
Abstract
Experimental Medicine studies in psychiatric populations test specific, mechanistic hypotheses related to the biology of mental illness, by combining well-characterized neurobiological probes and laboratory-based measures of behavioral performance and neurobiology. However, scientific inquiry through the acute administration of psychoactive drugs to patients with serious mental illness raises important ethical issues. These issues arise in studies in which the psychostimulant, amphetamine, is used as an Experimental Medicine probe in patients with schizophrenia. In this study, we summarize relevant aspects of our experience with acute, laboratory-based challenges of amphetamine in schizophrenia patients. Schizophrenia patients participated in one or more Experimental Medicine studies involving limited doses of amphetamine with clinical monitoring, over a 4-year period. Acute (within hours of ingestion; collective n = 53), subacute (three active doses over 4 weeks; n = 28), and long-term (mean = 17 months after ingestion; n = 19) effects of amphetamine ingestion were assessed. In antipsychotic (AP)-medicated schizophrenia patients, amphetamine was associated with no detrimental subjective, autonomic, or functional changes. Symptoms assessed acutely, subacutely, or long term were either unchanged or diminished. No adverse acute, subacute, or long-term consequences from the Experimental Medicine use of amphetamine in antipsychotic-medicated schizophrenia patients were detected. These findings do not address the safety or effectiveness of the use of amphetamine in unmedicated patients, or as an adjunctive treatment for schizophrenia. Indeed, it is important to distinguish evidence-based risks of symptom exacerbation in an Experimental Medicine setting vs. risks associated with long-term, daily clinical use or even misuse of amphetamine.
Similar content being viewed by others


Introduction
Etiological models and therapeutic approaches for schizophrenia have traditionally focused on brain dopamine systems [1,2,3,4]. Twenty-first century versions of the “dopamine hypothesis” of schizophrenia posit that schizophrenia reflects abnormalities in the levels of dopamine neurotransmission or sensitivity to dopamine receptor stimulation, in subcortical (primarily D2-family receptors) and cortical (primarily D1-family receptors) forebrain dopamine fields [5]. The basis for such forebrain dopamine system abnormalities has been attributed to in-utero perturbations in neurodevelopment, risk genes, inflammatory cascades, placental deficiency, or other epigenetic factors, perhaps combined with precipitating processes during childhood and adolescence, including recreational drug exposure and addiction [5,6,7]; presumably, heterogeneity in the clinical schizophrenia phenotype reflects the differential contributions of these factors in different individuals. Pharmacotherapies for schizophrenia have historically targeted dopaminergic transmission, primarily designed to block D2-family (among other) receptors [8], and more recent strategies have explored the use of D1-family agonists as pro-cognitive agents for schizophrenia [9]. Other models of schizophrenia pathophysiology focus on deficient neurotransmission of N-methyl-d-aspartate (NMDA) but have not yet yielded effective therapeutics.
For all of the elegant science conducted in order to explicate the biology and therapeutics of schizophrenia, we are still a long way from this goal. As we await further advances in psychiatric genomics, which to date have yielded over 100 genes that collectively account for about 7% of the genetic variance associated with the schizophrenia diagnosis [10], our field will continue to rely on experimental approaches that probe brain function and dysfunction in schizophrenia. Experimental Medicine—one potentially informative scientific approach to understanding the biology of schizophrenia—combines well-characterized pharmacological probes with laboratory-based measures of behavioral performance and neurobiology, to determine how specific chemical changes alter brain function in schizophrenia patients vs. healthy subjects.
A major challenge with the Experimental Medicine approach to studying schizophrenia is that probing patients with drugs designed to explicate dysfunction in specific neurochemical substrates—e.g., dopamine or NMDA neurotransmission—might destabilize or otherwise adversely impact those substrates, and thereby have deleterious clinical consequences. This concern has been raised with d-amphetamine—a well-understood and thus experimentally valuable pharmacologic probe for brain dopaminergic activity. Amphetamine has been used extensively in preclinical and clinical studies to understand the normal function of brain dopamine systems; it is a very powerful experimental “known” with which to probe the still largely “unknown” dopaminergic pathology in schizophrenia patients. However, much of the early foundation of the “dopamine hypothesis” of schizophrenia was based on the observation that some features of schizophrenia can be reproduced in healthy subjects after sustained, escalating, and high doses of amphetamine [11]. Even a single dose or limited dosing of psychoactive drugs, as typically used in an Experimental Medicine design, can theoretically produce long-term changes in brain function [12]. Thus, a rational concern is created by studies that administer a drug known—under specific conditions—to evoke [13] or increase the risk for the development of psychosis [14], to patients who already have a severe psychotic disorder, even within the highly controlled context of an Experimental Medicine study. Both ethically and scientifically, we must consider whether such exposure to amphetamine might adversely impact schizophrenia patients, either by triggering a psychotic exacerbation, creating other concerns via interactions with known health vulnerabilities of schizophrenia patients (e.g., stigmata of chronic antipsychotic exposure, including metabolic syndrome, and associated hypertension and risk of cardiovascular disease), or otherwise worsening the longitudinal course of their illness.
