
Abstract
The senescence-accelerated mouse prone-8 (SAMP8) model has been considered as a good model for aged-related cognitive decline and Alzheimer’s disease (AD). Since epigenetic alterations represent a crucial mechanism during aging, in the present study we tested whether the inhibition of the histone deacetylase sirtuin 2 (SIRT2) could ameliorate the age-dependent cognitive impairments and associated neuropathology shown by SAMP8 mice. To this end, the potent SIRT2-selective inhibitor, 33i (5 mg/kg i.p. 8 weeks) was administered to 5-month-old (early treatment) and 8-month-old (late treatment) SAMP8 and aged matched control, senescence-accelerated mouse resistant-1 (SAMR1) mice. 33i administration to 5-month-old SAMP8 mice improved spatial learning and memory impairments shown by this strain in the Morris water maze. SAMP8 showed hyperphosphorylation of tau protein and decrease levels of SIRT1 in the hippocampus, which were not altered by 33i treatment. However, this treatment upregulated the glutamate receptor subunits GluN2A, GluN2B, and GluA1 in both SAMR1 and SAMP8. Moreover, early SIRT2 inhibition prevented neuroinflammation evidenced by reduced levels of GFAP, IL-1β, Il-6, and Tnf-α, providing a plausible explanation for the improvement of cognitive deficits shown by 33i-treated SAMP8 mice. When 33i was administered to 8-month-old SAMP8 with a severe established pathology, increases in GluN2A, GluN2B, and GluA1 were observed; however, it was not able to reverse the cognitive decline or the neuroinflammation. These results suggest that early SIRT2 inhibition might be beneficial in preventing age-related cognitive deficits, neuroinflammation, and AD progression and could be an emerging candidate for the treatment of other diseases linked to dementia.
Similar content being viewed by others

Introduction
Aging is the main risk factor of many neurodegenerative diseases associated with cognitive deterioration, including the different types of dementia. In the past decades, there has been an extensive research on these age-related disorders and pathologies with a special focus on establishing the aetiology and developing therapies for their prevention and treatment [1]. Among these, epigenetic alterations represent a crucial mechanism behind the deterioration of cellular functions observed during aging and in age-related disorders [2, 3]. Mediated by enzymes, epigenetic information is reversible, suggesting the potential for future therapeutic interventions. Accordingly, understanding the epigenetic changes that happen during aging is a major ongoing area of study, which may potentially lead the way to the development of novel therapeutic approaches to delay aging and age-related diseases.
Among all age related neurodegenerative diseases, Alzheimer’s disease (AD) is the most common form of dementing illness with >45 million people suffering worldwide [4]. AD is predominantly a sporadic late-onset disease (sAD) with exponentially increasing prevalence starting at the age of 65 years, which exceeds 25% in those over the age of 90 years. Most of the cases are commonly caused by complex interactions regarding multiple genetic, epigenetic, and environmental factors. The heavy burden on patients, their family, and society prompts the need for new target research for possible AD therapeutics that can address the pathophysiology as well as the associated cognitive decline.
Among the epigenetic targets linked to cognitive function, histone acetylation has been found to have a critical role in memory acquisition and maintenance [5]. Histone acetylation is regulated by the opposing action of histone acetyl transferases (HATs) and histone deacetylases (HDACs). Interestingly, it has been demonstrated that this balance between HATs and HDACs is altered in aging. Further, deficits in age related memory acquisition are due to an increase in HDAC activity and therefore to a decrease in the transcription of several genes involved in learning [6]. Thus it is plausible that pharmacological manipulations directed to counteract these age-related epigenetic modifications could revert the cognitive deficits associated with aging and AD [7].
HDACs in mammals, are divided into four groups: the zinc-dependent class I, II, and IV HDACs and the NAD+-dependent class III HDACs, which are also known as sirtuins. Among all sirtuins, sirtuin 2 (SIRT2) expression is found strongest in the brain [8]. SIRT2 is located predominantly in the cytoplasm but it can also interact with nuclear proteins playing a role in transcriptional repression of genes that encode for DNA-binding proteins as well as transcription factors that participate in synaptic plasticity, cell proliferation, differentiation, and cell survival [9]. Although its biological functions are not well described yet, some studies suggest that excess of SIRT2 might be deleterious to neurons [10, 11]. In agreement with this hypothesis, an age-dependent accumulation of SIRT2 in the mouse brain and spinal cord has been shown [12] and reported that its upregulation is a specific feature associated with stress-induced premature senescence [13].
On the other hand, SIRT2 inhibition provided protection in different models of Parkinson disease and Huntington disease [14,15,16] and exerted antidepressant-like effects in different depression mouse models [17, 18].
Although less is known about the role of SIRT2 in AD [19,20,21], recent studies have suggested that inhibition of SIRT2 is a safe and promising neuroprotective target in both tau-associated frontotemporal dementia and AD [22,23,24,25]. Based on all these considerations, we aimed to explore the effects of SIRT2 inhibition in cognitive impairment and associated hippocampal neuropathology of the senescence-accelerated mouse prone-8 (SAMP8) mouse model. This mouse model manifests irreversible premature senescence and shares similar characteristics with aged humans such as a reduced lifespan, lordosis, hair loss, and reduced physical activity. On the other hand, its control, the senescence-accelerated mice resistant-1 (SAMR1) strain presents a normal aging pattern. Moreover, SAMP8 mice also exhibit age-related learning and memory deficits, as well as the key neuropathological features seen in AD patients, such as amyloid-β (Aβ) deposition, increased levels of hyperphosphorylated tau, oxidative stress (OS), and gliosis [26, 27]. The fact that the SAMP8 mouse is a spontaneous animal model and not a transgenic mice model of AD suggests that the underlying mechanisms must be linked to epigenetics (for a review, see [28]) and represents more closely the complex multifactorial nature of sAD [27]. In this sense, recent studies have described epigenetic alterations in SAMP8 mice [29, 30] and it has been proposed that epigenetic regulation at young ages give rise to significant beneficial effects at the molecular, cellular, and behavioural levels, particularly in the hippocampus [31, 32]. For these reasons, the SAMP8 mouse has been proposed as a suitable rodent model for studying the molecular mechanisms underlying cognitive impairment in aged subjects [26] and sAD [27].
Materials and methods
Drugs
The SIRT2-selective inhibitor 33i (2-{3-(3 fluorophenethyloxy)phenylamino}benzamide) was synthesized via modification of the procedure reported by Suzuki et al. [33] (see Supplementary Data) and prepared in physiological serum with Tween 80 (18%) and 5% dimethyl sulfoxide (DMSO) in saline.
Animals, treatments, and experimental design
Experiments were carried out in male SAMP8 (28–30 g) and SAMR1 (35–39 g). These animals were bred from founders provided by Dr. Pallas (University of Barcelona, Spain). Animals were housed (5 per cage) in constant conditions of humidity and temperature (22 ± 1 °C) with a 12/12-h light–dark cycle (lights on at 7:00 a.m.). Food and water were available ad libitum. Only males were used for the present study because female SAMP8 mice have less robust memory changes than male SAMP8 mice [34]. All the procedures followed in this work and animal husbandry were conducted according to the principles of laboratory animal care as detailed in the European Communities Council Directive (2013/53/EC) and were approved by the ethical committee of the University of Navarra. All efforts were made to minimize animal suffering and to reduce the number of animals used in the experiments.
