
Abstract
Bipolar disorder (BD) is a highly heritable mood disorder with intermittent episodes of mania and depression. Lithium is the first-in-line medication to treat BD, but it is only effective in a subset of individuals. Large-scale human genomic studies have repeatedly linked the ANK3 gene (encoding ankyrin-G, AnkG) to BD. Ank3 knockout mouse models mimic BD behavioral features and respond positively to lithium treatment. We investigated cellular phenotypes associated with BD, including dendritic arborization of pyramidal neurons and spine morphology in two models: (1) a conditional knockout mouse model which disrupts Ank3 expression in adult forebrain pyramidal neurons, and (2) an AnkG knockdown model in cortical neuron cultures. We observed a decrease in dendrite complexity and a reduction of dendritic spine number in both models, reminiscent of reports in BD. We showed that lithium treatment corrected dendrite and spine deficits in vitro and in vivo. We targeted two signaling pathways known to be affected by lithium using a highly selective GSK3β inhibitor (CHIR99021) and an adenylate cyclase activator (forskolin). In our cortical neuron culture model, CHIR99021 rescues the spine morphology defects caused by AnkG knockdown, whereas forskolin rescued the dendrite complexity deficit. Interestingly, a synergistic action of both drugs was required to rescue dendrite and spine density defects in AnkG knockdown neurons. Altogether, our results suggest that dendritic abnormalities observed in loss of function ANK3 variants and BD patients may be rescued by lithium treatment. Additionally, drugs selectively targeting GSK3β and cAMP pathways could be beneficial in BD.
Similar content being viewed by others

Introduction
Bipolar disorder (BD) refers to a group of severe and chronic mental disorders characterized by unusual shifts in mood, alternating between mania or hypomania and depression, and is often associated with cognitive impairment [1]. Affecting more than 1% of the world population [2], BD is associated with a 15-fold higher suicide risk than in the general population [3]. In addition, differences in brain morphology have been observed in BD patients: patients present reductions in white and gray matter volume [4] and show structural brain abnormalities, such as thinner cortices [5]. Postmortem human brain tissue analysis from BD patients shows a decrease in spine density and spine length in the cortex compared to controls [6]. Moreover, human-induced pluripotent stem cells (hiPSCs) derived from BD patients’ neurons also present a decrease in dendrite complexity and spine density [7, 8].
Lithium is a mood stabilizer commonly used to treat BD patients as a first-line intervention [9], and it has been extensively characterized as effective in reducing depression and mania [10]. In addition, lithium has a significant effect on suicide reduction in BD patients [11]. Interestingly, BD patients treated with lithium showed normalized dendritic spine density and dendrite length decreases [6, 7]. However, less than one-third of BD patients present a full response to lithium [12]. In addition, lithium treatment is associated with several secondary effects, including cognitive side effects, weight gain, or kidney malfunction [13,14,15] that contribute to poor compliance and impaired patient outcomes.
The ANK3 gene has emerged as robustly implicated in BD. Recent genome-wide association studies (GWASs) for BD have found multiple single nucleotide polymorphisms (SNPs) in ANK3’s upstream region that are associated with the disease [16, 17]. The ANK3 gene encodes multiple isoforms of AnkG, of which the 190, 270, and 480 kDa isoforms are the most prominent in the brain [18]. The giant 270 and 480 kDa isoforms have been extensively studied due to their key roles in the axon initial segment [19], in the nodes of Ranvier [20], and GABAergic synapses [21]. However, an emerging role for the 190 kDa isoform has recently been discovered in dendritic spine maintenance and long-term potentiation through subsynaptic nanodomains in dendritic spine heads and necks [22].
In addition to likely having a contributing role in BD, ANK3 has been implicated in modifying the lithium response. The responsiveness of BD patients to lithium treatment appears to be a heritable trait [23] linked to genetic markers in patients as evidenced in GWAS [24, 25]. SNPs within the ANK3 regulatory domains were identified and associated with lithium response in BP patients. Interestingly, lithium rescues several behavioral deficits related to BD in ANKG knockout (KO) mice [26,27,28]. Multiple BD models [29, 30] and patients [31] show impaired cortical neuroarchitecture, and lithium has been shown to rescue these deficits, which could contribute to improving behavioral outcomes. Thus, there appears to be a compelling link between dendritic dysfunction and BD, as well as the mechanism of lithium action. However, the role of Ank3 in regulating dendritic stability and the response to lithium has not been assessed. Moreover, the precise molecular targets of lithium are elusive.
Evidence suggests lithium acts directly through GSK3β by inhibiting enzymatic action [32] or indirectly by inducing GSK3β serine 9 phosphorylation and autoinhibition [33]. In addition, lithium appears able to phenocopy different activities induced by GSK3β in multiple organisms [32,33,34]. Previous works have shown that inhibition of GSK3β using CHIR99021 reduced hyperlocomotion and had antidepressant-like properties in mice [35]. Another pathway, the cyclic adenosine monophosphate (cAMP) signaling pathway, plays a central role in the lithium response by regulating biological functions [36]. The cAMP level is increased in the frontal cortex after lithium stimulation [37] suggesting that lithium may mediate its actions through the regulation of cAMP levels. Moreover, a reduction of expression of Cyclic-AMP Response Element Binding Protein (CREB) in the dorsolateral prefrontal cortex of BD patients [38].
In this work, we analyzed the effect of Ank3 expression reduction on dendritic component phenotype within in vivo and in vitro Ank3 disruption models and the interaction with lithium treatment. Then, we targeted two known pathways involved in the lithium response to replicate its action and narrow a specific mechanism.
Methods and materials
Extended methods are presented in the Supplement.