Our research program routinely uses Experimental Medicine to study effects of amphetamine and other drugs on neurophysiological and neurocognitive measures in schizophrenia patients. In the process, we have accumulated “naturalistic” data related to symptoms and function in schizophrenia patients as they enroll in these studies and, in some cases, over a long period after they have completed these studies. Importantly, these data were not collected for the purpose of assessing long-term safety and tolerability of the Experimental Medicine use of amphetamine in schizophrenia patients; nonetheless, they provide an opportunity to review this issue and thus potentially inform the field, albeit in a post-hoc fashion. The primary goal of this report is to describe subjective and psychotic symptoms that follow amphetamine administration to medicated schizophrenia patients in a placebo (PBO)-controlled, double-blind Experimental Medicine design.
Methods
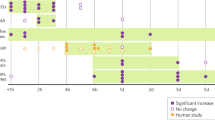
Studies were approved by the University of California, San Diego Institutional Review Board and posted on clinicaltrials.gov. Paid participants took part in one or more of three studies (Fig. 1), over an ~4-year period. Detailed methods for Study I are reported [15], whereas Study II and III remain in progress. All participants maintained stable AP doses for >1 month at the time of study enrollment. After it was determined that they met study inclusion criteria (based on current and past medical and psychiatric history, medication and recreational drug use, and family history of psychosis), subjects came to the laboratory for a screening visit. During that visit, subjects were informed of the potential risks and benefits of the study, read and signed a consent form for study participation, underwent a medical interview and structured diagnostic assessment (Mini International Neuropsychiatric Interview (M.I.N.I plus 6) for patients), symptom rating scales (PANSS [16]; Study I) or SANS and SAPS ([17, 18]; Study II and III), Brief Psychiatric Rating Scale (BPRS) psychosis subscales ([19]; “suspiciousness, hallucinations, unusual thoughts, conceptual disorganization”: Study II and III), Scale of Functioning (SOF [20]; Studies I, II, and III) and Global Assessment of Functioning (GAF [21]; Studies I, II, and III)), physical examination and electrocardiogram, and completed urine toxicology with exclusion for any recreational drug. Women underwent a urine-based pregnancy test. Neurocognition was assessed (MATRICS Comprehensive Cognitive Battery [22]), and subjects completed study-specific “baseline” behavioral and neurophysiological measures (reported previously [15, 23]). Participant information is found in Table 1.

Schedule of Study I, II, and III. To date, 28 subjects have completed Study II; 10 of these patients had previously completed Study I and 9 patients subsequently completed Study III. The potential adverse effects of amphetamine on both subjective experience, clinical symptoms, and autonomic function were assessed across the 4 weeks of testing within Study II (N = 28) and across the months-to-years separating the completion of two studies by a total of 19 patients. a Study 2 and 3 are in progress. b Study 3 ac ve drug = memantine [41] or tolcapone (n = 1; [42]); for the present report, only screening data from Study 3 are used, i.e. prior to any drug administration
Studies I and III involved two test days, ~7d apart, starting 7d after screening; Study II included four test days, ~7d apart, starting 7d after screening (Fig. 1). Testing was double-blind (pills were repackaged and coded by the hospital study pharmacist) and drug order was randomized. In Study I, pills were either PBO or 10 mg amphetamine; in Study II, pills were either PBO or 2.5, 5, or 10 mg amphetamine. Study III did not use amphetamine; data were used only to assess symptoms and function on the screen day—to gauge any potential long-term effects of Study I and/or II amphetamine exposure.
On test days, subjects were transported by hired drivers to arrive at the laboratory at 0830 h, ate a standardized breakfast, provided urine for toxicology (and pregnancy testing in women), and heart rate (HR) and blood pressure (BP) were determined (sitting position, brachial cuff). Subjects completed symptom rating visual analog scales (VAS) at designated intervals, starting before pill ingestion (baseline); in Study II, a pre-pill BPRS was completed, followed 300 min post-pill by a second BPRS. VAS scores assessed general somatic and psychological symptoms, and level of consciousness [24, 25]. Ratings assessed several states, but analyses were limited to three: “happy,” “drowsy,” and “anxious.” Details of these rating scales are found in Swerdlow et al. [26]. The study pill was administered at 0900 h. A licensed physician was present throughout the test day and subjects were carefully monitored; autonomic and subjective VAS ratings were continued until 430 min post-pill. Over the course of each test day, participants completed study-specific neurophysiological measures (reported in refs. [15, 23] or to be reported elsewhere). Subjects were paid on completion of each visit and then returned to their living setting by hired drivers.