In the first experiment, 2- and 9-month-old SAMP8 and SAMR1 animals (n = 7 animals per group) were sacrificed by cervical dislocation and the hippocampus, frontal cortex, and striatum were prepared for SIRT2 analysis by western blot.
The next sets of experiments were designed to evaluate whether SIRT2 inhibition with 33i treatment could prevent (early treatment) or reverse (late treatment) the age-dependent progression of the pathology in SAMP8 mice. For this, 5-month-old SAMP8, with a mild cognitive impairment and some neuropathological signs (early treatment), or 8-month-old SAMP8 mice, with an established cognitive impairment and severe neuropathological alterations (late treatment), were treated intraperitoneally (i.p.) once a day with 33i (5 mg/kg) or vehicle (18% Tween 80, 5% DMSO in saline) for 8 consecutive weeks. Aged-matched SAMR1 control mice followed the same experimental design. Animals were randomized for treatment. For the “early treatment” group, the final number of animals per group was: SAMR1-vehicle n = 10; SAMR1-33i n = 12; SAMP8-vehicle n = 8; SAMP8-33i n = 10. For the “late treatment” group, 10 animals were selected for each group, but 4 SAMP8-vehicle mice died during the first 3 weeks of treatment and 1 SAMR1 treated with 33i had to be removed from the experiment due to skin wounds and a poor general condition. Therefore, the final number of animals in each group that performed the behavioural tests was SAMR1-vehicle n = 10, SAMR1-33i n = 9, SAMP8-vehicle n = 6, and SAMP8-33i n = 10. Sample size for the studies were chosen following previous studies in our laboratory [35,36,37] and using one of the available interactive web sites (http://www.biomath.info/power/index.html).
The dose of 33i was chosen based on a previous study carried out in our laboratory [17]. Behavioural tests started at the beginning of the sixth week (Fig. 1a). During these days, 33i was given at the end of the behavioural tests. Mice were sacrificed 1 h after the last trial of the Morris water maze (MWM). The last drug injection took place 20–24 h before the sacrifice.

Early SIRT2 inhibition improves learning and memory deficits in SAMP8 mice. a Experimental design of 33i administration and the subsequent behavioural tests. Five-month-old (for the early treatment) or 8-month-old (for the late treatment) SAMR1 and SAMP8 mice were treated daily with 33i (5 mg/kg i.p.) or the vehicle used to prepare 33i (see “Materials and methods”) for 8 consecutive weeks. Behavioural tests started at the beginning of the sixth week. During these days, 33i or vehicle were given at the end of the behaviour test. Mice were sacrificed after the last trial of the Water Morris Maze. b SIRT2 protein (left) and Sirt2 gene expression (right) in 2- and 9-month-old SAMR1 and SAMP8 mice (n = 7) (For SIRT2 protein levels: F = 6.568, *p < 0.05, main effect of age, two-way ANOVA). c Effect of 33i treatment on Abca1 gene expression (n = 6–7) (F = 7.974, p < 0.05, main effect of treatment, two-way ANOVA). d Escape latency in the hidden platform phase. SAMP8 mice had significant higher escape latency than SAMR1 mice, an effect reversed by 33i. *p < 0.05 vs SAMR1 vehicle; #p < 0.05 vs SAMP8 vehicle. e In the retention phase, data are presented as percentage of time spent in the target quadrant. Note that, on the fourth and seventh day, 33i administration in SAMP8 mice increased the time in the target quadrant (Day 4: F = 9.138, p < 0.05; Day 7: F = 11.92, p < 0.05, two-way ANOVA followed by Tukey test) (Day 9: F = 7.507, p < 0.05, main effect of strain, two-way ANOVA). f Representative western blot and quantitative measurement of SIRT1 normalized to β-actin in the hippocampus of SAMR1 and SAMP8 treated with vehicle or 33i (n = 6) *p > 0,05, (F = 5.975, p < 0.05, main effect of strain, two-way ANOVA). g Representative western blot and quantitative measurement of hippocampal phosphorylated tau (AT8/Total Tau). 33i did not reverse the higher levels of tau phosphorylation that SAMP8 mice have compared to SAMR1 mice (n = 7) *p > 0.05, (F = 4.882, p < 0.05, main effect of strain, two-way ANOVA). No significant differences were observed in Aβ40 (h) and Aβ42 (i) levels in the hippocampus of 7-month-old SAMR1 and SAMP8 mice (n = 5–6). In all panels, results are shown as mean ± SEM
In order to evaluate the effect of 33i on acetylated histone 4 (AcH4), 3-month-old SAMP8 and SAMR1 animals (n = 6 animals per group) were treated with vehicle (18% Tween 80, 5% DMSO in saline) or 33i (5 mg/kg i.p. every 24 h, 5 days) and sacrificed 2 h after last administration.
Behavioural tests
Spontaneous locomotor activity
Locomotor activity was measured in an open field consisting of 8 black square arenas (43 × 50 × 45 cm3) using a video tracking system (Ethovision XT 11.5 plus multiple body point module) (Ethovision XT 11.5; Noldus Information Technology B.V, Wageningen, Netherlands) in a softly illuminated experimental room. One mouse was placed in each cage and distance travelled (cm) and speed (cm/s) was recorded during a 30-min period.
Rotarod
Motor coordination and balance were measured by rotarod test (LE8200 Panlab, Harvard Apparatus). The animals were evaluated for 3 trials on 2 consecutive days. In each session, all groups were placed on the rod that was scheduled to accelerate gradually (4–40 rpm) for 5 min. The time (s) that each mouse takes to fall was scored and then the mean of the three trails was obtained.
Marble burying
Exploratory behaviour usually present in normal mice behaviour was assessed with this test. Twelve marbles (1.5-cm diameter) were placed uniformly in a cage (45 × 28 × 20 cm3) containing a constant amount of sawdust (3 cm deep). Mice were placed in the center of the cage and left for 30 min. The number of marbles buried (2/3 covered with sawdust) after this period was recorded by two blind experimenters.
MWM test
The MWM test was used to test spatial memory and to evaluate the working and reference memory functions in response to treatment, as previously described [36].
The water maze was a circular pool (diameter of 145 cm) filled with water (21–22 °C) and virtually divided into four equal quadrants identified as northeast, northwest, southeast, and southwest. First, mice underwent visible-platform training for 6 trials in 1 day (habituation phase) in which a platform was located in the southwest quadrant raised above the water with an object placed on top to facilitate its location. In this phase, it is confirmed that all mice exhibit a normal swimming pattern and are able to reach the platform.
Next, hidden-platform training (acquisition phase) was conducted with the platform placed in the northeast quadrant 1 cm below the water surface over 8 consecutive days (4 trials/day). Noteworthy, due to the weakness shown by 10-month-old SAMP8, we decided to end the test 1 day before in that experiment. Several large visual cues were placed in the room to guide the mice to the hidden platform. Each trial was finished when the mouse reached the platform (escape latency) or after 60 s, whichever came first. This escape latency was used to evaluate the spatial learning and memory of mice. Mice failing to reach the platform were guided onto it. After each trial, mice remained on the platform for 15 s.
