
Abstract
Circular RNAs (circRNAs) are a covalently closed subclass of non-coding RNA molecules formed by back splicing of linear precursor RNA. These molecules are relatively stable and particularly abundant in the mammalian brain and therefore may participate in neural development and function. With the emergence of circRNAs activity in gene regulation, these molecules have been implicated in several biological processes, including synaptic plasticity, and we therefore suspect they may have a role in neurobehavioral disorders. Here, we profile cortical circRNAs expression in 35 postmortem cortical gray matter (BA46) schizophrenia and a non-psychiatric comparison group, using circRNA enrichment sequencing. While more than 90,000 circRNAs species were identified in the dorsolateral prefrontal cortex (DLPFC), we observed lower complexity and substantial depletion in subjects with the disorder. Although circRNAs expression was independent of their host gene transcription, alternative splicing rates were lower in samples from cases compared to controls. Gene set analysis of differentially expressed circRNAs host genes revealed significant enrichment of neural functions and neurological disorders. Many of these depleted circRNAs are also predicted to sequester miRNAs that were shown previously to be increased in the disorder, potentially exacerbating the functional impact of their dysregulation through posttranscriptional gene silencing. While this is the first reported exploration of circRNAs in schizophrenia, there is significant potential for dysregulation more broadly in other major mental illnesses and behavioral disorders. Given their capacity for modulating miRNA function, circRNA may play a significant role in the pathophysiology of disease and even be targeted for therapeutic manipulation.
Similar content being viewed by others


Introduction
Schizophrenia is a complex chronic brain disorder that disrupts normal function of mind and affects thoughts, behavior, feelings, and speech [1]. The symptoms of schizophrenia typically emerge in adolescence and early adulthood and have been shown to be accompanied by structural and functional changes in the brain that impact on neural connectivity [2]. While the molecular determinants and neuropathology driving the pathogenesis and pathophysiology appear to be multifaceted, we know that both genetic and epigenetic mechanisms play a significant role in diverse etiological pathways. Posttranscriptional dysregulation of gene expression, in particular, has been a consistent feature of these mechanisms with strong association of miRNA genes, such as MIR137 and its target genes, with schizophrenia [3]. The expression of miRNA regulation, more broadly, appear to be dysregulated in schizophrenia [4] and miRNA may be important epigenomic modulators of environmental exposures [5]. This is significant as these molecules have a dramatic influence on the traffic, stability, and temporospatially specific translation of protein-coding genes in the brain [6, 7]. In our exploration of miRNA expression in schizophrenia we observed an overall increase in miRNA expression, particularly in the cortical gray matter of the superior temporal gyrus (STG: BA22) and dorsolateral prefrontal cortex (DLPFC: BA9) [4]. While we observed schizophrenia-associated changes in components of the miRNA biogenesis pathway, there are several other mechanisms that affect their sequence, stability, subcellular distribution, and bioavailability, which could modify the levels or function of miRNA in the brain. For example, miRNA distribution and trafficking within the cell can be modified by ribonucleoproteins, such as FMRP (associated with schizophrenia), and the neural granules they form [8]. They can also be encapsulated within membrane-bound intraluminal vesicles of the multivesicular bodies and released from neurons within exosomes [9]. The cellular impact of miRNA can also be attenuated by association with antisense nucleic acid sponging sequences that sequester miRISC complexes and prevent them from interacting with translatable mRNA targets [10]. These sponging sequences are typically single stranded RNA, known generically as competing endogenous RNA (ceRNA), can reside within both coding and non-coding RNA transcripts and their expression can be specifically modified by cells to buffer the function of their target miRNA. An interesting form of miRNA sponging ceRNA has emerged recently in the discovery of circular RNA (circRNA) [11]. These covalently closed RNA molecules have a high capacity for miRNA sponging and have very high stability within the cell due to their circularity [11]. These non-coding RNAs are conserved between species, are highly expressed in the brain and differentially expressed in different tissues and disease states [12,13,14]. We suspected for a variety of reasons, that they may potentially be dysregulated in schizophrenia and possibly modify the posttranscriptional function of miRNA in the disorder.
CircRNAs are produced by back splicing of 3′ and 5′ ends of a transcript, generating a single stranded circular structure [15]. While they are often derived from longer linear transcripts, mostly protein-coding genes, they have significantly higher stability than their linear counterparts [11]. Biogenesis of circRNAs is catalysed by canonical spliceosome machinery and is affected by levels of ADAR (A-to-I RNA editing) [16] and the RNA binding protein Quaking (QKI) [17], both of which have been associated with schizophrenia [18,19,20]. Using publicly available CLIP-seq data of circRNAs-miRNA interaction [21] and setting a very high stringency (>5) and high cutoff for clipReadNum (>5000), we found substantial enrichment of miRNA previously associated with schizophrenia in postmortem brain; 334 interactions (41%) were related to schizophrenia-associated miRNA. This led us to hypothesize that aberrant cortical circRNAs expression patterns may further contribute to the pathogenesis of schizophrenia by modifying the bioavailability of schizophrenia-associated miRNA. To further explore this idea, we applied a high-throughput sequencing approach to systematically analyse circRNAs expression in postmortem samples from the DLPFC (BA 46) from subjects with schizophrenia and a non-psychiatric comparison group.