Study participants are eligible for a subsequent study after a 3–6-month testing hiatus (specific interval constraints vary slightly across protocols). Beginning 3–6 months after completion of Study I or Study II, subjects who either recontacted the laboratory on their own initiative, or were recontacted by our staff, were invited to participate in a new study. In each subsequent study, participants completed screening assessments that included measures of symptoms and function. These screening assessments were then compared with measures obtained prior to their last study amphetamine exposure. Validated conversion functions were applied to SANS, SAPS, and PANSS scores [27] to permit comparisons across studies.
Analyses of amphetamine effects at acute (same day), subacute (across 4 weeks with three active dose exposures), and long-term (after many months of exposure) intervals are seen in Table 2. Sample sizes provided appropriate power (β = 0.8–0.9) to detect small-to-medium effect size metrics of clinical deterioration (d = 0.3–0.4) with α = 0.05.
In total, these studies in chronic, AP-medicated schizophrenia patients yielded information related to their subjective and autonomic response to acute amphetamine challenge (Study I and II, collective n = 53); their subjective, autonomic, and clinical changes in response to subacute administration of three active doses of amphetamine over 4 weeks (Study II, n = 28); and their clinical changes ≈17 months after laboratory exposure to amphetamine (n = 19). Data presented herein have never previously been published.
Results
Participants (Table 1) were very impaired AP-medicated individuals with longstanding psychotic disorders. All subjects took part in one or more studies that involved ingestion of amphetamine and a subsequent physiological and clinical (symptom) assessment, although this exposure history took several different “pathways” (Fig. 1). In total, 28 subjects completed Study II; 10 of these subjects had previously completed Study I, 8 of these subjects subsequently completed Study III, 1 completed Study I followed by Study III (no amphetamine ingestion in Study III) prior to completing Study II, and 4 subjects completed all 3 studies in sequence (I–II–III).
Acute (same day) and subacute (over 4 weeks) effects of amphetamine (2.5, 5, and 10 mg) on subjective experience, autonomic function, and symptom ratings
We previously reported [15] that Study I participants detected no significant effects of 10 mg amphetamine on ratings of “drowsy,” “happy,” “anxious,” or “concentration,” whereas modest amphetamine effects were noted in healthy subjects (reduced “drowsy” and increased “happy”). Patients reported no amphetamine-related adverse experiences and correctly identified their pill only at chance levels [15]. This relative absence of acute subjective effects of amphetamine in schizophrenia patients was replicated in Study II (Fig. 2). Using an anchored 100 mm VAS (“Not At All” to “Extremely”), subjects rated themselves as “Happy” in the range of 40–50 mm, whereas ratings for both “Drowsy” and “Anxious” were in the range of 15–20 mm. Analysis of variance (ANOVAs) revealed a significant main effect of amphetamine dose only for “Happy” (F = 3.73, df 3,60, p < 0.016) reflecting a modest reduction in this self-rating for the 2.5 mg dose vs. PBO (p < 0.025) and vs. 10 mg amphetamine (p < 0.003). There were no significant main effects of amphetamine on “Drowsy” (F < 1) or “Anxious” (F = 1.87, df 3,66, NS). All metrics declined over the course of the test day, but there were no significant interactions of amphetamine dose × time of day. Conceivably, adverse effects of repeated amphetamine administration on these three metrics might emerge with repeated dosing over the 4 weeks of Study II; however, ANOVAs of each of these three VAS ratings using test week as a within-subject factor revealed no significant effects of test day (all F’s < 1; data not shown).

Study II subjective ratings using a 100 mm visual analog scale (VAS). Amphetamine effects on self-rated levels of “Happy,” “Drowsy,” and “Anxious” are shown as a function of amphetamine dose, collapsed over the seven ratings that span one test day. See text for explanation (*2.5 mg < 0 mg (p < 0.025) and 2.5 mg < 10 mg (p < 0.003)
Study I [15] also reported that compared with healthy subjects, antipsychotic-medicated schizophrenia patients exhibited blunted effects of amphetamine on HR and BP, although some modest increases in diastolic blood pressure were noted, compared with PBO. The effects of amphetamine on autonomic function across the four testing weeks in Study II are seen in Fig. 3. Consistent with Study I [15] and others [28], amphetamine had only modest effects on autonomic measures in AP-medicated schizophrenia patients: although 10 mg of amphetamine produced the arithmetically highest autonomic responses, these differences did not achieve statistical significance. ANOVAs of HR and diastolic BP revealed no significant main effect of amphetamine dose (F’s = 1.96 and 1.87, respectively; both not significant (NS)) or time × dose interactions (F’s = 1.52 and <1, respectively; both NS); ANOVA of systolic BP revealed a significant effect of dose (F = 3.02, df 3,72, p < 0.04), reflecting a modest reduction in systolic BP for 5 mg vs. PBO (p < 0.08) and a significant increase for 10 mg vs. 5 mg (p < 0.005).