To test memory retention, three probe trials were performed at the beginning of the days 4, 7, and 9 in the “early treatment experiment” and days 4, 6, and 8 in the “late treatment experiment”. In the probe trials, the platform was removed from the pool and mice were allowed to swim for 60 s. The percentage of time spent in the target quadrant was recorded. All trials were monitored by a video camera set above the centre of the pool and connected to a video tracking system (Ethovision XT 11.5; Noldus Information Technology B.V, Wageningen, Netherlands).
Western blot
Western blot analysis was carried out in hippocampal tissues as previously described [17].
Amyloid precursor protein (APP) carboxy-terminal fragments were analysed as previously described [38].
For determination of Aβ oligomers, a sample of hippocampus was homogenized in a buffer containing 2% sodium dodecyl sulfate, 10 mM Tris-HCl (pH 7.4), protease inhibitors (Complete™ Protease Inhibitor Cocktail, Roche), and phosphatase inhibitors (0.1 mM Na3VO4, 1 mM NaF). The homogenates were sonicated for 2 min and ultracentrifuged at 100,000 × g for 1 h at 4 °C. The proteins were separated in a 15% acrylamide gel under non-reducing conditions.
For ubiquitinated protein determination, hippocampal tissues were lysed with NP buffer (150 nM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol, 25 mM Tris-Cl pH 7.5) and protease inhibitor cocktail and left on ice for 20 min. After centrifugation at 16,000 rpm for 5 min, we collected the supernatant containing the NP-soluble fraction [39].
Membranes were probed overnight at 4 °C with the following primary antibodies: Rabbit polyclonal antibodies included anti-microtubule-associated protein 1A/1B-light chain 3 (LC3), anti-pro-brain-derived neurotrophic factor (anti-pro-BDNF), anti-Beclin1, anti-Lamin A/C, anti-Rho-associated protein kinase 2 (ROCK2), anti-postsynaptic density protein 95 (PSD95) (Cell Signaling Technology, Danvers, MA, USA), and anti-glutamate ionotropic receptor AMPA-type subunit 1 (GluA1), anti-glutamate ionotropic receptor NMDA-type subunit 2A (GluN2A), anti-acetyl-Histone (AcH4) (Merck Millipore), anti-activity-regulated cytoskeleton-associated protein (ARC) (H-300) (Santa Cruz Biotechnology, Inc, Dallas, TX, USA), anti-SIRT2, anti-tubulin (all of them at 1:1000 dilution), and anti C-terminal of APP (1:2000) (Sigma-Aldrich, St. Louis, MO, USA); rabbit monoclonal antibodies included anti-cyclic response element-binding protein (CREB) (48H2) and anti-myelin basic protein (MBP) (1:1000) (Cell Signaling Technology, Danvers, MA, USA); mouse monoclonal antibodies included anti-synaptophysin (SY338) (Abcam, Cambridge, UK), anti-glutamate ionotropic receptor NMDA-type subunit 2B (GluN2B), anti-p-CREB, anti-ubiquitin (P4D1) (Santa Cruz Biotechnology, Inc, Dallas, TX, USA), anti-actin, anti-SIRT1, anti-Tau (Sigma-Aldrich, St. Louis, MO, USA), anti-p-Tau (1:1000) (Thermo Fisher Scientific, Waltham, MA, USA), and purified anti-β-Amyloid 1–16 Antibody (6E10) (all of them at 1:1000, Covance). β-Actin or tubulin (1:10 000, Sigma-Aldrich, St. Louis, MO, USA) were used as internal control. Odyssey® goat anti-rabbit and anti-mouse secondary antibodies (1:5000; Odyssey, LI-COR®, Lincoln, USA) were used. Bands were visualized using Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Results were calculated as the optical density values of the control vehicle-treated SAMR1 mice.
RNA extraction and real-time reverse transcriptase-PCR
RNA extraction and real-time reverse transcriptase-PCR amplification assays for gene targets were performed as previously described [17]. Primers for Sirt2, Abca1, Glun2a, Glun2b, Glua1, interleukin 1β (Il-1β), interleukin 6 (Il-6) and tumour necrosis factor-alpha (Tnf-α) were used (Applied Biosystems, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase was used as internal control. Samples were analysed by a double delta CT (∆∆CT) method. Relative transcription levels (2−∆∆Ct) were expressed as a mean ± standard error of the mean.
Immunohistochemistry
The brains of three mice per experimental group were histologically processed to confirm the presence of reactive astrocytes/microglia in the hippocampus. For this purpose, one brain hemisphere was postfixed for 24 h with paraformaldehyde 4% after dissection and conserved in sucrose 30% for 1 week. Immunofluorescence was performed as previously described [37]. Sections were incubated with the primary antibody anti-glial fibrillary acidic protein (anti-GFAP; 1:500; Cell Signaling, Danvers, MA, USA) or CD11b (1:500; Novus Biologicals) overnight at 4 °C, washed with phosphate-buffered saline, and incubated with the secondary antibody (Alexa Fluor Anti-mouse 546, Thermo Fisher, Pittsburg, PA, USA) for 2 h at room temperature, protected from light. To ensure comparable immunostaining, sections were processed together under identical conditions. Fluorescence signals were detected with Nikon Eclipse E800 (Nikon, Shinagawa, Tokyo, Japan). Quantification of fluorescent signal in brain sample images was carried out using a plugin developed for Fiji7ImageJ, an open-source Java-based image processing software. The plugin was developed by the Imaging Platform of the Centre for Applied Medical Reseach (CIMA, Pamplona, Spain).
Quantification of Aβ by enzyme-linked immunosorbent assay
Total Aβ42 and Aβ40 burden was measured in the hippocampus of the animals as previously described [37]. High-sensitive enzyme-linked immunosorbent assay kits from Wako (cat#292–64501 for Aβ42 and cat#294-64701 for Aβ40, Wako Chemicals, Richmond, VA) were used following the manufacturer’s instructions.
Quantification of IL-1β in brain lysates
The forebrain was sonicated in a specific lysis buffer (Cell Lysis Buffer 2, R&D systems, cat# 895347) at 1:4 dilution, incubated on ice for 30 min, and centrifuged 12 min at 13,000 rpm at 4 °C. Fifty microlitres of the resulting supernatant was assayed for the levels of IL-1β using the Quantikine ELISA Kit (R&D systems, cat# MLB00C) following the manufacturer’s instructions. Each sample was analysed in duplicate.
Statistical analysis
Results are expressed as mean ± standard error of the mean. Data were analysed by two-way analysis of variance (ANOVA) (strain × treatment) followed by Tukey post hoc test except for the habituation and acquisition phase of the MWM, which were analysed with repeated measures. Post hoc test was applied only if F was significant. In the figure legends, F values represent the F of interaction followed by the p value of the corresponding post hoc test. In those cases where the F of interaction was not statistically significant, the F value shown represents the main effect observed of strain or treatment. Differences were considered statistically significant at p < 0.05. Data analyses were performed using GraphPad Prism 6 (GraphPad Software, Inc. La Jolla, CA, USA).