Materials and methods
Sample collection
Postmortem brain samples from DLPFC (BA46) from 20 subjects with schizophrenia and 20 subjects with no history of psychiatric diagnoses were obtained from New South Wales Tissue Resource Center, Sydney, Australia. Tissue preparation and total RNA extraction was performed as described by Weickert et al. [22]. A diagnosis of schizophrenia, in accordance with DSM-IV criteria, was confirmed by medical file review using the Item Group Checklist of the Schedules for Clinical Assessment in Neuropsychiatry and the Diagnostic Instrument for Brain Studies. For all subjects a written consent was gained from the next of kin, and individuals that had a history of drug or alcohol abuse, or other condition that could affect the agonal state were excluded. All the studied subjects were of Caucasian descent. Subjects with schizophrenia were matched for age, gender, brain hemisphere, pH, and postmortem interval (PMI) (Table S1).
RNA-seq library construction and sequencing
For total RNA sequencing, 12 μg of RNA from 20 schizophrenia and 20 controls were DNase treated (Ambion) before the depletion of ribosomal RNA with Ribominus kit (Invitrogen). Fragmentation and library preparation was carried out using the Total RNA-Seq kit (Invitrogen). Agilent 2100 Bioanalyzer chips were used to quality control libraries before 50 cycles of single end sequencing on the SOLiDv4 DNA sequencer (Life Technologies). For circRNAs enriched sequencing, 5 μg of RNA from 20 schizophrenia and 20 controls subjects were treated with DNase I (1 U/1 μg RNA, Thermo Scientific, USA) and then depleted for ribosomal RNA with Ribo-Zero rRNA Removal kit (epicenter, USA). In order to enrich for circRNAs species, RNase R (Illumina) treatment (3 U/μg) was performed at 37 °C for 15 min. Sequencing libraries were generated from schizophrenia and control subjects using Illumina TruSeq Stranded Total RNA Library Prep Kit LT (Illumina) according to the manufacturer’s protocol and then were checked for quality using microcapillary electrophoresis on an Agilent 2100 bioanalyzer. Libraries from schizophrenia and control tissues were evenly distributed in four pools to avoid bias in clustering and then separately run on NexSeq550 instrument with 150 cycles of single-end reads.
Computational pipeline for circRNAs prediction
We used two different methods to predict back-splice junction reads for circRNAs in our circRNAs enriched dataset; CIRCexplorer [23] and find_circ [11]. In CIRCexplorer pipeline, sequence reads were first aligned with TopHat 2.0.9 (parameters: -a 6 --microexon-search -m 2) to hg19 human reference genome. Unmapped reads were collected and aligned onto the reference genome using TopHat-Fusion (parameters: --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search). All reads mapped on the same chromosome, however, in a reverse order were collected as candidate back-spliced junctions. To identify the positions of the acceptor or donor sites of each back-spliced event, the back-spliced junction reads were remapped against gene annotations. Using a custom script, all junction reads having an alignment shift against canonical splice sites were corrected and reads mapped on non-canonical splice sites or different genes were filtered out.
In relation to the find_circ pipeline, we aligned the sequence reads to the human reference genome hg19 using Bowtie2 (parameters: -p16 --very-sensitive --phred64 --mm-M20 --score-min5C, −15, 0) and unmapped reads were extracted from BAM files. Reads were then split to obtain 20mer anchors at both ends using a custom script and mapped independently to the genome using Bowtie2, keeping their paired ordering (parameters: -p16 –reorder --mm -M20 --score-min5C, −15, 0). Anchors that mapped in the reverse ordering were extracted and the associated reads were remapped to collect the reads with full reverse alignment. The resulting reads were considered as back-splice junctions indicating circRNAs splicing. In the final step, the back-splice reads were run through a custom script to filter for minimal quality cutoffs and all candidates with at least two supporting reads were regarded as circRNAs.
Identification of circRNAs in total RNA sequencing was accomplished using a comprehensive database (over 90,000 unique circRNAs) created from our circRNA enriched dataset. This enabled compatibility between our circRNAs pipeline and color-space sequencing reads. To create the circRNAs database we used the genomic coordinates of all circRNA identified using the two pipelines and retrieved whole circRNA sequences using existing exon annotation with bedtools (getfasta –fi). Both ends of each read were split into 35-mer anchor and then concatenated to generate 70 bp back-splice junction reads (head-to-tail). This database was then used as an index for the alignment in Bowtie. All the sequencing reads were aligned to the reference genome hg19 with Tophat 2.0.10 (tophat2 -a 6 --microexon-search –m 2) and unmapped reads were extracted. These reads were then aligned to the circRNAs database using Bowtie 1.0.0 (parameters: bowtie -y --chunkmbs 128 -a --best --strata). Five samples were excluded from analysis after QC leaving 35 for group comparisons.
Expression analysis
To estimate the expression of circRNAs we quantified the number of reads spanning back-spliced junctions (circular reads). The relative expression of each circRNAs candidate was denoted as TPM (transcript per million mapped reads), where the effective length measured as: (sequencing length –2 * 6). To compare circRNAs expression between schizophrenia and controls two-tailed Wilcoxon rank-sum test was applied.