Study II autonomic measures (a heart rate, b systolic blood pressure, and c diastolic blood pressure). No statistically significant effects of amphetamine dose were detected on any measure, despite modest trends for increases at the highest (10 mg) dose
Symptoms of psychosis were either unchanged or reduced 5 h after pill administration and across the 4 weeks of Study II (which included three active doses of amphetamine). BPRS psychosis subscale scores were generally higher prior to pill administration compared with ~5 h post pill, for both PBO and all active doses. ANOVA with amphetamine dose, time (pre- vs. post-pill), and BPRS subscale as within-subject factors revealed no significant effect of amphetamine dose (F < 1), a significant effect of time (pre- vs. post-pill; F = 24.76, df 1,72, p < 0.0001) and subscale (F = 8.39, df 3,72, p < 0.0001), and a significant time × subscale interaction (F = 7.47, df 3,72, p < 0.0005) (Fig. 4a). The magnitude of BPRS response to amphetamine did not differ between groups who did vs. did not “self-select” to participate in more than one study: comparison of subjects who participated only in Study II (n = 9) vs. those who had also previously participated in Study I, or who subsequently completed Study III (n = 19), revealed very similar patterns (no significant effect of Group, amphetamine dose or Group × amphetamine dose interaction (F’s < 1, NS), a significant effect of time (p < 0.0001) but no significant Group × time interaction (F < 1); data not shown). Repeated amphetamine exposure was not associated with increased BPRS scores: collapsed across amphetamine dose, over the 4 consecutive weeks, subscale scores either remained relatively stable or declined slightly. ANOVA revealed no significant effect of test week (F = 1.01, df 3,72, NS), no interaction of test week × time (pre- vs. post pill) (F < 1), and no interaction of test week × time × subscale (F = 1.34, df 9,216, NS) (Fig. 4b). In other words, over the 4 weeks of testing, despite the fact that subjects had received cumulatively more amphetamine, psychotic symptoms did not become worse in these AP-medicated schizophrenia patients.

a Study II BPRS subscale scores prior to pill administration and 300 min later, by dose of amphetamine. Independent of dose (including placebo), ratings were significantly lower 300 min after pill administration (*p < 0.0001). b Study II BPRS subscale scores prior to pill administration and 300 min later, shown for the four consecutive test weeks of Study II, collapsed across dose. BPRS scores did not increase across the 4 weeks of testing, despite repeated dosing of amphetamine on 3 out of those 4 weeks. c Positive and negative symptoms (Global SAPS and SANS scores) measured during screening for two consecutive studies: either Study I followed by Study II or Study II followed by Study III. (*p < 0.009, reduced SAPS Global score ~17 months (on average) after exposure to either 10 or 17.5 mg of amphetamine. See text for Cohen’s d values for subjects tested three times [40]. d BPRS subscale scores prior to Study II and Study III, showing no increased (and, in some cases, a trend towards reduced) symptoms ~9 months (on average) after exposure to 17.5 mg amphetamine (“Halluc” = hallucinations; “Susp” = suspiciousness)
Long-term effects of amphetamine administration on symptom ratings and function ≈17 months later
Nineteen subjects participated in two studies: either Studies I and II or Studies II and III; four of these individuals participated sequentially in Studies I, II, and III, and thus had a cumulative amphetamine exposure of 27.5 mg (10 mg in Study I and 17.5 mg in Study II). The mean interval between amphetamine exposure and subsequent study screening was 17.2 months (range 3.9–66.1). We designated the screening prior to the first exposure to amphetamine as “Screen 1,” the screening prior to the subsequent study as “Screen 2,” and for four subjects the screening prior to the last study as “Screen 3.” Thus, Screen 2 ratings reflect the impact of exposure to 10 mg amphetamine during Study I (n = 10) or of exposure to 17.5 mg (2.5, 5, and 10 mg) amphetamine in Study II (n = 9). For the four subjects who participated sequentially in all three studies, Screen 3 ratings reflect exposure to both 10 mg (Study I) and 17.5 mg (Study 2) of amphetamine (total of 27.5 mg).