Conditional KO mice
Ank3-floxed mice (The Jackson Laboratory, USA, Cat#029797) (Ank3flox/flox) were crossed with Camk2a-Cre mice (The Jackson Laboratory, USA., Cat#005359) for forebrain-specific homozygous deletion of Ank3 (Ank3flox/flox, Camk2a-cre). The resulting mouse strain was called Ank3 cKO. To maintain the colonies, Ank3 cKO mice were mated with Ank3flox/flox mice. All experiments were performed following protocols approved by the Institutional Animal Care and Use Committee at Northwestern University.
Golgi-Cox staining
We used a modified Golgi staining method adapted from the protocols used by Bayram-Weston et al. [39]. For apical dendritic spine quantification, only the first branch of each apical dendrite more than 30 µm away from the cell body was included for the analysis of spine linear density (number of spines/10-μm dendritic length). Mounted sections of 120 µm thickness were imaged on a microscope (LSM 510; Carl Zeiss) using a 63x oil lens with AxioCam Imaging System software from Zeiss (Oberkochen, Germany). For dendrite analysis, images were acquired on a Nikon (Amsterdam, Netherlands) Ti2, wide-field microscope using a 20x lens over the full slice. Traces of dendrites were drawn and analyzed with Sholl analysis in ImageJ (open source).
Neuronal cell culture and transfection
Dissociated cultures of primary cortical neurons were prepared from E18 Sprague-Dawley rat (Charles River, Wilmington MA, USA) embryos as previously described [22]. Neurons were transfected at DIV21 with Lipofectamine® 2000 Transfection Reagent (Thermo Fisher Scientific, Waltham MA, USA, Cat#11668019).
Confocal microscopy
Confocal images of immunostained neurons were obtained with a Nikon (Amsterdam, Netherlands) C2 confocal microscope. Images of neurons were taken using the 63× oil-immersion objective (numerical aperture (NA) = 1.4) as z-series of 7–12 images, averaged two times, taken at 0.4-μm intervals, with 1024 × 1024 pixel resolution. Detector gain and offset were adjusted in the channel of each cell fill (GFP or mCherry) to include all spines and enhance edge detection. Intensity plot profiles for dendrite/spine localization were performed in ImageJ. A 4-µm line was drawn across 3–5 spines per neuron and averaged across neurons to produce average intensity plot profiles ± standard error of the mean (SEM). Spine density, width, and length were analyzed with ImageJ. Epifluorescence images were obtained with a 10× objective, and traces of dendrites were drawn and analyzed with Sholl analysis in ImageJ. In culture, pyramidal neurons conserve their polarity and develop a long dendrite, which we refer to as the apical dendrite, and shorter dendrites near the soma, which we refer to as basal dendrites for Sholl analysis.
Pharmacological treatments
Nine weeks old wild-type and cKO mice were fed with 0.2% lithium carbonate chow for 1 week followed by 2 weeks of 0.4% lithium carbonate chow (Envigo Teklad, Indianapolis IN, USA, TD.05573 and TD.05185. All mice were given supplemental drinking water with 0.9% (w/v) sodium chloride to counteract the potential toxicity of lithium for 3 weeks. Lithium blood concentration was accessed with Quadrupole ICP-MS (Thermo iCapQ) for 0.88mEq/L on average.
Cortical neuron cultures were treated with lithium chloride (Sigma-Aldrich, Saint-Louis MO, USA Cat#P6407) at 2 mM, CHIR99021 (LC Laboratories, Woburn MA, USA, Cat#C-6556) at 5 µM, Forskolin (Cell Signaling Technology, Danvers MA, USA Cat#3828) at 50 µM, or the corresponding vehicle for 24 h. The PKA inhibitor Rp-8-Br-cAMPS (Biolog Life Science Institute, Bremen, Germany, Cat# B 001) was used 30 min before lithium treatment at 100 μM.
Results
AnkG maintains dendritic spine density and dendrite complexity in pyramidal neurons in vivo and in vitro
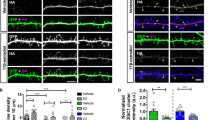
To investigate the role of AnkG expression on dendrite complexity and dendritic spine morphology, we performed Golgi-Cox staining in Ank3 cKO (Ank3flox/flox: Camk2a-Cre) mouse brain slices (Fig. 1A, D). In this model, Cre recombinase is expressed under a Camk2a promoter to drive the deletion of the Ank3 gene in the postnatal forebrain [28]. As expected, western blot and immunostaining experiments showed robust reductions in AnkG protein expression in the cortex of cKO mice (Supplementary Fig. 1A–D). Quantitative analysis revealed a decrease in spine density in the Ank3 cKO cortex (t(90) = 5.593, p < 0.0001, Fig. 1B, E, G), which supports our previous in vitro findings [22]. We also observed with Sholl analysis that Ank3 cKO mice had striking alterations in dendrite morphology with a significant decrease in basal dendritic complexity between 20 and 50 μm (Fig. 1C, F, H). To specifically determine whether the effects in dendrites were cell-autonomous or non-autonomous, we knocked down AnkG expression in cultured rat primary cortical neurons using a previously described AnkG RNAi, which induced a 50% decrease of AnkG staining in dendrites after 3 days [22]. In line with in vivo results, we observed reduced complexity in the basal dendritic arbor of AnkG knockdown pyramidal neurons (Fig. 1K), with no change in apical dendrites (Fig. 1L). AnkG-190 has an emerging role in regulating dendritic morphological structures [22]. To identify the Ank3 isoform involved in mediating this effect, we co-expressed RNAi-resistant constructs of different isoforms. In line with our previous reports, co-expression of an RNAi-resistant AnkG-190 construct restored dendritic complexity, whereas co-expression of an RNAi-resistant AnkG-270 construct produced no effect (Fig. 1I–L), supporting AnkG-190 as a key regulator of dendritic stability.