Total RNA sequencing, mapping, and mRNA expression analysis
RNA-seq reads were mapped to the reference genome with X-MATE recursive mapping pipeline according to Fillman et al. [24]. For junction alignment, 10 nucleotides were required to overlap the exon–exon boundary all the tag lengths ≥40 nt were required to have 10 nucleotides overlap with exon boundaries and five nucleotides for tag lengths < 40 nt. Exon junction libraries were created using Ref-Seq hg19 annotations using the scripts within X-MATE. The average of mapped reads per individual was 59,418,524 (43%). For mRNA expression analysis, results from the alignment were converted into an interval format and counts per exon were calculated using exon-annotated genome (hg19). The counts were assembled into 33,105 transcript isoforms and then analyzed using EdgeR (www.bioconductor.org/packages/2.3/bioc/html/edgeR.html) for differential gene expression between schizophrenia and control subjects. Fold change and multiple correction-adjusted P-values were fed into Ingenuity Pathway Analysis (www.ingenuity.com) for pathway analysis. The gene list was filtered keeping only those with a false discovery rate of <0.05.
cDNA synthesis and quantitative PCR (qPCR)
Validation of the expressed circRNAs was performed using qPCR. Total RNA treated with DNAse I (1 U/1 μg RNA, Thermo Scientific) was then used for reverse transcription using Superscript II Reverse Transcriptase (Invitrogen) with random hexamers according to the manufacturer’s instructions. To confirm the resistance of circRNAs to RNaseR, qPCR was carried out using RNA samples with and without RNaseR treatment. Specific divergent primers were designed for circRNAs to detect back-splice junctions. qPCR reactions were performed on an Applied Biosystems 7500 real-time PCR machine using Power SYBR green master mix (ThermoFisher) following the manufacturer’s instructions. GAPDH mRNA was also used as an internal reference for normalization. Expression values were determined using the ΔCt method. All reactions were performed in triplicate and presented as means ± SEM. The Student’s t-test was used to compare expression levels. Primer sequences for RT-PCR and RT-qPCR used were listed in Table S2.
Functional enrichment analysis
Gene Ontology enrichment analysis was carried out for circRNAs host genes, and the miRNA target genes using GO (http://geneontology.org) [25]. The pathway analysis of the interacting miRNA was performed using mirPath [26]. We also used ToppFun tool from ToppGene (https://toppgene.cchmc.org/) [27, 28] for disease enrichment analysis for DE circRNAs host genes.
Characterization of alternative back-splicing and novel circRNAs
All four basic types of alternative splicing events, including alternative 5′ splicing, alternative 3′ splicing, alternative cassette exon and intron retention were identified and quantitated in expressed circRNAs using RNase R RNA-seq dataset with relevant metrics according to Zhang et al. [29]. In brief, CIRCexplorer2 recruits Cufflinks to perform de novo assembly for circRNAs transcripts and the assembled results is used to characterizes alternative splicing events.
To detect novel transcripts for circRNAs, The Cufflinks reference annotation based transcript (RABT) assembly method [30] was used. Briefly, the RNase R sequencing reads that mapped to the reference using TopHat2 were assembled using filtered gene annotation (hg19 knownGene.txt) and then all reads with minimum two junction reads supporting all the isoform junctions were retained using Cufflinks 2.2.1 (parameters: -u -F 0 -j 0).
Potential miRNA targets and network construction
To predict miRNA binding sites within circRNAs, exonic sequence of all circRNAs were obtained using hg19 annotation. Then using TargetScan_60 and targetscan_61_context_scores Perl scripts (http://www.targetscan.org/vert_71/) [31,32,33,34] miRNA binding sites were determined. To filter for more reliable interactions, we set a context score threshold cutoff < −0.4. In addition, all miRNA targeted less than five times were also discarded. In order to generate the circRNAs-miRNA-mRNA network, we then determined which differentially expressed mRNAs in total RNAseq data, are targeted by these miRNAs. All the circRNAs-miRNA-mRNA networks were constructed using Cytoscape tool v.3.5.1 (http://www.cytoscape.org/) [35].
Results
circRNAs are downregulated in postmortem DLPFC in schizophrenia
In recent studies circRNAs molecules have been shown to display dynamic expression in the brain during development, including neural differentiation and maturation [36]. To investigate the possibility that these molecules are altered in the brain of people with schizophrenia, we profiled the expression pattern of circRNAs in postmortem DLPFC from subjects with a diagnosis of schizophrenia and a non-psychiatric comparison group using RNA-seq. CircRNAs transcript reads were extracted from rRNA-depleted total RNA sequencing (average 130 million reads per sample) and normalized against the density of RNA-seq mapped reads (transcripts per kilobase million (TPM)) in each sample (Table S3). A total of 35,257 distinct circRNAs were detected in combined schizophrenia and control groups, of which 10,541 (29.9%) were only observed in controls and 9003 (25.5%) were only observed in schizophrenia, indicating a smaller fraction of circRNAs were expressed in the schizophrenia samples. Case and control comparison identified 574 differentially expressed circRNAs (P-value < 0.05) (Fig. 1a, b, Table S4) with 390 circRNAs (68%) reduced in schizophrenia with expression completely absent for 240 species. By contrast, only 184 circRNAs were upregulated and most of these (120) were completely absent in controls, suggesting that there was an overall bias towards downregulation (Fig. 1a, b, Table S4).


