory diseases have been characterized by dysregulation in the NF-κB. NF-κB represents a key node in propagation of cellular signals, driving cell fate decisions toward proliferation, or apoptotic clearance.70 Loss-of-function mutations in the NFKB1 gene have been depicted in patients with CVID presenting with Epstein–Barr virus susceptibility; on the other hand, activation of NF-κB occurs in many autoinflammatory diseases. Here we will focus on recently identified mutations of the NF-κB pathway.
TNFAIP3 encodes A20, a ubiquitin-removing enzyme involved in NF-κB regulation. A20 expression is induced upon NF-κB activation but acts then as a negative feedback regulator of the NF-κB signal transduction. TNFAIP3 has been associated with multiple autoimmune diseases through GWAS studies, including SLE, type-I diabetes, IBD, ankylosing arthritis, Sjögren’s syndrome, and RA.71,72 Heterozygous mutations in TNFAIP3 leading to A20 haploinsufficiency has been shown in patients presenting with Behçet-like disease, developing early-onset fevers, arthralgia, oral and genital ulcers, and ocular inflammation.73
Similar to TNFAIP3, otulin is a deubiquitinase that negatively regulates NF-κB signaling.74 Four families with homozygous mutations of OTULIN have been described with autosomal-recessive inflammatory disease with recurrent nodular panniculitis with fever and neutrophil infiltration, lipodystrophy, diarrhea, joint swelling, and failure to thrive. Recent reports have shown that HSCT resolves the disease symptoms.75
Mutations in CECR1, encoding for ADA2 enzyme, have been identified in a patient cohort with PAN-like systemic vasculitis. ADA2 deficiency (DADA2) was first described as a vasculopathy with early-onset strokes accompanied by mild immunodeficiency mainly affecting B cells.76 ADA2 is expressed in the myeloid lineage and, once secreted, induces differentiation of monocytes into macrophages, possibly by binding proteoglycan-like structures on the cellular surface.77 The main clinical features are those due to vasculitis and stroke, along with occasional hematologic and immunologic dysregulation.78 Recent reports have extended the clinical spectrum of DADA2 beyond the typical clinical picture of systemic inflammation presented in the first papers. CECR1 mutations have been demonstrated in patients with autoimmunity, lymphoproliferation, and a CID as well as patients presenting primarily with CVID.79
Conclusion
IEIs present with inflammatory complications and autoimmunity as well as severe, recurrent, or unusual infections. Autoimmunity presenting within a multisystem disease spectrum as well as therapy resistance may alert clinicians considering IEI as a possible diagnosis. Understanding the involved pathways will provide insights and influence decision making process during the daily clinical practice of pediatricians. With the genomic precision medicine approach leading to personalized treatments, better outcomes and increase in quality of life of the patients are aimed. Knowledge of IEI and the autoinflammatory disorders will enable a better management of these patients.
References
Eyre, S., Orozco, G. & Worthington, J. The genetics revolution in rheumatology: large scale genomic arrays and genetic mapping. Nat. Rev. Rheumatol. 13, 421–432 (2017).
Schmidt, R. E., Grimbacher, B. & Witte, T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat. Rev. Rheumatol. 14, 7–18 (2017).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
McIntosh, L. A. et al. Genome-wide association meta-analysis reveals novel juvenile idiopathic arthritis susceptibility loci. Arthritis Rheumatol. 69, 2222–2232 (2017).
Ombrello, M. J. et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: clinical and therapeutic implications. Ann. Rheum. Dis. 76, 906–913 (2017).
Yasin, S. & Schulert, G. S. Systemic juvenile idiopathic arthritis and macrophage activation syndrome: update on pathogenesis and treatment. Curr. Opin. Rheumatol. 30, 514–520 (2018).
McAllister, K., Eyre, S. & Orozco, G. Genetics of rheumatoid arthritis: GWAS and beyond. Open Access Rheumatol. 3, 31–46 (2011).
Taylor, J. C. et al. Genome-wide association study of response to methotrexate in early rheumatoid arthritis patients. Pharmacogenomics J. 18, 528–538 (2018).
Fischer, A. et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 140, 1388.e8–1393.e8 (2017).
Liston, A., Enders, A. & Siggs, O. M. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation. Nat. Rev. Immunol. 8, 545–558 (2008).
Notarangelo, L. D. Functional T cell immunodeficiencies (with T cells present). Annu. Rev. Immunol. 31, 195–225 (2013).
Puck, J. M. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol. Rev. 287, 241–252 (2019).
Henderson, L. A. et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J. Allergy Clin. Immunol. 132, 969.e1-2–971.e1-2 (2013).
Kuijpers, T. W. et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood 117, 5892–5896 (2011).
Farmer, J. R. et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J. Allergy Clin. Immunol. Pract. 7, 1970–1985.e4 (2019).