Results
SIRT2 is increased in the hippocampus of 9-month-old SAMR1 and SAMP8 mice
For SIRT2 quantification, both bands (37/43 kDa) were quantified. As shown in Fig. 1b, a significant increase in hippocampal SIRT2 protein was observed in 9-month-old SAMR1 and SAMP8 mice compared to 2-month-old mice. This effect did not correlate with increases in Sirt2 gene expression. On the other hand, no significant differences were observed when the expression of SIRT2 was analysed in the frontal cortex and the striatum of the mice (Fig. S1A, B).
33i inhibits SIRT2 activity in the hippocampus
The inhibitory effect of 33i on SIRT2 activity was verified by measuring in the hippocampus the gene expression of ATP-binding cassette transporter Abca1 (a known transporter of cholesterol whose transcription is inhibited by SIRT2 [22]). Two-way analysis ANOVA revealed an increase in hippocampal gene expression Abca1 in 33i-treated SAMP8 and SAMR1 mice (Fig. 1c). This effect was further confirmed with an increase of AcH4 levels in a different set of animals sacrificed 2 h after the last administration of 33i (Fig. S1C).
Early treatment: effect of SIRT2 inhibition in 5-month-old SAMP8 mice
Behavioural tests
Regarding the spontaneous motor activity, the SAMP8 mice showed a reduction in the distance travelled (Fig. S2A). Moreover, their duration in the rotarod apparatus was significantly shorter than that of SAMR1 mice. These effects were not reversed by the 33i treatment (Fig. S2B).
On the other hand, the mean number of marbles buried by SAMR1 mice was higher than the corresponding values of vehicle-treated SAMP8 mice (8.6 and 4.125, respectively). Noteworthy, 33i was able to reverse SAMP8’s altered normal exploratory behaviour (Fig. S2C).
The effect of 33i on the cognitive decline observed in SAMP8 mice was evaluated using the MWM test. In the habituation phase, there was no difference in swimming speed across the four groups (Fig. S2D). Moreover, although strain differences were observed regarding the escape latencies (Fig. S2E), no significant differences were found between saline and 33i-treated animals, which enabled us to exclude any potential anxiolytic effect of the 33i compound or motivational and sensorimotor factors on animal learning and memory performance.
As shown in Fig. 1d, in the acquisition phase, the escape latencies of SAMR1 are lower than the ones of SAMP8, indicating the existence of learning deficits in these mice. 33i-treated SAMP8 mice showed a marked improvement in their behavioural performance as their escape latencies were significantly shorter than SAMP8 mice treated with vehicle from the first day of acquisition until last day. No significant differences were found between vehicle or 33i-treated SAMR1 mice.
At the beginning of the fourth, seventh, and ninth day, all mice were subjected to a probe trial in which they swam in the pool with the platform removed as a putative measurement of memory retention (Fig. 1e). On days 4 and 7, 33i-treated SAMP8 spent significant more time in the target quadrant than SAMP8 mice treated with vehicle. No differences were observed between vehicle or 33i-treated SAMR1 mice. On day 9, only a main effect of strain was found, with SAMP8 mice spending less time in the target quadrant than SAMR1.
Effect of 33i on SIRT1 expression
SAMP8 mice had significantly lower levels of SIRT1 than SAMR1. However, no differences were observed between both vehicle and 33i-treated animals (Fig. 1f).
Effect of 33i on neuropathological hallmarks of AD
Western blot analysis revealed that phosphorylated tau levels (AT8 antibody) normalized to total tau (detected by T46 antibody) were significantly increased in SAMP8 mice compared with that in SAMR1 mice. No differences were observed between both saline and 33i-treated animals (Fig. 1g).
Regarding Aβ pathology, no significant differences were observed across all four groups in hippocampal Aβ40 (Fig. 1h) and Aβ42 levels (Fig. 1i), APP processing (Fig. S3A), and Aβ oligomers (Fig. S3B).
Effect of 33i on autophagy and ubiquitin–proteasome system
Although no differences were detected in Beclin 1 and ROCK2 levels (Fig. 2a, b), SAMP8 mice have higher levels of LC3-II compared to SAMR1 (Fig. 2c). However, no significant differences were observed between vehicle and 33i-treated animals.

SIRT2 inhibition upregulates GluN2A, GluN2B, and GluA1 expression in 7-month-old SAMR1 and SAMP8 mice. No differences were found concerning hippocampal protein levels of Beclin 1 (a) and ROCK2 (b) (n = 7). c SAMP8 show higher levels of LC3-II (n = 7). d Representative image and quantitative measurement of hippocampal ubiquitinated proteins (n = 7) (F = 6.454, p < 0.05, main effect of strain, two-way ANOVA). Effect of 33i on hippocampal GluN2A (e), GluN2B (f), and GluA1 (g) expression (n = 6–8). *p < 0.05 (F = 8.724, p < 0.05; F = 6.487, p < 0.05; F = 30.40, p < 0.05, respectively, main effect of treatment, two-way ANOVA). Results show that 33i treatment increases the gene expression of h Glun2a (F = 6.436, p < 0.05, main effect of treatment) and i Glun2b (F = 6.496, p < 0.05, main effect of treatment) but not of Glua1 j (n = 5–8). Results are shown as mean ± SEM
Moreover, SAMP8 mice presented significantly more accumulation of ubiquitinated proteins than SAMR1, which was not reverted by 33i treatment (Fig. 2d).
Effect of 33i on myelination
Taking into account that SIRT2 is highly expressed in oligodendrocytes [8], we next evaluated whether SIRT2 inhibition would have any effect on MBP levels. As show in Fig. S3C, no differences were observed among all the four groups.
Effect of SIRT2 inhibition on learning and memory-related proteins
No differences were observed in the hippocampal expression of phospho-CREB, CREB, pro-BDNF, PSD95, synaptophysin, and ARC across all the four groups (Fig. S4A–E).
However, GluN2A, GluN2B, and GluA1 proteins increased in both SAMR1 and SAMP8 mice treated with 33i compared to control vehicle mice (Fig. 2e–g). Noteworthy, a significant increase was observed in the hippocampal gene expression of Glun2a and Glun2b (Fig. 2h, i). However, no differences were found in the gene expression of Glua1 (Fig. 2j).
Early 33i treatment prevents the neuroinflammation in 7-month-old SAMP8 mice
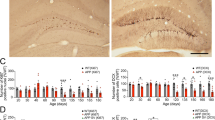
Our data showed a significant increase in hippocampal GFAP immunoreactivity in SAMP8 mice compared to that in SAMR1 mice (Fig. 3a, b). This increase was reverted by 33i treatment in SAMP8 mice. No differences were observed between vehicle or 33i-treated SAMR1 mice.

Early 33i treatment reduces the neuroinflammation in 7-month-old SAMP8 mice. 33i (5 mg/kg i.p. every 24 h) or vehicle was administered to 5-month-old SAMR1 and SAMP8 for 8 weeks. Mice were sacrificed 24 h after the last administration. a Representative image and quantitative measurement of hippocampal GFAP protein levels. Note that SAMP8 show increased levels of GFAP, which are reversed by 33i treatment (n = 7) (F = 34.87, p < 0.05). This effect is also seen in the quantification of the immunofluorescence images (b) (n = 3 mice per group) (F = 15.80, p < 0.05) Scale bar = 200 µm. c IL-1β protein levels measured by ELISA (F = 11.90, p < 0.05). Gene expression of Il-1β (F = 16.61, p < 0.05) (d), Il-6 (F = 14.29, p < 0.05) (e), and Tnf-α (F = 19.26, p < 0.05) (f) (n = 5–7). Note that 33i treatment reversed the increased levels of these proinflammatory cytokines shown by SAMP8 mice. GAPDH was used as an internal control. Results are shown as mean ± SEM. *p < 0.05. Two-way ANOVA followed by Tukey test
Moreover, the expression of IL-1β protein and gene expression of Il-1, Il-6, and Tnf-α were increased in vehicle-SAMP8 compared to that in SAMR1, an effect reverted by 33i treatment (Fig. 3c–f). However, a significant increase in Il-6 and Tnf-α was observed in 33i-treated SAMR1 (Fig. 3e, f).