Differential expression of circRNAs and their validation in schizophrenia and enrichment analysis for the DE circRNAs host genes. a Volcano plots indicating −log10 (P value) vs. log2 fold difference in circRNA abundance (TPM) between schizophrenia and control subjects from total RNA dataset. Red dots denote significant upregulated and green dots downregulated circRNAs (P < 0.05) with fold-change cutoff at 1.5. b Heatmap of circRNA normalized read counts between schizophrenia and control subjects from total RNA dataset. c Volcano plots indicating −log10 (P value) vs. log2 fold difference in circRNA abundance (TPM) between schizophrenia and control subjects from circRNA enriched dataset. Red dots denote significant upregulated and green dots downregulated circRNAs (P < 0.05) with fold-change cutoff at 1.5. d Heatmap of circRNA normalized read counts between schizophrenia and control subjects from circRNA enriched dataset. e Gene ontology and disease pathways enrichment analyses for the genes producing DE circRNAs. f The expression levels of eight circRNAs were validated by qPCR in post-mortem cortex of schizophrenia (SZ) patients (n = 16) vs. healthy controls (CN) (n = 16). Data are shown as mean ± SEM. CircRNAs expression was normalized using GAPDH as control. Statistical significance was assessed by Student’s t-test (one-tailed). *P < 0.05; **P < 0.01; ***P < 0.001. g Expression levels of the eight validated circRNAs measured with RNA sequencing
Given that circRNAs may display coordinated expression pattern dictated by their host gene, we performed differential expression analysis for schizophrenia vs. control by considering all the circRNAs within the same gene as one event. As expected, these results showed the same trend as individual circRNAs analysis with 173 genes downregulated out of 252 undergoing differential circRNAs biogenesis (P-value < 0.05). We also found that 116 circRNAs are spliced from the same locations as the circRNAs differentially expressed in the individual analysis, suggesting there are particular loci actively producing multiple circRNAs species that are altered in schizophrenia.
As the circRNAs read count data was extracted from total RNA sequencing, we wanted to determine if RNA-samples depleted of linear RNA would also display a difference between cases and controls. This was achieved using read count data from circRNAs-enriched cDNA libraries (>10-fold with respect to total RNA) produced by exoribonuclease R (RNaseR) depletion of linear RNA (Table S3). The results again revealed a reduction in the expression of circRNAs in the schizophrenia samples, with 72% of differentially expressed circRNAs (818) downregulated compared to 28% upregulated (324) (P-value < 0.05) (Fig. 1c, d, Table S4). Again, when circRNAs expression was consolidated with respect to each gene, 71.4% of the 273 loci undergoing circRNAs-splicing were downregulated, compared to 28.6% which were upregulated (P-value < 0.05). CircRNAs altered at 115 genes also accorded with circRNAs with differential expression in individual analysis.
To further explore the genomic distribution of differential circRNA expression, we mapped them to their chromosomal positions and found they were broadly distributed across all chromosomes. Chr1, chr2 and chr7, were over represented in both sequencing data, with ~30% of circRNA molecules being derived from these three chromosomes (Fig. S1A, S1B).
In preliminary functional analysis of the differentially expressed circRNAs host genes, we found that 30% of the host genes (in each dataset) have previously been associated with schizophrenia (Table S5). We then performed gene ontology enrichment for the DE circRNAs host genes which suggested that the genes are enriched with function categories associated with brain and behavior (adjusted P-value < 0.05, Fig. 1e, Table S5) including neurogenesis, dendritic morphogenesis, dendritic development, neuron differentiation, and brain development. These genes were also enriched with genes associated with cognitive and behavioral disorders including autism spectrum disorders, mental retardation and intellectual disability, share genetic risk and pathological pathways with schizophrenia (Fig. 1e, Table S5).
Experimental validation of circRNAs differential expression
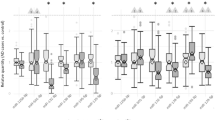
In order to validate the circRNA sequencing results, we selected eight circRNAs that showed significant alteration in schizophrenia in both total and circRNA enriched datasets and then ran qPCR using specific divergent primers to analyze the expression levels. Our data confirmed that all the selected circRNAs including circ-GPR137B-3, circ-PPP2CA-3, circ-VCAN-2, circ-LONP2–6, circ-TOP1–10, circ-MYO9A-66, circ-HP1BP3–7, and circ-ZNF236–2 were significantly differentially expressed in schizophrenia samples relative to healthy controls (Fig. 1f). The data indicated that RNA-seq analysis was consistent with qPCR results regarding the expression levels of the tested circRNAs (Fig. 1f, g).
Schizophrenia-associated circRNAs relationship with host gene expression
Given the variation in cortical circRNAs expression in schizophrenia, we wanted to determine if this change was independent or linked with changes the host genes transcription. This was achieved by cross-referencing the differentially expressed circRNAs with the expression of their corresponding linear RNAs. While more than half had discordant expression change with respect to their linear counterparts, observed in the scatter plots of circRNAs (TPM case/control), downregulation in circRNAs was correlated with a change in host gene mRNA (P-value 0.022) (Fig. 2a, S2). However, despite this relationship, when analyzed globally for all the identified circRNAs, only relatively small changes in linear isoforms were observed in comparison to their circRNA isoforms (Fig. 2b).