Volk, T. et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum. Mol. Genet. 24, 7361–7372 (2015).
Whitmore, K. V. & Gaspar, H. B. Adenosine deaminase deficiency - more than just an immunodeficiency. Front. Immunol. 7, 314 (2016).
Gokturk, B. et al. CD3G gene defects in familial autoimmune thyroiditis. Scand. J. Immunol. 80, 354–361 (2014).
Rowe, J. H. et al. Patients with CD3G mutations reveal a role for human CD3gamma in Treg diversity and suppressive function. Blood 131, 2335–2344 (2018).
Poliani, P. L., Fontana, E., Roifman, C. M. & Notarangelo, L. D. zeta Chain-associated protein of 70 kDa (ZAP70) deficiency in human subjects is associated with abnormalities of thymic stromal cells: implications for T-cell tolerance. J. Allergy Clin. Immunol. 131, 597–600 e591-592 (2013).
Chan, A. Y. et al. A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. J. Exp. Med. 213, 155–165 (2016).
Perniola, R. Twenty years of AIRE. Front. Immunol. 9, 98 (2018).
Constantine, G. M. & Lionakis, M. S. Lessons from primary immunodeficiencies: autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 287, 103–120 (2019).
Barzaghi, F. et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J. Allergy Clin. Immunol. 141, 1036.e5–1049.e5 (2018).
Gambineri, E. et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front. Immunol. 9, 2411 (2018).
Cepika, A. M. et al. Tregopathies: monogenic diseases resulting in regulatory T-cell deficiency. J. Allergy Clin. Immunol. 142, 1679–1695 (2018).
Vignoli, M. et al. CD25 deficiency: a new conformational mutation prevents the receptor expression on cell surface. Clin. Immunol. 201, 15–19 (2019).
Matsuoka, K. I. Low-dose interleukin-2 as a modulator of Treg homeostasis after HSCT: current understanding and future perspectives. Int. J. Hematol. 107, 130–137 (2018).
von Spee-Mayer, C. et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 75, 1407–1415 (2016).
Schubert, D. et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 20, 1410–1416 (2014).
Schwab, C. et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J. Allergy Clin. Immunol. 142, 1932–1946 (2018).
Slatter, M. A. et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J. Allergy Clin. Immunol. 138, 615.e1–619.e1 (2016).
Lo, B. et al. Autoimmune disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349, 436–440 (2015).
Gamez-Diaz, L. et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J. Allergy Clin. Immunol. 137, 223–230 (2016).
Lo, B. et al. CHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiency. Blood 128, 1037–1042 (2016).
Candotti, F. Clinical manifestations and pathophysiological mechanisms of the Wiskott-Aldrich Syndrome. J. Clin. Immunol. 38, 13–27 (2018).
Volpi, S. et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J. Allergy Clin. Immunol. 143, 2296–2299 (2019).
Gutierrez, M. et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol. Cell Endocrinol. 473, 166–177 (2018).
Fabre, A. et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J. Allergy Clin. Immunol. Pract. 7, 1958–1969.e9 (2019).
Mogensen, T. H. IRF and STAT transcription factors - from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front. Immunol. 9, 3047 (2018).
Weinacht, K. G. et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J. Allergy Clin. Immunol. 139, 1629–1640 e1622 (2017).
Seidel, M. G. et al. The European Society for Immunodeficiencies (ESID) Registry working definitions for the clinical diagnosis of inborn errors of immunity. J. Allergy Clin. Immunol. Pract. 7, 1763–1770 (2019).
Odnoletkova, I. et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J. Rare Dis. 13, 201 (2018).
Gutierrez, M. J., Sullivan, K. E., Fuleihan, R., Consortium, U. & Bingham, C. O. 3rd Phenotypic characterization of patients with rheumatologic manifestations of common variable immunodeficiency. Semin. Arthritis Rheum. 48, 318–326 (2018).
Jorgensen, S. F., Fevang, B. & Aukrust, P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J. Clin. Immunol. 39, 30–36 (2019).
El-Sayed, Z. A. et al. X-linked agammaglobulinemia (XLA): phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ. J. 12, 100018 (2019).
Jhamnani, R. D., Nunes-Santos, C. J., Bergerson, J. & Rosenzweig, S. D. Class-switch recombination (CSR)/hyper-IgM (HIGM) syndromes and phosphoinositide 3-kinase (PI3K) defects. Front. Immunol. 9, 2172 (2018).
Azizi, G. et al. Autoimmunity in primary antibody deficiencies. Int. Arch. Allergy Immunol. 171, 180–193 (2016).
Kang, E. M. et al. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 127, 1319–1326 (2011). Quiz 1327–1328.