A–F Bright-field images of Golgi-Cox staining in somatosensory cortical slices from (A) wild-type (WT) and (D) Ank3 cKO mice, scale = 100 µm. Dendrite pictures (B and E, scale = 5 µm) and representative traces of basal dendrites (C and F, scale = 20 µm) from pyramidal neurons from cortical layer 2/3 (G) Scatter plot of spine density from WT and AnkG cKO mice (46 cells, 3 brains per group, t-test, ± SEM, ***p ≤ 0.001). H Graph showing Sholl analysis for basal dendrites (20 neurons, 3 brains per group, 2-way ANOVA with Sidak’s post-test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM. (I) Representative traces of cultured rat neurons expressing control or AnkG RNAi with GFP (no AnkG), GFP-AnkG-190, GFP-AnkG-270. Scale = 100 µm. Graphs showing Sholl analysis for total (J), basal (K), and apical (L) dendrites (15–22 neurons, 2-way ANOVA with Dunnett’s post-test and row repeated measures, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM).
Lithium treatment rescues dendrite and spine deficits in AnkG–deficient pyramidal neurons in the somatosensory cortex in Ank3 cKO mice
Lithium treatment attenuates anxiety behaviors [26] and other psychiatric-related behaviors [27] in Ank3-deficient mouse models. Moreover, lithium corrects mania-like behavior in the Ank3 cKO mouse [28]. To determine the neuronal mechanism of these behavioral effects, we assessed lithium’s ability to rescue morphological deficits (Fig. 2A, C) in Ank3 cKO mice. As expected, vehicle-treated Ank3 cKO animals were found to have a significant decrease in spine density and dendritic complexity within the L2/3 pyramidal neurons in the cortex. However, lithium treatment in Ank3 cKO mice increased dendritic spine density within these neurons (Fig. 2A, B). Lithium-treated Ank3 cKO mice also showed recovery in the proximal portion of the basal dendrites (Fig. 2C, D). These results indicate that lithium can rescue dendritic abnormalities linked to loss of Ank3 and that dendritic morphological recovery may play a role in the ability of lithium to induce behavioral recovery. Interestingly, no significant effects on WT neuron spine density were observed (Fig. 2B). Similarly, lithium treatment failed to evoke alterations in basal dendrite complexity in WT mice (Fig. 2D). It appears that loss of AnkG modifies the neuronal response to lithium in Ank3 cKO animals.

A Bright-field dendrite pictures of Golgi-Cox staining from somatosensory cortical pyramidal neurons from cortical layer 2/3 of wild-type and Ank3 cKO mice treated with or without lithium carbonate (scale = 20 µm). B Scatter plot of spine density from WT and AnkG cKO mice (37–45 cells, 3 brains per group, 2-way ANOVA with Dunnett’s post-test, ±SEM, #p ≤ 0.0001). C Representative traces of basal dendrites (scale = 20 µm) from somatosensory cortical pyramidal neurons from cortical layer 2/3. D Graph showing Sholl analysis for basal dendrites (23–29 neurons, 3 brains per group, 2-way ANOVA with Dunnett’s post-test and row repeated measures, ***p ≤ 0.001, ±SEM).
Lithium treatment rescues dendritic complexity and spine deficits in AnkG knockdown neuronal culture
We tested whether lithium could reverse dendrite and dendritic spine deficits caused by reduced AnkG expression in vivo. Previous studies from our group showed that 24 h or less of different pharmacological treatments was enough to modify significantly the dendrite complexity in DIV21 mature cultures [40, 41]. Therefore, we treated control neurons and AnkG knockdown neurons with either the vehicle or lithium for 24 h and assessed spine density and dendrite morphology measurements. As previously published, then consistent with cKO animals (Fig. 1), AnkG knockdown induced a decrease in dendritic spine density and size (Fig. 3A–D). Lithium treatment rescued the dendritic spine deficits in apical dendrites, resulting in the normalization of linear spine density, as well as a significant increase in spine width and length in AnkG knockdown neurons (Fig. 3E–H). These in vitro data corroborate the rescue effects performed in vivo (Fig. 1) and provide a pliable system to assess the mechanism of lithium action.

A Confocal images of mCherry from cultured rat neurons transfected for three days with control or AnkG RNAi and treated with lithium chloride, scale = 10 µm. B–D Scatter plots showing spine density, spine length, and spine head width (15–44 neurons from 3 independent experiments, 2-way ANOVA with Tukey’s post-test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM). E Representative traces of cultured rat neurons expressing control or AnkG RNAi, ±lithium chloride, scale = 100 µm. F–H Sholl analysis graph for total, basal, and apical dendrites (28–74 neurons from 3 independent experiments, 2-way ANOVA with Dunnett’s post-test row repeated measures, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM).
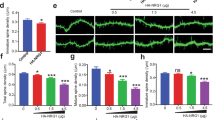
Inhibiting glycogen synthase kinase 3 beta (GSK3β) rescues spine morphology, but not spine density or dendritic complexity
It is unclear whether GSK3β is the main driver of the lithium effect, or if GSK3β can recapitulate the effects of lithium on ANK3-induced dendritic morphology. We decided to use CHIR99021 to study the involvement of GSK3β in the lithium-mediated recovery of Ank3 knockdown morphological deficits. We incubated control neurons and AnkG knockdown cortical neurons with CHIR99021 for 24 h and analyzed dendritic spines and dendrite complexity. Using CHIR99021 to inhibit GSK3β significantly reduced dendritic spine density, width, and length in the control condition (Fig. 4A–D). Surprisingly, CHIR99021 produced a different effect in AnkG knockdown neurons, rescuing dendritic spine width and length, but not density (Fig. 4A–D). In addition to dendritic spines, CHIR99021 induced a reduction in basal dendritic complexity in the control condition; however, CHIR99021 failed to alter dendrite complexity following the knockdown of AnkG (Fig. 4E–H). Those results indicate that inhibition of GSK3β induces a reduction in dendrite complexity and dendritic spine size in control neurons. However, this response is altered following AnkG knockdown, allowing the rescue of AnkG-mediated spine loss. To validate that Lithium acts through GSK3β inhibition, we overexpressed the GFP-GSK3βSA9 construction [42] to prevent the phosphorylation of serine 9 by lithium (Fig. 6). GFP-GSK3βSA9 did not affect dendrite complexity rescue by lithium (Fig. 6E, F). However, rescues of spine dendritic features affected previously by CHIR99021 in Fig. 4, were prevented in presence of the construct (Fig. 6C, D).