Comparison between circRNAs and their linear isoforms in schizophrenia and controls. a Relative quantification of DE circRNA and their linear isoforms in schizophrenia vs. control and density plot comparing changes of DE circRNA (TPM) and the corresponding mRNAs (TPM). b Density plot for all circRNAs vs. their mRNA in schizophrenia and controls
Interestingly, when we examined circRNAs expression at genes with differential mRNA expression in schizophrenia, there appeared to be a depletion of circRNAs expression. More specifically, the analysis of mRNA expression using EdgeR revealed 798 differentially expressed transcripts in schizophrenia compared to controls including 537 upregulated and 261 downregulated (adjusted P-value < 0.05, Table S4). Our analysis showed that only 37 of these genes produced circRNAs and only circ-PTGES3 was differentially expressed. Similarly, in the circRNAs enriched sequencing, only 104 differentially regulated mRNAs generated circRNAs, most of which had low expression.
CircRNA-miRNA-mRNA interactions and schizophrenia
Given the enormous diversity of circRNAs expression in the human DLPFC, we suspect this may have a variety of functions and even contribute to the pathophysiology of schizophrenia. These are relatively stable molecules and a large number of them have high levels of homology with miRNA sequences including those known to be altered in schizophrenia and other psychiatric diseases. Indeed, their high capacity for miRNA sponging has been reported in the literature and can be predicted using miRNA target finding algorithms. Moreover, the alteration of miRNA function-induced by miRNA sponging can be quantified for a phenotype, to some extent, by examining the associated change in mRNA expression. We used this approach to investigate the impact of schizophrenia-associated circRNAs-miRNA interactions through the expression of mRNAs and generated a network of circRNAs-miRNA-mRNA interactions before further characterizing the molecular pathways involved. Using TargetScan custom script, we scanned the DE circRNAs (n = 574) for miRNA binding sites and performed a context score cutoff < −0.4 to improve the detection quality. In addition, to make the candidates more reliable we chose overrepresented miRNAs by filtering out all the targeted miRNAs implicated less than five times. In total, 241 miRNAs were identified to interact with DE circRNAs. In order to explore the circRNA-miRNA-mRNA axis in schizophrenia, we determined the potential targets of the overrepresented miRNAs, using the stringent interaction criteria described above. Interestingly, we found 306, out of 595, differentially expressed mRNA in the same cohort were targeted by the circRNA-interacting miRNAs, including 217 with multiple miRNA associations (Fig. 3a, Table S5). When we selected the top 45 circRNAs, we found an even stronger interaction network between miRNAs and cognate mRNAs, which included 99 miRNAs and 224 differentially expressed mRNAs (Fig. 3b).

Prediction of interaction network of circRNA-miRNA-mRNA. a Potential interactions of all DE circRNAs with DE mRNAs via targeting miRNAs. b Interaction networks constructed for the top 45 DE circRNAs. c Potential interaction of all DE circRNA with previously schizophrenia-associated miRNAs. Purple Circles represent circRNA, red rectangles miRNA, and green triangles mRNA
Using this approach, we also examined the possible interaction between the DE circRNAs and miRNAs that were previously shown to be implicated in schizophrenia pathogenesis. This analysis supported our hypothesis that these interactions are important in the neuropathology of the disorder with 42 miRNAs associated with schizophrenia found to have interactions with these differentially expressed circRNAs (Fig. 3c, Table S5).
Alternative circRNAs splicing is reduced in schizophrenia
Considering the large number of circRNAs identified in the DLPFC, we further analyzed the diversity of alternative back-splicing utilized in this tissue and in respect to the disease phenotype. Our results showed that ~35–76% of back-splice sites are alternatively selected across the samples (Fig. 4a), suggesting that alternative back-splicing is very common for circRNAs expressed in the human brain. In addition to the alternative back-splicing that generate circRNAs, these molecules frequently have multiple exons and display alternative splicing during their biogenesis. In order to detect four types of alternative splicing events including cassette exons, retained introns and alternative 5′/3′ splice sites, we mapped reads from the circRNAs enriched dataset by de novo assembly using Cufflinks to characterize various alternative circRNA splicing sites. Interestingly, three types of alternative circRNA splicing were detected at significantly lower levels in the schizophrenia cohort compared to controls. This direction was in terms of total number of alternative splice sites and total number of unique alternative splice sites in each cohort (Fig. S3A-D). To quantify these alternative splicing events, we used Percent Splice-site Usage (PSU) for alternative 5′/3′ splice site selection, Percent Spliced In (PSI) for cassette exon selection and Percent Intron Retention (PIR) for intron retention according to Zhang et al. [29]. As shown in Fig. 4b–e, the results revealed an obvious trend, with the schizophrenia cases producing a lower level of alternatively spliced circRNAs and this reduction was detectable in all quantification categories. As illustrated in Fig. 4f, these results are not affected by sequencing libraries as the sequencing depth was similar between schizophrenia and control cohorts when they were compared.

Analysis of alternative (back-) splicing in circular RNAs. a Schematic diagrams of two types of alternative back-splicing (5′ and 3′), and number and percent of alternative back-splicing events by sample and diagnosis. Red polylines show canonical splicing and red arc lines indicate back-splicing (circularization). b–e Schematic diagram of four basic types of alternative splicing in circRNAs and their quantifications from schizophrenia and control groups. Alternative splicing events were measured by and PSU for alternative 5′ splice site selection (b), alternative 3′ splice site selection (c), PSI for cassette exon inclusion/exclusion (d), and PIR for intron retention (e). f Comparison of total number of mapped reads from schizophrenia and control cohorts
Using de novo assembly of the unmapped reads from the RNase R treated RNA-seq dataset and CIRCexplorer2 pipeline, we detected many alternative 5′/3′ back-splice sites in circRNAs from the exons not previously annotated in RefSeq, UCSC Known Genes, or Ensembl (Table S6). In total, 2658 circRNAs from previously unannotated exons were detected among all brain samples, with 40% could be identified in multiple brain samples. The results showed a lower number of novel back-splice sites in schizophrenia cases (n = 1539) compared to controls (n = 1781).