For the 19 subjects tested two or more times over the course of Studies I, II, and III, positive and negative symptoms were either unchanged or reduced on their follow-up visit (months after receiving amphetamine) compared with their initial visit (prior to first receiving amphetamine) (Fig. 4c). Global SAPS scores declined from Screen 1 to Screen 2 (F = 8.82, df 1,18, p < 0.009); for the four subjects who participated in all three studies (cumulative amphetamine exposure of 27.5 mg), Global SAPS scores declined 40%, from 10.5 to 6.25 (d = 0.93) from Screen 1 to Screen 3. Global SANS scores were relatively unchanged across testing, from Screen 1 to Screen 2 (means 8.84 vs. 7.68, respectively; F < 1); for the four subjects who completed all three studies in sequence, Global SANS scores declined 41%, from 12.25 to 7.75 (d = 1.11) from Screen 1 to Screen 3. Similarly, measures of function were either unchanged or reduced on follow-up visits: GAF scores did not differ significantly between Screen 1 and Screen 2 (F = 3.64, df 1,18, NS), but SOF total scores increased significantly (from 45.77 to 48.88, d = 2.55; F = 7.87, df 1,16, p < 0.015). This overall pattern of symptom stability or reduction over time was not a consequence of increased antipsychotic medication: Screen 1 vs. Screen 2 daily chlorpromazine (CPZ) equivalents were 646.12 mg vs. 531.64 mg (F = 2.25, df 1,18, NS); CPZ equivalents declined for 6 subjects by an average of 411 mg/d, increased in 2 subjects by an average of 146 mg/d, and were unchanged in 11 subjects. Among the subjects who received 3 doses of amphetamine (17.5 mg total) over 4 weeks (12 subjects who completed Study II and then were subsequently screened for Study III), BPRS subscale scores did not change significantly, 9.2 months later (on average) (Fig. 4d). ANOVA revealed no significant effect of Screen order (F < 1) or Screen order × subscale interaction (F = 1.44, df 3,33, NS).
Factors associated with amphetamine response
Exploratory analyses failed to detect any consistent relationships between patient factors (sex, age, age of onset, smoking status, CPZ equivalents, body weight, baseline symptom or function ratings) or experimental factors (number of active drug exposures (1 vs. 3), dose order, and number of studies completed) and acute, subacute, or long-term changes in symptom or function scores.
Discussion
In this study, AP-medicated schizophrenia patients did not exhibit any evidence of acute (hours), subacute (weeks) or long-term (≈1.5 years) adverse effects of exposure to 10 mg p.o. on 1 day and/or a total of 17.5 mg amphetamine p.o. divided over 3 weekly sessions. Metrics of amphetamine effects included acute and subacute subjective, symptomatic, and autonomic changes, as well as long-term symptomatic and functional changes. Both acutely and over 4 weekly tests (three of which involved active doses of amphetamine), doses of amphetamine up to 10 mg did not elevate symptoms of psychosis. Changes in HR and BP after an acute dose of amphetamine were modest, consistent with a longstanding literature of AP-induced blockade of amphetamine’s autonomic effects in these patients [28]. Among a smaller sample of individuals (n = 19) who were available for long-term follow-up, there was either no significant change or an actual reduction in psychotic symptoms and antipsychotic medication requirements, and either no change (GAF) or an increase (SOF) in function about 17.2 months (on average) after exposure to 10–17.5 mg of amphetamine. Thus, the aggregate findings of this review support the general safety of this use of amphetamine in AP-medicated schizophrenia patients within an Experimental Medicine design.
The lack of deleterious changes after acute amphetamine challenge is consistent with three published reports showing no symptom deterioration in antipsychotic-medicated schizophrenia patients in the hours after a single pill of amphetamine [29,30,31] (n = 32)). Of these studies, only Barch and Carter [29]) conducted clear pre- vs. post-pill structured clinical ratings (n = 10). To our knowledge, there are no other reports in antipsychotic-medicated schizophrenia patients that quantify clinical symptoms acutely, pre- and post-amphetamine, in a within-subject multi-dose design. As reviewed by Lindenmayer et al. [32], studies of psychostimulant effects in schizophrenia have focused on unmedicated patients, primarily using methylphenidate as a provocative agent. Although we are also unaware of any studies assessing subacute and long-term effects of single or repeated doses of amphetamine in schizophrenia patients, the observed lack of clinical deterioration is consistent with the positive clinical impact in schizophrenia patients of open-label daily amphetamine dosing for 10 weeks [33].
There are clear limitations to the “naturalistic” collection and post-hoc analysis of these data. Studies I–III were not conceived prospectively as interconnected parts of a longitudinal design to facilitate the type of exploration described in this report and several design issues complicate interpretation of the results—particularly as they relate to the long-term effects of amphetamine exposure in these patients. First, as there was no a priori goal to recruit subjects for longitudinal measures, the sample size for participants with long-term follow-up is modest. Nineteen subjects in a within-subject comparison provides power of 0.80 to detect a small-to-medium effect size (d = 0.35) long-term deterioration of symptoms. As the only evidence of symptom change was actually one of improvement (SAPS, SOF), it does not seem parsimonious to suggest that a larger sample would have revealed statistically significant changes in the “opposite direction.”