A Confocal images of mCherry from cultured rat neurons were transfected for three days with control or AnkG RNAi and treated with CHHIR99021 (scale = 10 µm). B–D Scatter plots showing spine density, spine length, and spine head width (21 neurons from 3 independent experiments, 2-way ANOVA with Dunnett’s post-test, *p ≤ 0.05, ***p ≤ 0.001, ±SEM). E Representative traces of cultured rat neurons expressing control or AnkG RNAi, ±CHHIR99021, scale = 100 µm. F–H Sholl analysis graph for total, basal, and apical dendrites (27–30 neurons from 3 independent experiments, 2-way ANOVA with Dunnett’s post-test and row repeated measures, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM).
The adenylyl cyclase activator, forskolin, rescues dendritic complexity
We assessed the role of cAMP in Ank3-mediated dendritic structural abnormalities, using forskolin to activate adenylate cyclase and increase intracellular levels of cAMP [43]. After 24 h of treatment on cultured neurons, increased cAMP was found to induce morphological alterations, distinct and complementary to CHIR99021: Forskolin treatment resulted in no effect on the morphology or linear density of dendritic spines in either control neurons or AnkG knockdown neurons (Fig. 5A–D). However, forskolin completely rescued basal dendritic complexity deficits observed in knockdown neurons and enhanced the complexity of basal dendrites in the control as compared to the condition with the vehicle (Fig. 5E–H). These results suggest that cAMP induction results in a recovery of dendritic complexity following a loss of AnkG. Interestingly, despite reports of both cAMP signaling and GSK3β contributing to lithium efficacy, the effects of cAMP induction were different from those of GSK3β, suggesting diverse mechanisms of lithium action on dendritic morphologies. Lithium has been described to increase PKA activity in rat frontal cortex [44] and cortical neuron culture [45]. To assess the necessity of cAMP activation during the lithium rescue, we experimented with the presence of the PKA inhibitor Rp-8-Br-cAMPS we studied neuronal morphology effects of lithium in the presence of the PKA inhibitor Rp-8-Br-cAMPS (Fig. 6). As hypothesized, inhibition of PKA blocked the dendritic complexity rescue by lithium supporting the role of PKA in the lithium pathway response (Fig. 6E, F).

A Confocal images of mCherry from cultured rat neurons transfected for three days with control or AnkG RNAi and treated with or without forskolin or forskolin + CHIR99021, scale = 10 µm. B–D Scatter plots showing spine density, spine length, and spine head width (23–30 neurons from 3 independent experiments, 2-way ANOVA with Dunnett’s post-test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM). E Representative traces of cultured rat neurons expressing control or AnkG RNAi, ±forskolin or forskolin + CHIR99021, scale = 100 µm. F–H Sholl analysis graph for total, basal, and apical dendrites (41–46 neurons from 3 independent experiments, 2-way ANOVA with Dunnett’s post-test row repeated measures, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ±SEM).

A Confocal images of GFP from cultured rat neurons transfected for three days with control or AnkG RNAi with or without HA-GSK3βS9A and treated with or without lithium chloride and Rp-8-Br-cAMPS, scale = 10 µm. B–D Scatter plots showing spine density, spine length, and spine head width (20 neurons from 2 independent experiments, 1-way ANOVA with Dunnett’s post-test, **p < 0.01, #p < 0.0001, ±SEM). E Representative traces of cultured rat neurons expressing control or AnkG RNAi, +/- HA-GSK3βS9A and treated with or without lithium chloride and Rp-8-Br-cAMPS, scale = 100 µm. F–H Sholl analysis graph for total, basal, and apical dendrites (41–46 neurons from 3 independent experiments, 2-way ANOVA with Dunnett’s post-test row repeated measures, *p < 0.05, #p < 0.0001, ±SEM).
CHIR99021 and forskolin synergize to rescue dendritic spine density
As explained above, lithium was found to result in a complete reversal of dendritic morphological deficits, both a recovery in dendritic spine density and size as well as basal dendrite complexity. Specific targeting of each pathway failed to emulate the entire lithium response. However, the ability of GSK3β inhibition to recover dendritic spine deficits and cAMP induction through forskolin treatment to rescue basal dendrite complexity suggests that lithium may act synergistically through these pathways. We set out to assess whether dual pathway regulation may result in a complete recovery of dendritic morphological abnormalities in AnkG knockdown neurons. Following 24-h treatment of cultured neurons with a combination of CHIR99021 and forskolin, both dendritic spine density/morphology (Fig. 5A–D) and basal dendrite complexity (Fig. 5E–H) were none significantly different suggesting forskolin could compensate for the CHIR99021 dendritic spines and dendrites deficits observed in Fig. 4. As expected, the combination of the two drugs acted as their sum, rescuing dendritic spine size and dendritic complexity in AnkG knockdown neurons. Moreover, the combined treatment normalized the spine density in AnkG knockdown neurons (Fig. 5B) which neither of the drugs could do alone, implying a possible synergy between the two pathways. Blocking individually each pathway prevented the spine density rescue (Fig. 6B).
Taken together, these data suggest that dual GSK3β inhibition and cAMP activation are sufficient to replicate the lithium response, and rescue dendritic deficits observe in Ank3 knockdown neurons (Supplementary Fig. 2).