The analysis by western blot and immunofluorescence of hippocampal CD11b levels revealed no significant differences across all four groups (Fig. S5A, B).
Late treatment: effect of SIRT2 inhibition in 8-month-old SAMP8 mice
Behavioural tests
33i did not improve the performance of 10-month-old SAMP8 mice in the rotarod (data not shown). However, 33i significantly reversed SAMP8’s altered normal exploratory behaviour in the marble burying test (Fig. S6A).
In the habituation phase of the MWM test, the swimming speed of SAMP8 mice was significantly lower than that of the SAMR1 (Fig. S6B). Accordingly, a main effect of strain was observed when the escape latencies of this phase were analysed (data not shown). However, since no treatment effect was found, we used the escape latency for evaluation of the spatial learning and memory in the mice.
In the acquisition phase of the MWM, the learning curve of 33i-treated SAMP8 showed a trend towards improvement when compared with vehicle SAMP8. However, the intra-group comparisons revealed that there were no statistical differences between the days showing that neither of the two groups learned the location of the platform (Fig. 4a). When testing memory retention, a strain effect on days 6 and 8 was found, whereas no significant differences were found between vehicle or 33i-treated animals (Fig. 4b).

Effect of a late treatment with 33i on 10-month-old SAMP8 and SAMR1 mice. 33i (5 mg/kg i.p. every 24 h) or vehicle was administered to 8-month-old SAMR1 and SAMP8 for 8 weeks. Behavioural tests started at the beginning of the sixth week. a Escape latency in the hidden platform phase of the Morris water maze (Acquisition). b The probe trial data presenting the percentage of time spent in the target quadrant. Results are shown as mean ± SEM (SAMR1-vehicle n = 10; SAMR1–33i n = 9; SAMP8-vehicle n = 6; SAMP8-33i n = 10). *p < 0.05 (Day 6: F = 5.253, p < 0.05; Day 8: F = 16.46, p < 0.05, main effect of strain, two-way ANOVA). c Representative western blot and quantitative measurement of hippocampal phosphorylated tau (AT8/Total Tau). 33i did not reverse the higher levels of tau phosphorylation observed in SAMP8 mice (n = 7) (F = 40.56, p < 0.05, main effect of strain). SAMP8 mice had increased hippocampal levels of Aβ40 (d) (n = 6) (F = 31.17, p < 0.05, main effect of strain) and Aβ42 (e) (n = 7–8) (F = 7.861, p < 0.05, main effect of strain). Results are shown as mean ± SEM. *p < 0.05, two-way ANOVA
Neurochemical analysis
Ten-month-old SAMP8 mice showed increased levels of phospho-Tau (Fig. 4c) and amyloid pathology evidenced by increased levels of Aβ40 and Aβ42 (Fig. 4d, e) and decreased levels of full-length APP (Fig. S6D), which were not affected by 33i treatment. Moreover, 33i did not modify Aβ oligomerization (Fig. S6C), levels of MBP (Fig. S6E), autophagy pathway, or the expression of ARC, CREB, pro-BDNF, or PSD95 (data not shown). However, GluN2A, GluN2B, and GluA1 proteins increased in both SAMR1 and SAMP8 mice treated with 33i compared to control vehicle mice (Fig. 5a–d).

Effect of 33i treatment on GluN2A, GluN2B, and GluA1 expression and neuroinflammation in 10-month-old SAMR1 and SAMP8 mice. 33i (5 mg/kg i.p. every 24 h) or vehicle was administered to 8-month-old SAMR1 and SAMP8 for 8 weeks. Mice were sacrificed 24 h after the last administration. SIRT2 inhibition upregulates GluN2A (a), GluN2B (b) and GluA1 (c) expression in 10-month-old SAMR1 and SAMP8 mice (n = 6–8). *p < 0.05 main effect of treatment (F = 4.518, p < 0.05; F = 7.019, p < 0.05; F = 4.652, p < 0.05, respectively, two-way analysis of variance (ANOVA)). d Representative western blot images showing the increase of GluN2A, GluN2B, and GluA1 in 33i-treated mice. e, f Representative images and quantitative measurement of hippocampal GFAP protein levels measured by western blot (n = 6–7) and immunofluorescence (n = 3) *p < 0.05 main effect of strain (F = 16.06, p < 0.05; F = 5.809, p < 0.05, respectively). Scale bar = 200 µm. g IL-1β protein levels measured by ELISA (n = 6) (F = 24.70, p < 0.05, main effect of strain). Effect of 33i on gene expression of h Il-1β (F = 5.492, p < 0.05, main effect of strain), i Il-6 (F = 16.77, p < 0.05, main effect of strain), and j Tnf-α in the hippocampus of 10-month-old SAMP8 and SAMR1 mice (n = 6–7). Note that 33i treatment did not reverse the increased expression of these proinflammatory cytokines shown by SAMP8 mice. GAPDH was used as an internal control. Results are shown as mean ± SEM. Two-way ANOVA
On the other hand, SIRT2 inhibition did not reduce the increased expression of GFAP (Fig. 5e, f), IL-1β protein, Il-1β, or Il-6 (Fig. 5g–i) shown by 10-month-old SAMP8 mice. Moreover, no significant differences were observed across all four groups when microglial reactivity (Fig. S7A, B) or Tnf-α (Fig. 5j) were analysed.
Discussion
Aging, a natural biological process associated with physiological decline, both physically and cognitively, is considered the main risk factor of many neurodegenerative diseases. Therefore, the pharmacological treatment of these pathologies must be understood in the context of the molecular biology of the aging process. In these sense, in the past years, epigenetic alterations have been revealed as a crucial mechanism behind the deterioration of cellular functions observed during aging and in age-related disorders [2, 3, 28].
Among the epigenetic targets linked to cognitive function, histone acetylation has been found to have a critical role in memory acquisition and maintenance [5]. Interestingly, the NAD+-dependent class III HDACs, known as sirtuins, have emerged as master regulators of metabolism and longevity [40]. However, their role in aging and cellular senescence remains controversial. Since an excess of SIRT2 might be deleterious to neurons [11], SIRT2 inhibition has been proposed as a therapeutic strategy for neurodegenerative diseases. Accordingly, previous studies have shown that pharmacological inhibition of SIRT2 exerts neuroprotective effects in diverse models [14, 15, 22, 23]. Based on these considerations, we have explored the effects of SIRT2 inhibition in the SAMP8 mouse, considered as a good model for aged-related cognitive decline and sAD.