Second, although the acute and subacute effects of active doses of amphetamine in Study II could be compared with a within-subject PBO control response, no such parallel “placebo” group of schizophrenia patients (i.e., who received PBO on all test days) could be used to interpret the long-term changes, or lack thereof, in the amphetamine-exposed patients. Although study participants exhibited no evidence of clinical deterioration—and indeed, exhibited some evidence of clinical improvement—in the many months after amphetamine exposure, it is possible that these same subjects might have experienced even more improvement, had they received one to three pills of PBO (instead of amphetamine) as a study intervention. However, the likelihood that a cohort of schizophrenia patients would have exhibited greater spontaneous clinical improvement (compared with the stable SANS scores and significant reductions in SAPS scores (p < 0.009) and gains in SOF scores (p < 0.015)) over the course of ~17 months after one to three pills of PBO is not easily reconciled with the known natural history of this illness.
Third, many uncontrolled factors other than amphetamine ingestion might have contributed to changes in symptoms or function in the intervals between Study I and II, or Study II and III, which were not monitored or detected by our review. Thus, although we found no significant changes in antipsychotic doses (chlorpromazine equivalents) between studies that might have reflected or caused a change in symptoms, other events—such as hospitalizations, recreational drug use, or changes in relationships or living settings—might certainly have gone unreported. We can say with some confidence that the reduction in psychotic symptoms during the months after amphetamine exposure did not reflect an increase in antipsychotic medication dose, as these doses generally remained unchanged or declined during that period.
Fourth, returning participants (i.e., Study I participants who returned for Studies II and III, and those from Study II who returned for Study III) were self-selected and might have represented a particularly resilient group, compared with those who did not participate in subsequent studies. This fact could not have impacted the findings from acute single-dose (Study 1) or acute and subacute multi-dose (Study 2) studies, but conceivably might have impacted the findings of long-term clinical changes (or lack thereof) among “returning” participants. Notably, very comparable acute and subacute responses to amphetamine were detected among Study II participants who did vs. did not return for a second study (see Results), suggesting that “self-selection bias” did not impact these long-term results.
Fifth, acute effects of amphetamine based on pre- vs. post-pill assessments are systematically confounded by time-of-day and measurement order, as all pills were administered at 0900 h; thus, the reduction in BPRS scores post- vs. pre-pill (including PBO) might reflect circadian differences, or simply the order effects related to habituation to the laboratory settings over the 5 h from pre- to -post-pill measurements. Similarly, reduced symptoms across multiple test visits might certainly reflect increased familiarity and comfort with the study procedures. Lastly, all negative findings—particularly the failure to detect an adverse effect—must be interpreted with caution.
The primary goal of the present analyses was to assess the effects of amphetamine, as administered within an Experimental Medicine design, on symptoms and function in antipsychotic-medicated schizophrenia patients. The specialized conditions of these Experimental Medicine studies prevent generalized conclusions regarding the use of amphetamine in clinical trials or other study designs with schizophrenia patients. It is possible that amphetamine exposure might have deleterious effects on patients with schizophrenia if it is administered earlier in the illness course, over sustained periods, with higher doses, in sensitive populations (e.g., based on sex, genotype, or substance use history), or in tandem with particular antipsychotic regimens (e.g., partial agonists). Several levels of safety precautions were utilized in the present studies; e.g., participants were transported to and from the laboratory each day by hired drivers, and were medically monitored throughout the test day. On each screen day and test day, study participants were confirmed to have negative urine toxicology results; this distinguishes our Experimental Medicine cohort from a general population of schizophrenia patients who might be actively using recreational drugs including methamphetamine. Conversely, much of the concern regarding the use of amphetamine in Experimental Medicine designs with schizophrenia patients might be based on adverse effects associated with amphetamine use that is quite distinct from the Experimental Medicine designs in our studies, e.g., with higher [34, 35], escalating [36], or sustained daily doses [33], without antipsychotics [34, 37], in first-episode or high-risk populations [14], or via intravenous routes of administration [35].
Amphetamine is known to stimulate presynaptic release of not only dopamine but also both serotonin and norepinephrine, in some cases at both central and peripheral sites. Most antipsychotics block neurotransmission at not only dopamine receptors (primarily D2-family receptors) but also at receptors for both serotonin and norepinephrine [38]. Thus, it would be parsimonious to suggest that, to the degree that antipsychotics “protect” patients from adverse effects of acute amphetamine, they might do so via actions that prevent increased activity of norepinephrine or serotonin at the central or peripheral end organs. As study subjects were treated with a variety of antipsychotics in combination, it was impossible to distinguish specific antipsychotics as being either more or less “protective” against potentially deleterious effects of amphetamine administration.