Discussion
In this study, we demonstrate a role for AnkG in the arborization of pyramidal neuronal dendrites and dendritic spine density in vivo. Multiple Ank3 KO mouse models have severe behavioral deficits related to psychiatric disorders [26,27,28,29,30,31]. Our data support that those phenotypes may, at least in part, depend on alterations of dendrite and spine architecture. Our findings strongly support the role of the AnkG-190 isoform as a part of the cell-autonomous maintenance of basal dendrite arborization in pyramidal neurons. AnkG-190 has been previously described as enriched in dendrites and dendritic spines whereas AnkG-270 was enriched in axons [22]. Moreover, AnkG-190 expression and dendritic spines density decreased in Ank3 W1989R mutant mice [46], and Ank3 mutant mice disrupting specifically AnkG-270 and AnkG-480 isoforms induce overexpression of AnkG-190 associated with an increase in dendritic spine density [47]. Together, as the L2/3 somatosensory cortex is implicated in BD and mood disorders [48] and excitatory neurons in this region are reported to display morphological impairments in BD patients [6], ANK3-mediated dendritic dysfunction may contribute to excitatory dysfunction in BD. Moreover, excitatory deficits in this region are likely to rely on the selective expression of the AnkG-190 isoform within glutamatergic neuronal dendrites, where the loss of AnkG-190 can mimic BD patient neuronal structural deficits. Altered neuronal morphology modifies neuronal functional properties and activity, and has a central role in neuropsychiatric diseases [49]. BD patient brain tissue in postmortem studies [6] and hiPSCs derived from the neurons [7, 8] both present reduced dendritic length and spine density. Multiple risk factors including genetic models, environmental manipulation, or drug injections have been studied in animal models and reproduced behavioral aspects of human BD [50]. GWAS studies have identified 65 genes (including ANK3) associated with SNPs enriched in BP [16] and several of them exhibit alterations in dendrite length or complexity, dendritic spine density or size, or both. In addition to genetic origin, other BD models show a modification in dendritic spines or dendrite lengths like sleep deprivation [51] amphetamine sensitization [52], and chronic social defeat [53]. Thus, dendritic morphology and spine density alterations appear to be hallmarks of BD.
The ability of lithium to provide relief to patients, alongside a normalization of cortical dendritic phenotypes in BD [6, 7], suggests that dendritic structural abnormalities may be an important facet of BD etiology. Lithium treatment at therapeutic doses does not present a significant clinical effect to stabilize mood variability in normal human subjects [54] and responsiveness to lithium depends in part on genetic factors [23]. One of the strongest factors in BD is CNVs at the 22q13.3 locus, duplicating the expression of SHANK3. In line with this, Shank3-overexpression animals display manic-like behavior but present an increase in dendritic spines in cultured hippocampal pyramidal neurons [55]. Interestingly, lithium will not rescue behaviors of this mouse model, suggesting that lithium may selectively recover dendritic spine loss [56]. Indeed, Shank3 deletion mice show a reduction in dendritic spine density in the hippocampus similar to the Ank3 cKO model and respond to lithium [57]. Together, these results suggest that there is a strong correlation between the mechanisms of BD, concerning dendritic morphology, and the response to lithium. It is also interesting to note that lithium has been shown to rescue mood-related behavioral deficits in different Ank3 KO mouse models [26, 27] including the same cKO mouse we use in this our work [28], without affecting wild-type mice, suggesting that ANK3 expression levels may affect the lithium response. Indeed, following Ank3 cKO and AnkG knockout, lithium selectivity increased spine densities, an effect absent in WT animals. Together, these observations suggest that lithium efficacy may selectively recover phenotypes associated with dendritic spine loss and reduced dendritic complexity and that AnkG-190 levels play a role in dictating the presence of these phenotypes in AnkG CKO animals. Moreover, AnkG-190 levels may regulate the ability of lithium to regulate excitatory dendritic abilities.
However, the precise targets and mechanisms of lithium action are elusive, with multiple signaling pathways affected by lithium treatment. A recent study observed a reduction in the expression of KIF5B and other proteins involved in the axonal structure and transport and glutamatergic neurotransmission in Ank3± mice leading to a potential action of lithium-induced GSK3β inhibition on axon and synaptic deficits [27]. Later, it was demonstrated that specific inhibition of GSK3β was able to rescue EB3 expression increase and tubulin polymerization decrease in Ank3 knockout mice [58]. Interestingly, despite this, GSK3β KO mice and mice treated by GSK3β inhibitor present a decrease in dendritic spine density and head volume in the hippocampus seemingly at odds with structural deficits observed in BD patients, and the observed effect of lithium [59]. In our in vitro experiments, we observed the same decrease in dendritic spine metrics. However, our results revealed that AnkG-190 loss modified both the lithium and GSK3β response on dendritic phenotypes in excitatory neurons. Indeed, GSK3β treatment mimicked lithium recovery of spine morphology in AnkG knockdown, further supporting GSK3B as an important mediator of the lithium response, and AnkG as a regulator of these responses. Despite this, GSK3b inhibition was unable to rescue all dendritic abnormalities, suggesting the presence of distinct signaling pathways affected by lithium. In previous work, Gottschalk et al. showed with mass spectrometry than Ank3 mutant mice have altered protein expression in the hippocampus compared to the wild-type mouse. Therefore, the specificity of GSK3β inhibitor action could be genotype-dependent and be explained by the difference in protein expression causing aberrant signaling in Ank3 mutant neurons [27]. Moreover, another pathway may be recruited during lithium treatment in the control condition and not in the AnkG KD condition to compensate for GSK3β inhibition which can’t be observed during the use of GSK3β inhibitor alone.