We first measured hippocampal SIRT2 expression in 2- and 9-month-old SAMR1 and SAMP8 mice. Our study shows that 9-month-old SAMR1 and SAMP8 have higher hippocampal levels of SIRT2 than 2 month-old mice. No significant differences were found between both strains, supporting a recent study that has shown increased SIRT2 plasma levels in AD subjects and aged-matched healthy controls compared to healthy young controls but no differences between AD and aged-matched controls [41]. These results suggest that SIRT2 might be a good biomarker of the aging process.
However, in contrast to Maxwell et al. [12], no significant differences were observed with aging in the frontal cortex or striatum. These apparent discrepancies may be due to the fact that different species and ages (19 vs 9 months) have been analysed in each study. Moreover, we found that hippocampal gene expression of Sirt2 was not increased with age, suggesting that the increase observed in SIRT2 in this brain area is due to a protein accumulation but not to an increase in its synthesis. Noteworthy, in agreement with Maxwell et al. [12], the increase in SIRT2 levels is not associated with significant changes in overall levels of its main substrates, acetylated tubulin or AcH4 (data not shown), which suggests the presence of other acetyltransferases counteracting the activity of SIRT2 [42] and questions the physiological relevance of this increase.
The compound 33i represents a new class of SIRT2-selective inhibitors [33]. We have recently demonstrated that the same dose of 33i used for the present study successfully reached the brain and inhibited SIRT2 deacetylase function. This effect was evidenced by increased AcH4 and an upregulation of Abca1 gene expression [17]. Accordingly, the inhibitory effect of 33i on SIRT2 activity in the present study was confirmed by measuring hippocampal gene expression of Abca1 and AcH4, which were significantly higher in both 33i-treated SAMR1 and SAMP8 mice.
Consistent with previous reports [35, 36], 7-month-old SAMP8 mice presented learning and memory impairments in the MWM. Noteworthy, this effect was markedly improved by early 33i treatment. This finding supports a recent study [43] that demonstrates how sodium butyrate, an HDAC inhibitor, ameliorates SIRT2-induced memory impairment in the novel object recognition test (NORT). In addition, our data are in agreement with another study where higher doses of another SIRT2 inhibitor (AK-7, 20 mg/kg) reversed the long-term memory impairment in two different transgenic mouse models in the NORT [23]. However, to our knowledge, this is the first study demonstrating the beneficial effects of SIRT2-specific inhibition in learning and memory impairments shown by SAMP8 in the MWM.
To fully figure out the mechanisms underlying these effects, different hypotheses were evaluated:
One possibility is that the inactivation of SIRT2 could lead to a compensatory activation of SIRT1 [44], which has been reported to reverse memory impairments in the SAMP8 model [45]. In agreement with what was previously described [29], our results show a decrease of SIRT1 in SAMP8 saline mice when compared to that in SAMR1 mice. Nevertheless, we did not find any differences in SIRT1 expression in the hippocampus of 33i-treated mice.
We next focused our study on the main neuropathological hallmarks of AD. Consistent with previous studies [36], SAMP8 mice showed an increased phosphorylation of tau at Ser202/Ser205 (AT8 epitope). However, in agreement with a previously reported study [22], SIRT2 inhibition did not affect tau hyperphosphorylation. Following the hypothesis that SIRT2 may affect APP metabolism [23] or Aβ aggregation [24], we also quantified hippocampal Aβ42 and Aβ40 levels, Aβ oligomerization, and APP processing. However, at the age of 7 months, no significant differences were observed across all four groups, suggesting that, in the SAMP8 mouse model, it is too early to test this hypothesis. In addition, it should be noted that, due to the absence of APP mutations, the SAMP8 model does not show a severe amyloid pathology. Therefore, it might not be the best animal model to investigate the effects of SIRT2 inhibition on APP processing.
Recently, SIRT2 was reported to interfere with autophagy [39, 46, 47] and to increase ubiquitinated aggregates in vitro [39]. Thus we next sought to investigate whether autophagy modulation by SIRT2 inhibition could be one of the mechanisms underlying 33i beneficial effects. However, 33i treatment did not have an effect on autophagy, suggesting that further experiments with a higher dose of 33i or in other animal models are necessary to fully understand the role of SIRT2 on age-related autophagy and ubiquitin–proteasome system alterations.
Since long-term memories require gene expression, the epigenetic mechanisms that modulate transcription have a critical role in memory. Accordingly, inhibitors of HDAC activity enhance histone acetylation, synaptic plasticity, learning, and memory [48].
N-methyl-d-aspartate (NMDA) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) glutamate receptors are crucial for synaptic plasticity and modulate long-term potentiation (LTP) in hippocampus, considered the basis for spatial learning and memory [49]. Interestingly, our results showed an increased in GluN2A, GluN2B, and GluA1 expression in the hippocampus of 33i-treated animals, providing a plausible explanation for the improvement of the cognitive deficits shown by SAMP8 mice. Previous research has shown that, out of all the NMDA receptor subunits, the GluN2B subunit is the most affected by the aging process [50]. Therefore, by increasing GluN2B expression in old mice, spatial memory will be enhanced [51]. Interestingly, our results demonstrate that SIRT2 inhibition results in an increase of hippocampal gene expression of Glun2a and Glun2b and, hence, in their synthesis. In line with these studies, we have also shown that SIRT2 inhibition increases, in SH-SY5Y cells, the expression of other genes involved in synaptic plasticity [52]. Although SIRT2 is mostly known as a cytosolic deacetylase [8], these studies support also the role of SIRT2 in the nucleus influencing gene expression. It is important to note that preliminary experiments obtained by chromatin immunoprecipitation assays with the antibody against AcH4 suggest that the increased gene expression found in these proteins is independent of AcH4 (unpublished results). Thus we acknowledge that further studies should elucidate the mechanisms underlying SIRT2 inhibition-induced increased expression of these NMDA receptor subunits.
Regarding the AMPA receptor, it has been previously established that stabilizing GluA1 surface expression in the hippocampus of aged rats attenuated age-related impairments of LTP [53]. Noteworthy, no significant differences were observed in Glua1 gene expression, which suggests that SIRT2 is not affecting the synthesis but its degradation. Interestingly, a recent study has shown that SIRT2 acts as GluA1 deacetylase regulating AMPA receptor proteostasis [54]. Hence, SIRT2 inhibition by 33i could increase GluA1 acetylation reducing AMPA degradation, confirming its potential regulatory role in synaptic plasticity and brain function. Moreover, this result further evidences the inhibitory effect of the compound 33i on hippocampal SIRT2 activity.