The human prefrontal cortex expresses a rich matrix of circRNAs
To explore circRNAs diversity in the human DLPFC at greater depth, we further analyzed the RNaseR-enriched read count data, and identified 95,232 unique circRNAs candidates in human BA46, correspond to 15,357 genes. We then compared these circRNAs with all previously detected in the human neuronal context in circBase [37] and circRNADb [38], and found that a large proportion (59%), had no previous annotation (Fig. 5a). Also, 53% of the brain-expressed circRNAs were identified as novel when compared with all those previously identified in other tissues in human (Fig. 5b). Analysis of the genes producing circRNAs showed that protein-coding genes harbor the majority of circRNAs (85%), however a considerable number of circRNAs (9,680) were generated from non-coding RNAs including lncRNAs and miRNAs, as shown in Fig. 5c. In addition, we detected over 5000 circular intronic RNA (ciRNA) that are spliced from intronic regions of genes, even though a high preference was clear for coding sequence (CDS) and UTRs exons (Fig. 5d). Further annotations of the brain circRNAs showed that 30% of circRNAs are 200–400 bp long and the splice sites span one to over ten exons, however, two-exon circRNAs were the most common type (Fig. S4A, S4B). Analysis of the exon’s position displayed that nearly all of the circRNAs were back-spliced from middle exons and only 567 were from either first or last exon (Fig. S4C). Also, one-exon circRNAs tend to be spliced from longer exons (Fig. S4D), suggesting that longer exons are more likely to form circRNAs due to easier folding and circularization. Given that different genes generated different numbers of circRNAs (Fig. 5e), we asked if there was a relationship between the length of genes and production of circRNAs from the same gene. While we observed correlation (cor = 0.46, P-value < 2.2e-16) between the number of exons per gene and the number of circRNAs spliced (Fig. S4E), other factors, such as intronic flanking sequence that induce stem-loops are known to enhance circRNA biogenesis. To further support the circularity of back-splice fusion read count analysis, we selected 15 circRNA candidates with a range of expression levels, from high to very low expressed circRNAs and tested their resistance to the exonuclease RNase R using qPCR with divergent primers that are incompatible on linear templates. The results showed all 15 circRNAs were at least ten-fold more stable than linear transcripts following RNase R treatment (Fig. S5).

Detection and classification of circRNAs. a Overlap between circRNAs identified from this study and previous human neuronal circRNAs. b Overlap between circRNAs from this study and previous human circRNAs. c Classification of genes producing circRNAs. d Genome annotation of the predicated circRNAs. e Number of unique circRNAs generated per gene
Discussion
Our analysis of RNA-sequencing in postmortem brain samples revealed remarkable diversity of circRNAs expression in the human DLPFC with over 95,000 different molecules, including half of which have not been previously reported (Fig. 5). This supports previous investigations that suggest there is very significant enrichment of circRNAs expression in the brain [11, 36, 39]. We performed differential analysis of these molecules in schizophrenia using both ribosomal RNA depleted and linear RNA depleted samples, which suggested there is an alteration of the biogenesis of these molecules characterized by a global reduction compared to the control group. There was also a reduction in the total number of unique circRNAs discovered in schizophrenia and a reduction of circRNAs expression at the gene level with substantial overlap between the two. Our analysis of differentially expressed circRNAs host genes revealed an enrichment of neurological processes that that are known to be targeted by risk factors for schizophrenia, such as neuronal differentiation and maturation [40].
Differential back-splicing could give rise to intragenic expression changes through various mechanisms. For example, circRNAs may interact with miRNA targeting its host gene and support its expression [41]. Alternatively, a change in back-splicing may directly alter the abundance of other splice variants of the host gene [39, 42, 43]. In view of the possibility of this relationship we explored the level of expression correlation between differentially expressed circRNAs and their linear isoforms. Surprisingly, more than half of the circRNAs displayed an inverse relationship with their linear RNA, suggesting the two forms of the transcript may sometimes antagonize each other’s biosynthesis. In view of this observation it was interesting that circRNAs spliced from genes with differentially expressed mRNA displayed a relatively low rate of back-splicing, suggesting that circRNAs biogenesis might be in direct competition with the schizophrenia-associated mRNAs as demonstrated in Drosophila S2 and HEK292 cells [42]. It is also likely that circRNAs act in trans by associating with other RNAs or genes to affect their mRNA expression. In this regard, it is well known that circRNAs are efficient ceRNA that interact and sponge miRNA preventing them from engaging in gene silencing. This is exemplified by ciRS-7 which was shown to negatively modulate miR-7 and miR-671 in mouse and human brain resulting in dysregulation of mRNA targets and behavioral phenotype [44, 45]. Our exploration of circRNAs-miRNA interactions (involving 226 unique miRNA) further supported this hypothesis and predicted more than 50% of changes in differentially expressed mRNAs in schizophrenia with high redundancy in the same data set. Interestingly, 73% of the interacting circRNAs had a corresponding change in expression direction as their target mRNAs, which supports an association between these molecules mediated by miRNA and their ceRNA network. Analysis of the top 45 differentially expressed circRNAs revealed an even stronger interaction network with 99 miRNAs which further corresponded with 224 differentially expressed mRNAs. Moreover, we predicted that the differentially expressed circRNAs could interact with more than 50 miRNA previously associated with schizophrenia. As many of these miRNA were observed to be up regulated in the disorder, a reduction of circRNAs regulation on these molecules would further exacerbate the miRNA induced gene silencing. Many of these miRNA are capable of reducing the stability of mRNA transcripts or reducing their translational efficiency, contributing to changes in gene expression at either, or both, the RNA and protein level, which have been observed in the neuropathology of schizophrenia. These findings suggest that the dysregulated circRNAs could be implicated in schizophrenia development by compounding the effect of elevated miRNA (Fig. S6).