SNPs of cAMP signaling pathway genes have been associated with BD [60]. Moreover, several downstream partners of cAMP are implicated in dendritic complexity and dendritic spine morphology like PKA and EPAC2 [41, 61], making cAMP a potential target to rescue phenotype observed in our AnkG knockdown. Forskolin is used during acute stimulation to induce long-term potentiation [62] and has been shown to rescue some Ank3 cKO mouse behavioral phenotypes in acute treatment [63] but a few are now about longer stimulation. After 24 h of incubation, we didn’t see any increase in dendritic spine metrics in both control and AnkG knockdown (Fig. 5B, C). It was not expected for the control condition since LTP induces an increase in spine size [64] but it can underline a compensation mechanism over time. For the dendritic complexity, forskolin rescued the deficit observed in AnkG knockdown neurons and even increased dendritic complexity in the control (Fig. 5E–H). Those data suggest that the forskolin effect is independent of the presence or absence of AnkG. Finally, during the drug combination, we observed the rescue of spine size implying forskolin didn’t interfere with GSK3B inhibition action (Fig. 5C, D). However, both drugs seem to act in different pathways affecting dendritic complexity and leading to a normalization of the different metrics close to the control condition suggesting additive effects. Surprisingly, the drug combination was able to rescue the dendritic spine density revealing a synergy between those two pathways and their importance in lithium rescue.
It became important to find early markers to complement this approach and help to design the appropriate treatment for the appropriate patient. One of those methods is the analysis of mRNA expression as blood biomarkers for neuropsychiatric diseases [65] including BD [66] which associates change in ANK3 mRNA with BD and SZ [67]. In addition, ANK3 is associated with mood regulation, stress response, and survivability and, is possibly linked to the antidepressant mianserin response[68]. Unfortunately, multiple GWAS found few genetic markers in BP associated with lithium response [24, 25]. However, ANK3 is a part of a glutamate receptor network with SNPs associated with lithium response [69] and is included in an overrepresented gene set of variants responding to lithium [70]. Our data support Ank3 expression reduction is associated with a reduction in dendritic features in the cortex. Importantly, lithium was ineffective in WT mice in altering neuroarchitecture, suggesting a role for Ank3 expression in the lithium response efficiency. Finally, we showed that drugs selectively targeting GSK3β and cAMP pathways recapitulate lithium action in neurons in vitro, and blocking those pathways will prevent lithium action. We showed here that those two pathways were necessary and sufficient to rescue all dendritic abnormalities and revealed a synergistic regulation that could be beneficial in BD therapeutic interventions. However, while facilitating drug and neuron analysis, our in vitro model remains limited, especially on the treatment timing Even though lithium can affect molecular pathways in vivo during the first hours [71], it is currently unknown whether lithium treatment would impact dendrite and spine morphology in this time frame. Thus, the chronic lithium treatment in vivo could recruit other pathways to lead to the modification observed in this work. Therefore, further analysis of those pathways in vivo in Ank3 and other BD models, especially behavioral rescues, will help to strengthen this hypothesis.
References
Cotrena C, Branco LD, Kochhann R, Shansis FM, Fonseca RP. Quality of life, functioning and cognition in bipolar disorder and major depression: a latent profile analysis. Psychiatry Res. 2016;241:289–96.
Alonso J, Petukhova M, Vilagut G, Chatterji S, Heeringa S, Ustun TB, et al. Days out of role due to common physical and mental conditions: results from the WHO World Mental Health surveys. Mol Psychiatry. 2011;16:1234–46.
Harris EC, Barraclough B. Suicide as an outcome for mental disorders. A meta-analysis. Br J Psychiatry. 1997;170:205–28.
Angelescu I, Brugger SP, Borgan F, Kaar SJ, Howes OD. The magnitude and variability of brain structural alterations in bipolar disorder: a double meta-analysis of 5534 patients and 6651 healthy controls. J Affect Disord. 2021;291:171–6.
Abe C, Ekman CJ, Sellgren C, Petrovic P, Ingvar M, Landen M. Cortical thickness, volume and surface area in patients with bipolar disorder types I and II. J Psychiatry Neurosci. 2016;41:240–50.
Konopaske GT, Lange N, Coyle JT, Benes FM. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 2014;71:1323–31.
Tobe BTD, Crain AM, Winquist AM, Calabrese B, Makihara H, Zhao WN, et al. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci USA. 2017;114:E4462–E71.
Ishii T, Ishikawa M, Fujimori K, Maeda T, Kushima I, Arioka Y, et al. In vitro modeling of the bipolar disorder and schizophrenia using patient-derived induced pluripotent stem cells with copy number variations of PCDH15 and RELN. ENEURO.0403-18.2019.
Malhi GS, Gessler D, Outhred T. The use of lithium for the treatment of bipolar disorder: Recommendations from clinical practice guidelines. J Affect Disord. 2017;217:266–80.
Geddes JR, Burgess S, Hawton K, Jamison K, Goodwin GM. Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am J Psychiatry. 2004;161:217–22.
Cipriani A, Hawton K, Stockton S, Geddes JR. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646.
Garnham J, Munro A, Slaney C, Macdougall M, Passmore M, Duffy A, et al. Prophylactic treatment response in bipolar disorder: results of a naturalistic observation study. J Affect Disord. 2007;104:185–90.
Zill P, Malitas PN, Bondy B, Engel R, Boufidou F, Behrens S, et al. Analysis of polymorphisms in the alpha-subunit of the olfactory G-protein Golf in lithium-treated bipolar patients. Psychiatr Genet. 2003;13:65–9.
Rybakowski JK, Abramowicz M, Szczepankiewicz A, Michalak M, Hauser J, Czekalski S. The association of glycogen synthase kinase-3beta (GSK-3beta) gene polymorphism with kidney function in long-term lithium-treated bipolar patients. Int J Bipolar Disord. 2013;1:8.
Dunner DL. Optimizing lithium treatment. J Clin Psychiatry. 2000;61:76–81.
Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021;53:817–29.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803.
Zhang X, Bennett V. Restriction of 480/270-kD ankyrin G to axon proximal segments requires multiple ankyrin G-specific domains. J Cell Biol. 1998;142:1571–81.
Leterrier C, Dargent B. No Pasaran! Role of the axon initial segment in the regulation of protein transport and the maintenance of axonal identity. Semin Cell Dev Biol. 2014;27:44–51.