Finally, we next examined whether 33i had an effect on different neuroinflammatory markers. In accordance with previous reports [55, 56], we observed a significant astrocyte activation in 7-month-old SAMP8 mice, which was significantly reversed by 33i, providing a possible mechanism underlying the memory improvement observed in this strain. Noteworthy, many studies have demonstrated the association between reduction in neuroinflammation and learning and memory amelioration in the SAMP8 model [37, 55, 57,58,59]. Moreover, our results support a previous study that showed the efficacy of SIRT2 inhibition to prevent the activation of astrocytes in rat primary cultures primed with Aβ42 [60]. We also assessed the effect of SIRT2 inhibition on other well-established markers of neuroinflammation: Il-1β, Il-6, and Tnf-α. In agreement with several studies that showed anti-inflammatory effects after SIRT2 inhibition [54, 61, 62], gene expression of Il-1β, Il-6, and Tnf-α in 33i-treated SAMP8 mice was significantly reduced. Interestingly, although only neuroinflammation was assessed, the possible implication of a peripheral anti-inflammatory effect after SIRT2 inhibition, as shown by [63], cannot be ruled out. On the other hand, 33i significantly increased Il-6 and Tnf-α gene expression in SAMR1 mice, supporting other studies that suggested a role for SIRT2 in inhibiting the inflammatory response [64, 65]. In any case, these increases in Il-6 and Tnf-α do not seem to have relevant consequences in the control strain. Noteworthy, no significant differences were found in GFAP, CD11b, and IL-1β between vehicle and 33i-treated SAMR1, evidencing a lack of neuroinflammation.
These apparently contradictory results regarding the role of SIRT2 in inflammation indicate that SIRT2 could play complex roles in certain biological processes. Accordingly, multiple studies have suggested that SIRT2 could produce contrasting roles also in cell death [66, 67] and OS [61, 64, 68] under different conditions, Thus whether and how SIRT2 regulates inflammation in the brain still remains unclear.
The positive results obtained when SIRT2 inhibitor was administered in an early stage of the disease allowed us to ask whether the administration of 33i to 8-month-old SAMP8 mice would be able to reverse the already established and severe cognitive deterioration and neuropathological signs. Interestingly, 33i treatment increased survival in SAMP8 strain (4 vehicle-treated SAMP8 died spontaneously while no 33i-treated SAMP8 died until they were sacrificed at the end of the experiment) and improved the natural marble-burying behaviour. In agreement with these results, a previous study have shown that SIRT2 inhibition reduced mortality in a mouse model of lethal septic shock [63]. However, although 33i treatment increased the expression of GluN2A, GluN2B, and GluA1 subunits, it was not able to reverse neither the memory dysfunction nor the neuroinflammation, the tau, or amyloid pathologies. Based on these results, the anti-inflammatory properties observed after early SIRT2 inhibition seem to be relevant for 33i-induced beneficial effects.
In conclusion, the results of this study indicate that early SIRT2 inhibition, through modulation of glutamate receptors and neuroinflammation, could be an ideal novel target to prevent age-related cognitive decline and neurodegeneration, specifically sAD. Noteworthy, 33i treatment was effective when SIRT2 was inhibited in animals with an early pathology, which highlights the importance of the early diagnosis to maximize any therapeutic effect.
Funding and disclosure
None of the authors have any actual or potential conflict of interest including any financial, personal or other relationships with other people or organizations that could inappropriately influence their work. The research leading to these results has received funding from "la Caixa" Banking Foundation. This work was also supported by the Spanish Ministry of Economy and Competitiveness (SAF2011-27910 and SAF2017-87595-R, Spanish Government).
References
Partridge L. Intervening in ageing to prevent the diseases of ageing. Trends Endocrinol Metab 2014;25:555–7.
Akbarian S, Beeri MS, Haroutunian V. Epigenetic determinants of healthy and diseased brain aging and cognition. JAMA Neurol 2013;70:711–8.
Brunet A, Berger SL. Epigenetics of aging and aging-related disease. J Gerontol Ser A Biol Sci Med Sci 2014;69:S17–20.
Guzman-Martinez L, Maccioni RB, Farías GA, Fuentes P, Navarrete LP. Biomarkers for Alzheimer’s disease. Curr Alzheimer Res. 2019;16;518–28.
Gräff J, Tsai L-H. The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharm Toxicol 2013;53:311–30.
Sharma M, Shetty MS, Arumugam TV, Sajikumar S. Histone deacetylase 3 inhibition re-establishes synaptic tagging and capture in aging through the activation of nuclear factor kappa B. Sci Rep 2015;5:16616.
Mastroeni D, et al. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging 2011;32:1161–80.
Jayasena T, et al. Application of targeted mass spectrometry for the quantification of sirtuins in the central nervous system. Sci Rep 2016;6:35391.
Eskandarian HA, et al. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 2013;341:1238858.
Suzuki K, Koike T. Mammalian Sir2-related protein (SIRT) 2–mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience 2007;147:599–612.
Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE 2008;3:e4090.
Maxwell MM, et al. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum Mol Genet 2011;20:3986–96.
Anwar T, Khosla S, Ramakrishna G. Increased expression of SIRT2 is a novel marker of cellular senescence and is dependent on wild type p53 status. Cell Cycle 2016;15:1883–97.
Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007;317:516–9.
Chopra V, et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep 2012;2:1492–7.
de Oliveira RM, et al. The mechanism of sirtuin 2–mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol 2017;15:e2000374.
Erburu M, et al. SIRT2 inhibition modulate glutamate and serotonin systems in the prefrontal cortex and induces antidepressant-like action. Neuropharmacology 2017;117:195–208.
Muñoz-Cobo I, Belloch FB, Díaz-Perdigón T, Puerta E, Tordera RM. SIRT2 inhibition reverses anhedonia in the VGLUT1+/- depression model. Behav Brain Res 2017;335:128–31.
Polito L, et al. The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease in two Caucasian case-control cohorts. Alzheimers Dement 2013;9:392–9.
Wei W, et al. The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease: a meta-analysis. Neuromolecular Med 2014;16:448–56.
Cacabelos R, et al. Sirtuins in Alzheimer’s disease: SIRT2-related genophenotypes and implications for pharmacoepigenetics. Int J Mol Sci 2019;20:1249.
Spires-Jones TL, et al. Inhibition of sirtuin 2 with sulfobenzoic acid derivative AK1 is non-toxic and potentially neuroprotective in a mouse model of frontotemporal dementia. Front Pharmacol 2012;3:42.
Biella G, et al. Sirtuin 2 inhibition improves cognitive performance and acts on amyloid-β protein precursor processing in two Alzheimer’s disease mouse models. J Alzheimers Dis 2016;53:1193–207.
Silva DF, Esteves AR, Oliveira CR, Cardoso SM. Mitochondrial metabolism power SIRT2-dependent deficient traffic causing Alzheimer’s-disease related pathology. Mol Neurobiol 2017;54:4021–40.
Esteves AR, et al. Acetylation as a major determinant to microtubule-dependent autophagy: relevance to Alzheimer’s and Parkinson disease pathology. Biochim Biophys Acta. 2018. https://doi.org/10.1016/j.bbadis.2018.11.014.
Tomobe K, Nomura Y. Neurochemistry, neuropathology, and heredity in SAMP8: a mouse model of senescence. Neurochem Res 2009;34:660–9.
Pallas M, et al. From aging to Alzheimer’s disease: unveiling "the switch" with the senescence-accelerated mouse model (SAMP8). J Alzheimers Dis 2008;15:615–24.
Griñán-Ferré C, et al. Understanding epigenetics in the neurodegeneration of Alzheimer’s disease: SAMP8 mouse model. J Alzheimers Dis 2018;62:943–63.
Cosín-Tomás M, et al. Epigenetic alterations in hippocampus of SAMP8 senescent mice and modulation by voluntary physical exercise. Front Aging Neurosci 2014;6:51.