Recent research has suggested that circRNAs may play a role in cellular homeostasis by regulating different aspects of cellular growth, differentiation, and apoptosis. They have also been implicated in drug resistance and the immune-response pathways [46, 47]. For example, Circ-RasGEF1B can activate antimicrobial response by stabilizing mature ICAM1 mRNA, a cell surface adhesion molecule involved in attracting lymphocytes and aiding immune responses [48]. HeLa cells transfected with circRNAs displayed increased expression of 84 innate immunity-related genes and reduced susceptibility to viral infection [49]. Pro-inflammatory cytokines such as interleukin (IL)-1 and tumor necrosis factor (TNF) were also positively correlated with circ-CER expression [50]. We observed many of the stress-related circRNAs such as Circ-FOXO3 [51] to be present in our postmortem DLPFC samples suggesting that circRNAs expression may also be altered in response to cellular stress. Several circRNAs downregulated in schizophrenia, including circ-ARID1A, circ-PHC3, and circ-CDC73, are associated in stress metabolism through forming circ-IMP3 (IGF2BP, insulin-like growth factor 2 binding protein 3) complexes [52]. When we performed pathways analysis for the DE circRNA-interacting miRNAs and their target genes, we observed a significant enrichment of the GO terms associated with inflammation and stress, such as Toll-like receptor signalling, immune system process, acute inflammatory response, and response to stress (Table S5). These findings suggest that alterations in cortical circRNA expression may be related to neuroinflammatory or other stress response mechanisms associated with schizophrenia and other psychiatric disorders.
While little is known about the mechanism driving the alteration of circRNAs biogenesis in the brain, there is some suggestion that expression levels of two key proteins, ADAR1 and QKI can be modified by environmental influences in other tissues. ADAR1, through its RNA editing activity, reduces circRNA biogenesis by melting stem-loops that help to drive the circularization of mRNA transcripts [16]. It has been reported to regulate endoplasmic reticulum stress and is essential for intestinal homeostasis, stem cell maintenance [53] and has been previously implicated in the neuropathology of schizophrenia [18]. ADAR1 is also involved in the cellular response to viral infection, e.g., in hepatitis C virus, lymphocytic choriomeningitis virus, and polyomavirus [54], but also function as a viral enhancer, e.g., in influenza virus, measles virus, and human immunodeficiency virus-1 [54] which suggest it may be modulated by infection. Similarly, the splicing factor QKI has also been shown to have a critical role in response to inflammatory diseases by promoting monocyte differentiation into pro-atherogenic macrophages [55]. QKI has a significant impact on circRNAs biogenesis by enhancing cis-RNA interactions and circularization [17] and have been previously implicated in the neuropathology of schizophrenia [20]. In earlier work we showed that the activity splicing co-factor QKI is also modulated by schizophrenia-associated changes in the highly expressed lincRNA known as Gomafu (MIAT) in postmortem cortex [56]. Gomafu contains several repeats of the QKI consensus motif and can sequester the protein in the nucleus and reduce its capacity for organizing splicing and circRNA biogenesis.
circRNAs expression has recently been shown to be altered after neural activity and may be influential in the regulation of synaptic plasticity [39]. A number of these circRNAs were also expressed in the DLPFC, including circHomer1, circKlhl2 circMpped1, and circNell2. Interestingly, circNEll2, which was upregulated following the induction of synaptic plasticity, was found to be downregulated in schizophrenia. These data suggest that circRNAs biogenesis could be regulated in response to neural activity. Interestingly, the QKI-binding lncRNA Gomafu was also shown to be downregulated in human iPSC neurons in response to potasium induced depolarisation [56]. An activity-associated reduction in Gomafu would increase QKI and circRNA biogenesis. Another plausible mechanism for this neural activity-associated back splicing is RNA methylation. Recent studies have shown N6-adenosine methylation (m6A), is dynamically regulated in the prefrontal cortex where induction of m6A level promoted consolidation of fear memory [57]. Similarly, discrete sequence-specific m6A modification was also shown to be widespread in circRNAs, modify their stability [58] and also enhance their potential for cap-independent translation [59]. M6A methylation may modify circRNA biogenesis by altering pre-mRNA splicing [60]. Several of these m6A-containing circRNAs (n = 75) were observed in the DLPFC in the current study, including two (circ-GLB1 and circ-MAST3) that were differentially expressed in schizophrenia, suggesting this mechanism may also alter circRNA levels and function in the disorder.