Nelson AD, Jenkins PM. Axonal membranes and their domains: assembly and function of the axon initial segment and node of Ranvier. Front Cell Neurosci. 2017;11:136.
Tseng WC, Jenkins PM, Tanaka M, Mooney R, Bennett V. Giant ankyrin-G stabilizes somatodendritic GABAergic synapses through opposing endocytosis of GABAA receptors. Proc Natl Acad Sci USA. 2015;112:1214–9.
Smith KR, Kopeikina KJ, Fawcett-Patel JM, Leaderbrand K, Gao R, Schurmann B, et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 2014;84:399–415.
Grof P, Duffy A, Cavazzoni P, Grof E, Garnham J, MacDougall M, et al. Is response to prophylactic lithium a familial trait? J Clin Psychiatry. 2002;63:942–7.
Hou L, Heilbronner U, Degenhardt F, Adli M, Akiyama K, Akula N, et al. Genetic variants associated with response to lithium treatment in bipolar disorder: a genome-wide association study. Lancet 2016;387:1085–93.
Perlis RH, Smoller JW, Ferreira MA, McQuillin A, Bass N, Lawrence J, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009;166:718–25.
Leussis MP, Berry-Scott EM, Saito M, Jhuang H, de Haan G, Alkan O, et al. The ANK3 bipolar disorder gene regulates psychiatric-related behaviors that are modulated by lithium and stress. Biol Psychiatry. 2013;73:683–90.
Gottschalk MG, Leussis MP, Ruland T, Gjeluci K, Petryshen TL, Bahn S. Lithium reverses behavioral and axonal transport-related changes associated with ANK3 bipolar disorder gene disruption. Eur Neuropsychopharmacol. 2017;27:274–88.
Zhu S, Cordner ZA, Xiong J, Chiu CT, Artola A, Zuo Y, et al. Genetic disruption of ankyrin-G in adult mouse forebrain causes cortical synapse alteration and behavior reminiscent of bipolar disorder. Proc Natl Acad Sci USA. 2017;114:10479–84.
Liu C, Zhang L, Wu J, Sui X, Xu Y, Huang L, et al. AnkG hemizygous mice present cognitive impairment and elevated anxiety/depressive-like traits associated with decreased expression of GABA receptors and postsynaptic density protein. Exp Brain Res. 2017;235:3375–90.
van der Werf IM, Van Dam D, Missault S, Yalcin B, De Deyn PP, Vandeweyer G, et al. Behavioural characterization of AnkyrinG deficient mice, a model for ANK3 related disorders. Behav Brain Res. 2017;328:218–26.
Lopez AY, Wang X, Xu M, Maheshwari A, Curry D, Lam S, et al. Ankyrin-G isoform imbalance and interneuronopathy link epilepsy and bipolar disorder. Mol Psychiatry. 2017;22:1464–72.
Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–9.
Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–77.
Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–8.
Pan JQ, Lewis MC, Ketterman JK, Clore EL, Riley M, Richards KR, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology. 2011;36:1397–411.
Roux M, Dosseto A. From direct to indirect lithium targets: a comprehensive review of omics data. Metallomics. 2017;9:1326–51.
Wiborg O, Kruger T, Jakobsen SN. Region-selective effects of long-term lithium and carbamazepine administration on cyclic AMP levels in rat brain. Pharm Toxicol. 1999;84:88–93.
Ren X, Rizavi HS, Khan MA, Bhaumik R, Dwivedi Y, Pandey GN. Alteration of cyclic-AMP response element binding protein in the postmortem brain of subjects with bipolar disorder and schizophrenia. J Affect Disord. 2014;152-154:326–33.
Bayram-Weston Z, Olsen E, Harrison DJ, Dunnett SB, Brooks SP. Optimising Golgi-Cox staining for use with perfusion-fixed brain tissue validated in the zQ175 mouse model of Huntington’s disease. J Neurosci Methods. 2016;265:81–8.
Blizinsky KD, Diaz-Castro B, Forrest MP, Schurmann B, Bach AP, Martin-de-Saavedra MD, et al. Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc Natl Acad Sci USA. 2016;113:8520–5.
Srivastava DP, Woolfrey KM, Jones KA, Anderson CT, Smith KR, Russell TA, et al. An autism-associated variant of Epac2 reveals a role for Ras/Epac2 signaling in controlling basal dendrite maintenance in mice. PLoS Biol. 2012;10:e1001350.
Kirshenboim N, Plotkin B, Shlomo SB, Kaidanovich-Beilin O, Eldar-Finkelman H. Lithium-mediated phosphorylation of glycogen synthase kinase-3beta involves PI3 kinase-dependent activation of protein kinase C-alpha. J Mol Neurosci. 2004;24:237–45.
Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA. 1981;78:3363–7.
Mori S, Tardito D, Dorigo A, Zanardi R, Smeraldi E, Racagni G, et al. Effects of lithium on cAMP-dependent protein kinase in rat brain. Neuropsychopharmacology. 1998;19:233–40.
Liang MH, Wendland JR, Chuang DM. Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol Cell Neurosci. 2008;37:440–53.
Nelson AD, Caballero-Floran RN, Rodriguez Diaz JC, Hull JM, Yuan Y, Li J, et al. Ankyrin-G regulates forebrain connectivity and network synchronization via interaction with GABARAP. Mol Psychiatry. 2020;25:2800–17.
Jenkins PM, Kim N, Jones SL, Tseng WC, Svitkina TM, Yin HH, et al. Giant ankyrin-G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci USA. 2015;112:957–64.
Kropf E, Syan SK, Minuzzi L, Frey BN. From anatomy to function: the role of the somatosensory cortex in emotional regulation. Braz J Psychiatry. 2019;41:261–9.
Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci. 2018;19:215–34.