It is worth noting that the doses used in the present studies (2.5–10 mg po) are relatively low compared with other studies of amphetamine effects in healthy adults, but are consistent with doses used in Experimental Medicine studies in schizophrenia patients and thus are appropriate for the primary goal of this study. It is also important to note that these doses are adequate to produce gains in both learning (5 and 10 mg) and sensorimotor gating (10 mg) in schizophrenia patients in the present Experimental Medicine studies [15, 23, 39]. Thus, even these lower doses can provide valuable neurophysiological and behavioral information, to advance our understanding of brain function in schizophrenia; the present study supports the contention that such doses are not only informative but also safe when used in an Experimental Medicine design in these patients.
It is essential that scientists use an abundance of caution when engaging vulnerable human subjects in experimentation, even when pursuing knowledge that will ultimately benefit those subjects, their families, or the larger population of individuals with mental illness. This caution should reflect the best available objective data that confirm a true risk of a specific experimental intervention, rather than an extrapolation based on the effects of different interventions, in different populations, or based on hypothesized models. Our present findings identify no acute, subacute, or long-term deleterious effects of the use of limited, low doses of amphetamine in an Experimental Medicine design in AP-medicated schizophrenia patients. Given the potential value of using well-characterized pharmacological probes and neurobiological measures as tools for understanding the biology of mental illness, and the relative paucity of alternative experimental avenues, it is important to maintain an objective view of the risks and benefits of the Experimental Medicine approach, and to reach conclusions based on empirical data.
Funding and disclosure
These studies were supported by MH059803, MH094320, and by a Distinguished Investigator Award to NRS by the Brain & Behavior Research Foundation. GAL has served as a consultant for Astellas, Boehringer Ingelheim, Heptares, Merck, NeuroSig, Neuroverse, and Takeda. The authors declare no competing interests.
References
Friedhoff AJ, VanWinkle E. Conversion of dopamine to 3,4-Dimethoxyphenylacetic acid in schizophrenia patients. Nature. 1963;199(Sep 28):1271–2.
van Rossum JM. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Arch Int Pharm Ther. 1966;160:492–4.
Snyder SH. Amphetamine psychosis: a “model” schizophrenia mediated by catecholamines. Am J Psychiatry. 1973;130:61–7.
Stevens J. An anatomy of schizophrenia? Arch Gen Psychiatry. 1973;29:177–89.
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III-the final common pathway. Schizophr Bull. 2009;35:549–62.
Swerdlow NR. Are we studying and treating schizophrenia correctly? Schizophr Res. 2011;130:1–10.
Ursini G, Punzi G, Chen Q, Marenco S, Robinson JF, Porcelli A, et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med. 2018;24:792–801.
Creese I, Burt DR, Snyder SH. Dopamine receptors and average clinical doses. Science. 1976;192:481–3.
Girgis RR, Van Snellenberg JX, Glass A, Kegeles LS, Thompson JL, Wall M, et al. A proof-of-concept, randomized controlled trial of DAR-0100A, a dopamine-1 receptor agonist, for cognitive enhancement in schizophrenia. J Psychopharmacol. 2016;30:428–35.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7
Randrup A, Munkvad I. Influence of amphetamines on animal behaviour: stereotypy, functional impairment and possible animal-human correlations. Psychiatr Neurol Neurochir. 1972;75(May-Jun):193–202.
Antelman SM, Gershon S. Clinical application of time-dependent sensitization to antidepressant therapy. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:65–78.
Wallis GG, McHarg JF, Scott OC. Acute psychosis caused by dextro-amphetamine. Br Med J. 1949;2:1394.
Moran LV, Ongur D, Hsu J, Castro VM, Perlis RH, Schneeweiss S. Psychosis with methylphenidate or amphetamine in patients with ADHD. N Engl J Med. 2019;380:1128–38.
Swerdlow NR, Tarasenko M, Bhakta SG, Talledo J, Alvarez AI, Hughes EL, et al. Amphetamine enhances gains in auditory discrimination training in adult schizophrenia patients. Schizophr Bull. 2017;43:872–80.
Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–76.
Andreasen NC. The scale for the assessment of negative symptoms (SANS). Iowa City, IA: University of Iowa; 1984a.
Andreasen NC. The scale for the assessment of positive symptoms (SAPS). Iowa City, IA: University of Iowa; 1984b.
Overall JE, Gorham DR. The brief psychiatric rating scale. Psychol Rep. 1962;10:799–812.
Rapaport MH, Bazetta J, McAdams LA, Patterson J, Jeste DV. Validation of the scale of functioning in older outpatients with schizophrenia. Am J Geriatr Psychiatry. 1996;4:218–28.
Hall RC. Global assessment of functioning. A modified scale. Psychosomatics. 1995;36:267–75.
Nuechterlein KH, Green MF, Kern RS, Baade LE, Barch DM, Cohen JD, et al. The MATRICS Consensus Cognitive Battery, part 1: test selection, reliability, and validity. Am J Psychiatry. 2008;165:203–13.