Cosín-Tomás M, et al. Temporal integrative analysis of mRNA and microRNAs expression profiles and epigenetic alterations in female SAMP8, a model of age-related cognitive decline. Front Genet 2018;9:596.
Griñan-Ferré C, et al. Environmental enrichment modified epigenetic mechanisms in SAMP8 mouse hippocampus by reducing oxidative stress and inflammaging and achieving neuroprotection. Front Aging Neurosci 2016;8:241.
Griñan-Ferré C, et al. Environmental enrichment improves behavior, cognition, and brain functional markers in young senescence-accelerated prone mice (SAMP8). Mol Neurobiol 2016;53:2435–50.
Suzuki T, et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J Med Chem 2012;55:5760–73.
Flood JF, Farr SA, Kaiser FE, Morley JE. Age-related impairment in learning but not memory in SAMP8 female mice. Pharm Biochem Behav 1995;50:661–4.
Orejana L, Barros-Miñones L, Aguirre N, Puerta E. Implication of JNK pathway on tau pathology and cognitive decline in a senescence-accelerated mouse model. Exp Gerontol 2013;48:565–71.
Orejana L, Barros-Miñones L, Jordán J, Puerta E, Aguirre N. Sildenafil ameliorates cognitive deficits and tau pathology in a senescence-accelerated mouse model. Neurobiol Aging 2012;33:625.e11–20.
Orejana L, et al. Sildenafil decreases BACE1 and cathepsin B levels and reduces APP amyloidogenic processing in the SAMP8 mouse. J Gerontol Ser A Biol Sci Med Sci 2015;70:675–85.
García-Osta A, Cuadrado-Tejedor M. Advanced assay monitoring APP-carboxyl-terminal fragments as markers of APP processing in Alzheimer disease mouse models. Methods Mol Biol 2016;1303:117–23.
Gal J, Bang Y, Choi HJ. SIRT2 interferes with autophagy-mediated degradation of protein aggregates in neuronal cells under proteasome inhibition. Neurochem Int 2012;61:992–1000.
Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron 2008;58:10–14.
Wongchitrat P, et al. Alterations in the expression of amyloid precursor protein cleaving enzymes mRNA in Alzheimer peripheral blood. Curr Alzheimer Res 2018;16:29–38.
Wloga D, Joachimiak E, Fabczak H. Tubulin post-translational modifications and microtubule dynamics. Int J Mol Sci 2017;18:2207.
Yoo DY, et al. Sodium butyrate, a histone deacetylase Inhibitor, ameliorates SIRT2-induced memory impairment, reduction of cell proliferation, and neuroblast differentiation in the dentate gyrus. Neurol Res 2015;37:69–76.
Theendakara V, et al. Neuroprotective Sirtuin ratio reversed by ApoE4. Proc Natl Acad Sci USA 2013;110:18303–8.
Porquet D, et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age (Dordr) 2013;35:1851–65.
Zhao Y, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 2010;12:665–75.
Inoue T, et al. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J 2014;281:2623–37.
Fonseca R. The aging memory: modulating epigenetic modifications to improve cognitive function. Neurobiol Learn Mem 2016;133:182–4.
Traynelis SF, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharm Rev 2010;62:405–96.
Magnusson KR, Nelson SE, Young AB. Age-related changes in the protein expression of subunits of the NMDA receptor. Brain Res Mol Brain Res 2002;99:40–45.
Brim BL, et al. Memory in aged mice is rescued by enhanced expression of the GluN2B subunit of the NMDA receptor. Behav Brain Res 2013;238:211–26.
Muñoz-Cobo I, et al. Nucleocytoplasmic export of HDAC5 and SIRT2 downregulation: two epigenetic mechanisms by which antidepressants enhance synaptic plasticity markers. Psychopharmacology (Berl) 2018;235:2831–46.
Luo Y, et al. Reversal of aging-related emotional memory deficits by norepinephrine via regulating the stability of surface AMPA receptors. Aging Cell 2015;14:170–9.
Wang B, et al. SIRT2 plays significant roles in lipopolysaccharides-induced neuroinflammation and brain injury in mice. Neurochem Res 2016;41:2490–2500.
Moreno LCGEI, et al. Effect of the oral administration of nanoencapsulated quercetin on a mouse model of Alzheimer’s disease. Int J Pharm 2017;517:50–57.
Rancán L, et al. Protective effect of xanthohumol against age-related brain damage. J Nutr Biochem 2017;49:133–40.
Cheng J, et al. Vitamin D combined with resveratrol prevents cognitive decline in SAMP8 mice. Curr Alzheimer Res 2017;14:820–33.
Sun CY, et al. Neurobiological and pharmacological validity of curcumin in ameliorating memory performance of senescence-accelerated mice. Pharm Biochem Behav 2013;105:76–82.
Chen Z-J, Yang Y-F, Zhang Y-T, Yang D-H. Dietary total prenylflavonoids from the fruits of Psoralea corylifolia L. prevents age-related cognitive deficits and down-regulates Alzheimer’s markers in SAMP8 mice. Molecules 2018;23:196.
Scuderi C, et al. Sirtuin modulators control reactive gliosis in an in vitro model of Alzheimer’s disease. Front Pharmacol 2014;5:89.
Lee AS, et al. SIRT2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem Biophys Res Commun 2014;450:1363–9.
Chen H, Wu D, Ding X, Ying W. SIRT2 is required for lipopolysaccharide-induced activation of BV2 microglia. Neuroreport 2015;26:88–93.
Zhao T, et al. Selective inhibition of SIRT2 improves outcomes in a lethal septic model. Curr Mol Med 2015;15:634–41.
Pais TF, et al. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J 2013;32:2603–16.
Yuan F, et al. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-κB p65 acetylation and activation. J Neurochem 2016;136:581–93.
Li Y, et al. SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 2011;16:34–45.
He X, et al. SIRT2 activity is required for the survival of C6 glioma cells. Biochem Biophys Res Commun 2012;417:468–72.
Lynn EG, McLeod CJ, Gordon JP, Bao J, Sack MN. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3 zeta and BAD in H9c2 cells. FEBS Lett 2008;582:2857–62.
Acknowledgements
The authors are grateful to Ms. Sandra Lizaso and Mr. Mikel Aleixo for their excellent technical assistance. They would also like to thank “Amigos de la Universidad de Navarra” for a fellowship to TD-P and BB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Diaz-Perdigon, T., Belloch, F.B., Ricobaraza, A. et al. Early sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsychopharmacol. 45, 347–357 (2020). https://doi.org/10.1038/s41386-019-0503-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0503-8
This article is cited by
-
Alzheimer’s Disease-Related Epigenetic Changes: Novel Therapeutic Targets
Molecular Neurobiology (2024)
-
SIRT2 counteracts primate cardiac aging
Nature Aging (2023)
-
SIRT2 Inhibition Rescues Neurodegenerative Pathology but Increases Systemic Inflammation in a Transgenic Mouse Model of Alzheimer’s Disease
Journal of Neuroimmune Pharmacology (2023)
-
The sirtuin family in health and disease
Signal Transduction and Targeted Therapy (2022)
-
Insulin Deficiency Increases Sirt2 Level in Streptozotocin-Treated Alzheimer’s Disease-Like Mouse Model: Increased Sirt2 Induces Tau Phosphorylation Through ERK Activation
Molecular Neurobiology (2022)