Beyer DKE, Freund N. Animal models for bipolar disorder: from bedside to the cage. Int J Bipolar Disord. 2017;5:35.
Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife. 2016;13424.001
Arroyo-Garcia LE, Tendilla-Beltran H, Vazquez-Roque RA, Jurado-Tapia EE, Diaz A, Aguilar-Alonso P, et al. Amphetamine sensitization alters hippocampal neuronal morphology and memory and learning behaviors. Mol Psychiatry. 2021;26:4784–94.
Colyn L, Venzala E, Marco S, Perez-Otano I, Tordera RM. Chronic social defeat stress induces sustained synaptic structural changes in the prefrontal cortex and amygdala. Behav Brain Res. 2019;373:112079.
Barton CD Jr, Dufer D, Monderer R, Cohen MJ, Fuller HJ, Clark MR, et al. Mood variability in normal subjects on lithium. Biol Psychiatry. 1993;34:878–84.
Han K, Holder JL Jr, Schaaf CP, Lu H, Chen H, Kang H, et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature. 2013;503:72–7.
Tatavarty V, Torrado Pacheco A, Groves Kuhnle C, Lin H, Koundinya P, Miska NJ, et al. Autism-associated Shank3 is essential for homeostatic compensation in rodent V1. Neuron. 2020;106:769–77 e4.
Song TJ, Lan XY, Wei MP, Zhai FJ, Boeckers TM, Wang JN, et al. Altered behaviors and impaired synaptic function in a novel rat model with a complete Shank3 deletion. Front Cell Neurosci. 2019;13:111.
Garza JC, Qi X, Gjeluci K, Leussis MP, Basu H, Reis SA, et al. Disruption of the psychiatric risk gene Ankyrin 3 enhances microtubule dynamics through GSK3/CRMP2 signaling. Transl Psychiatry. 2018;8:135.
Ochs SM, Dorostkar MM, Aramuni G, Schon C, Filser S, Poschl J, et al. Loss of neuronal GSK3beta reduces dendritic spine stability and attenuates excitatory synaptic transmission via beta-catenin. Mol Psychiatry. 2015;20:482–9.
McDonald ML, MacMullen C, Liu DJ, Leal SM, Davis RL. Genetic association of cyclic AMP signaling genes with bipolar disorder. Transl Psychiatry. 2012;2:e169.
Jones KA, Sumiya M, Woolfrey KM, Srivastava DP, Penzes P. Loss of EPAC2 alters dendritic spine morphology and inhibitory synapse density. Mol Cell Neurosci. 2019;98:19–31.
Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, et al. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–62.
Yoon S, Piguel NH, Khalatyan N, Dionisio LE, Savas JN, Penzes P. Homer1 promotes dendritic spine growth through ankyrin-G and its loss reshapes the synaptic proteome. Mol Psychiatry. 2021;26:1775–89.
Fifkova E, Van Harreveld A. Long-lasting morphological changes in dendritic spines of dentate granular cells following stimulation of the entorhinal area. J Neurocytol. 1977;6:211–30.
Niculescu AB, Le-Niculescu H, Roseberry K, Wang S, Hart J, Kaur A, et al. Blood biomarkers for memory: toward early detection of risk for Alzheimer disease, pharmacogenomics, and repurposed drugs. Mol Psychiatry. 2020;25:1651–72.
Le-Niculescu H, Case NJ, Hulvershorn L, Patel SD, Bowker D, Gupta J, et al. Convergent functional genomic studies of omega-3 fatty acids in stress reactivity, bipolar disorder and alcoholism. Transl Psychiatry. 2011;1:e4.
Wirgenes KV, Tesli M, Inderhaug E, Athanasiu L, Agartz I, Melle I, et al. ANK3 gene expression in bipolar disorder and schizophrenia. Br J Psychiatry. 2014;205:244–5.
Rangaraju S, Levey DF, Nho K, Jain N, Andrews KD, Le-Niculescu H, et al. Mood, stress and longevity: convergence on ANK3. Mol Psychiatry. 2016;21:1037–49.
Higgins GA, Allyn-Feuer A, Barbour E, Athey BD. A glutamatergic network mediates lithium response in bipolar disorder as defined by epigenome pathway analysis. Pharmacogenomics. 2015;16:1547–63.
Stone W, Nunes A, Akiyama K, Akula N, Ardau R, Aubry JM, et al. Prediction of lithium response using genomic data. Sci Rep. 2021;11:1155.
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–5.
Acknowledgements
This work was supported by NIH R01MH107182 to PP. We thank Mrs. Lili Hamedi for her generous support. Floxed Ank3 mice were generously provided by Prof. Van Bennett from Duke University. Wide-field microscope imaging work was performed at the Northwestern University Center for Advanced Microscopy, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Metal analysis was performed at the Northwestern University Quantitative Bio-element Imaging Center generously supported by NASA Ames Research Center NNA06CB93G. We are grateful to members of the Penzes lab for helpful discussions, especially Dr. Marc P. Forrest, Dr. Marc Dos Santos, and Dr. Euan Parnell for their help on the manuscript. All experiments involving animals were performed according to the Institutional Animal Care and Use Committee of NU.
Author information
Authors and Affiliations
Contributions
NHP performed and analyzed cellular, confocal imaging and Golgi staining imaging, and led the project. SY performed conditional knockout mouse breeding and Golgi staining studies. RG and KEH generated and maintained neuronal cultures. JCG and TLP advised on the project. KRS performed cellular experiments. PP supervised the project. NHP, SY, JCG, TLP, KRS, and PP contributed to the writing and/or critical revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Piguel, N.H., Yoon, S., Gao, R. et al. Lithium rescues dendritic abnormalities in Ank3 deficiency models through the synergic effects of GSK3β and cyclic AMP signaling pathways. Neuropsychopharmacol. 48, 1000–1010 (2023). https://doi.org/10.1038/s41386-022-01502-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01502-2