Swerdlow NR, Bhakta SG, Talledo JA, Franz DM, Hughes EL, Rana BK, et al. Effects of amphetamine on sensorimotor gating and neurocognition in antipsychotic-medicated schizophrenia patients. Neuropsychopharmacology. 2018a;43:708–17.
Bond AJ, James DC, Lader MH. Physiological and psychological measures in anxious patients. Psychol Med. 1974;4:364–73.
Norris H. The action of sedation on brain-stem oculomotor systems in man. Neuropharmacology. 1971;10:181–91.
Swerdlow NR, Eastvold A, Karban B, Ploum Y, Stephany N, Geyer MA, et al. Dopamine agonist effects on startle and sensorimotor gating in normal male subjects: time course studies. Psychopharmacol (Berl). 2002;161(May):189–201.
van Erp TG, Preda A, Nguyen D, Faziola L, Turner J, Bustillo J, et al. Converting positive and negative symptom scores between PANSS and SAPS/SANS. Schizophr Res. 2014;152:289–94.
Modell W, Hussar AE. Failure of dextroamphetamine sulfate to influence eating and sleeping patterns in obese schizophrenic patients: clinical and pharmacological significance. JAMA. 1965;193:275–8.
Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophr Res. 2005;77:43–58.
Goldberg TE, Bigelow LB, Weinberger DR. Cognitive and behavioral effects of the coadministration of dextroamphetamine and haloperidol in schizophrenia. Am J Psychiatry. 1991;148:78–84.
Pietrzak RH, Snyder PJ, Maruff P. Use of an acute challenge with d-amphetamine to model cognitive improvement in chronic schizophrenia. Hum Psychopharmacol. 2010;25:353–8.
Lindenmayer JP, Nasrallah H, Pucci M, James S, Citrome L. A systematic review of psychostimulant treatment of negative symptoms of schizophrenia: challenges and therapeutic opportunities. Schizophr Res. 2013;147(Jul):241–52.
Lasser RA, Dirks B, Nasrallah H, Kirsch C, Gao J, Pucci ML, et al. Adjunctive lisdexamfetamine dimesylate therapy in adult outpatients with predominant negative symptoms of schizophrenia: open-label and randomized-withdrawal phases. Neuropsychopharmacology. 2013;38:2140–9.
Angrist B, Rotrosen J, Gershon S. Differential effects of amphetamine and neuroleptics on negative vs. positive symptoms in schizophrenia. Psychopharmacol (Berl). 1980;72:17–9.
van Kammen DP, Bunney WE Jr, Docherty JP, Marder SR, Ebert MH, Rosenblatt JE, et al. d-Amphetamine-induced heterogeneous changes in psychotic behavior in schizophrenia. Am J Psychiatry. 1982;139(Aug):991–7.
Segal DS, Kuczenski R. An escalating dose “binge” model of amphetamine psychosis: behavioral and neurochemical characteristics. J Neurosci. 1997;17:2551–66.
Abi-Dargham A, van de Giessen E, Slifstein M, Kegeles LS, Laruelle M. Baseline and amphetamine-stimulated dopamine activity are related in drug-naïve schizophrenic subjects. Biol Psychiatry. 2009;65:1091–93.
Nasrallah HA. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiatry. 2008;13:27–35.
Swerdlow NR, Franz DM, Benster L, Talledo J, Bhakta SG, Light GA. Early Auditory Information Processing in Schizophrenia as a Therapeutic Target: Preliminary Dose-Response Findings With Amphetamine. Proc Am Coll Neuropsychopharmacology 57th Annual Meeting 2018b, 6 December 2018. https://doi.org/10.1038/s41386-018-0268-5.
Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed. Hillsdale, NJ: L Erlbaum Assoc.; 1988.
Swerdlow NR, Bhakta S, Chou HH, Talledo JA, Balvaneda B, Light GA. Memantine effects on sensorimotor gating and mismatch negativity in patients with chronic psychosis. Neuropsychopharmacol. 2016;41:419–30.
Bhakta SG, Light GA, Talledo JA, Balvaneda B, Hughes E, Alvarez A, et al. Tolcapone-enhanced neurocognition in healthy adults: neural basis and predictors. Int J Neuropsychopharmacol. 2017;20:979–87.
Acknowledgements
Ms. Maria Bongiovanni assisted in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Swerdlow, N.R., Bhakta, S.G., Talledo, J. et al. Lessons learned by giving amphetamine to antipsychotic-medicated schizophrenia patients. Neuropsychopharmacol. 44, 2277–2284 (2019). https://doi.org/10.1038/s41386-019-0495-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0495-4
This article is cited by
-
Cross-diagnostic determinants of cognitive functioning: the muscarinic cholinergic receptor as a model system
Translational Psychiatry (2023)
-
Clinical correlation but no elevation of striatal dopamine synthesis capacity in two independent cohorts of medication-free individuals with schizophrenia
Molecular Psychiatry (2022)