Dunogue, B. et al. Chronic granulomatous disease in patients reaching adulthood: a nationwide study in France. Clin. Infect. Dis. 64, 767–775 (2017).
Gardiner, G. J. et al. A role for NADPH oxidase in antigen presentation. Front. Immunol. 4, 295 (2013).
Errante, P. R., Perazzio, S. F., Frazao, J. B., da Silva, N. P. & Andrade, L. E. Primary immunodeficiency association with systemic lupus erythematosus: review of literature and lessons learned by the Rheumatology Division of a tertiary university hospital at Sao Paulo, Brazil. Rev. Bras. Reumatol. Engl. Ed. 56, 58–68 (2016).
Lopez-Lera, A. et al. Complement as a diagnostic tool in immunopathology. Semin. Cell Dev. Biol. 85, 86–97 (2019).
Ricklin, D., Reis, E. S. & Lambris, J. D. Complement in disease: a defence system turning offensive. Nat. Rev. Nephrol. 12, 383–401 (2016).
de Jesus, A. A., Canna, S. W., Liu, Y. & Goldbach-Mansky, R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu. Rev. Immunol. 33, 823–874 (2015).
Ozen, S. & Bilginer, Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat. Rev. Rheumatol. 10, 135–147 (2014).
Manukyan, G. & Aminov, R. Update on pyrin functions and mechanisms of familial mediterranean fever. Front. Microbiol. 7, 456 (2016).
Waite, A. L. et al. Pyrin and ASC co-localize to cellular sites that are rich in polymerizing actin. Exp. Biol. Med. (Maywood) 234, 40–52 (2009).
Xu, H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014).
Sag, E., Bilginer, Y. & Ozen, S. Autoinflammatory diseases with periodic fevers. Curr. Rheumatol. Rep. 19, 41 (2017).
Hoang, T. K. & Albert, D. A. Novel presentations of periodic fever syndromes: discrepancies between genetic and clinical diagnoses. Eur. J. Rheumatol. 6, 12–18 (2019).
Laccetta, G., Tutera, M., Miccoli, M. & Consolini, R. Effects of anakinra on health-related quality of life in a patient with 1129G>A/928G>A mutations in MVK gene and heterozygosity for the mutation 2107C>A in CIAS1 gene. Front. Pediatr. 5, 128 (2017).
Drenth, J. P. et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat. Genet. 22, 178–181 (1999).
Hull, K. M. et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 81, 349–368 (2002).
Canna, S. W. et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 46, 1140–1146 (2014).
Romberg, N. et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 46, 1135–1139 (2014).
Crow, Y. J. & Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440 (2015).
Oda, H. & Kastner, D. L. Genomics, biology, and human illness: advances in the monogenic autoinflammatory diseases. Rheum. Dis. Clin. North Am. 43, 327–345 (2017).
Uggenti, C. & Crow, Y. J. Taking the STING out of inflammation. Nat. Rev. Rheumatol. 14, 508–509 (2018).
Hoeger, B., Serwas, N. K. & Boztug, K. Human NF-kappaB1 haploinsufficiency and Epstein-Barr virus-induced disease-molecular mechanisms and consequences. Front. Immunol. 8, 1978 (2017).
Dieguez-Gonzalez, R. et al. Analysis of TNFAIP3, a feedback inhibitor of nuclear factor-kappaB and the neighbor intergenic 6q23 region in rheumatoid arthritis susceptibility. Arthritis Res. Ther. 11, R42 (2009).
Vereecke, L., Beyaert, R. & van Loo, G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem. Soc. Trans. 39, 1086–1091 (2011).
Berteau, F. et al. Autosomic dominant familial Behcet disease and haploinsufficiency A20: a review of the literature. Autoimmun. Rev. 17, 809–815 (2018).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Damgaard, R. B. et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 11, e9324 (2019). pii.
Navon Elkan, P. et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N. Engl. J. Med. 370, 921–931 (2014).
Carmona-Rivera, C. et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 134, 395–406 (2019).
Ozen, S. What’s new in autoinflammation? Pediatr. Nephrol. https://doi.org/10.1007/s00467-018-4155-4 (2018).
Schepp, J. et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol. 69, 1689–1700 (2017).
Bousfiha, A. et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J. Clin. Immunol. 38, 129–143 (2018).
Author information
Authors and Affiliations
Contributions
S.K.B., S.O. and K.B. wrote and revised the manuscript. J.P. prepared the figures and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Köstel Bal, S., Pazmandi, J., Boztug, K. et al. Rheumatological manifestations in inborn errors of immunity. Pediatr Res 87, 293–299 (2020). https://doi.org/10.1038/s41390-019-0600-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0600-8
This article is cited by
-
Genetic Disorders in Pediatric Rheumatology Clinic: When to Suspect, and Why?
Indian Journal of Pediatrics (2023)