In summary, our study provides further evidence of the tremendous diversity of circRNAs expression in the human brain and suggests that their biogenesis or stability is reduced in schizophrenia. With their potential for regulation through intracellular miRNA sponging and even cap-independent protein translation, these molecules are potentially an important part of the molecular fabric and regulatory landscape in the brain. Moreover, they could be important in the etiology of schizophrenia and other neurodevelopmental disorders. Further investigations of these molecules in the context of psychiatric disorders is therefore warranted given that they may represent novel targets for therapeutic manipulation or provide new biomarkers for further insight into disease status.
Funding and disclosure
Postmortem human tissues were received from the New South Wales Tissue Resource Center at the University of Sydney, which is supported by the National Health and Medical Research Council of Australia, Schizophrenia Research Institute, National Institute of Alcohol Abuse and Alcoholism (NIH (NIAAA) R24AA012725). This research was supported by an NHMRC project grants (1067137) and (1147644). EM, JA, and MG were supported by University of Newcastle RHD scholarships. JA was also supported by an Emlyn and Jennie Thomas Postgraduate Medical Research Scholarship. MC is supported by an NHMRC Senior Research Fellowship (1121474). CSW is funded by the NSW Ministry of Health, Office of Health and Medical Research, and is supported by NHMRC Principal Research Fellowship (1117079). The authors report no biomedical financial interests or potential conflicts of interest.
References
Trivedi JK. Cognitive deficits in psychiatric disorders: current status. Indian J Psychiatry. 2006;48:10–20.
Karlsgodt KH, Sun D, Cannon TD. Structural and functional brain abnormalities in Schizophrenia. Curr Dir Psychol Sci. 2010;19:226–31.
Mahmoudi E, Cairns MJ. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry. 2017;22:44–55.
Beveridge NJ, Gardiner E, Carroll AP, Tooney PA, Cairns MJ. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol Psychiatry. 2010;15:1176–89.
Hollins SL, Cairns MJ. MicroRNA: small RNA mediators of the brains genomic response to environmental stress. Prog Neurobiol. 2016;143:61–81.
Kiebler MA, Bassell GJ. Neuronal RNA granules: movers and makers. Neuron. 2006;51:685–90.
Glock C, Heumuller M, Schuman EM. mRNA transport & local translation in neurons. Curr Opin Neurobiol. 2017;45:169–77.
Wang B, Pan L, Wei M, Wang Q, Liu WW, Wang N, et al. FMRP-mediated axonal delivery of miR-181d regulates axon elongation by locally targeting map1b and calm1. Cell Rep. 2015;13:2794–807.
Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. 2009;11:1143–9.
Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–6.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta. 2016;1859:163–8.
Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205–11.
Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32:453–61.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE. 2012;7:e30733.
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10:170–7.
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125–34.
Aberg K, Saetre P, Lindholm E, Ekholm B, Pettersson U, Adolfsson R, et al. Human QKI, a new candidate gene for schizophrenia involved in myelination. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:84–90.
Aberg K, Saetre P, Jareborg N, Jazin E. Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc Natl Acad Sci USA. 2006;103:7482–7.
Silberberg G, Lundin D, Navon R, Ohman M. Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum Mol Genet. 2012;21:311–21.
Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–9.
Weickert CS, Sheedy D, Rothmond DA, Dedova I, Fung S, Garrick T, et al. Selection of reference gene expression in a schizophrenia brain cohort. Aust N Z J Psychiatry. 2010;44:59–70.
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–47.
Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2013;18:206–14.
The Gene Ontology C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–D338.
Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015;43:W460–6.
Chen J, Aronow BJ, Jegga AG. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinforma. 2009;10:73.
Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinforma. 2007;8:392.
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016;26:1277–87.
Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–9.
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20.
Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–85.
Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–70.
Chen X, Han P, Zhou T, Guo X, Song X, Li Y. circRNADb: a comprehensive database for human circular RNAs with protein-coding annotations. Sci Rep. 2016;6:34985.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18:603–10.
Ahmad R, Sportelli V, Ziller M, Spengler D, Hoffmann A. Tracing early neurodevelopment in Schizophrenia with induced pluripotent stem cells. Cells. 2018;7:140–65.
Li F, Zhang L, Li W, Deng J, Zheng J, An M, et al. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget. 2015;6:6001–13.
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55–66.
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792–806.
Piwecka M, Glazar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science. 2017;357:eaam8526. pii:
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8.
Fischer JW, Leung AK. CircRNAs: a regulator of cellular stress. Crit Rev Biochem Mol Biol. 2017;52:220–33.
Haque S, Harries LW. Circular RNAs (circRNAs) in Health and Disease. Genes. 2017;8:E353.
Ng WL, Marinov GK, Liau ES, Lam YL, Lim YY, Ea CK. Inducible RasGEF1B circular RNA is a positive regulator of ICAM-1 in the TLR4/LPS pathway. RNA Biol. 2016;13:861–71.
Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, et al. Sensing self and foreign circular RNAs by intron identity. Mol Cell. 2017;67:228–38 e5.
Liu Q, Zhang X, Hu X, Dai L, Fu X, Zhang J, et al. Circular RNA related to the chondrocyte ECM regulates MMP13 expression by functioning as a MiR-136 ‘Sponge' in human cartilage degradation. Sci Rep. 2016;6:22572.
Du WW, Yang W, Chen Y, Wu ZK, Foster FS, Yang Z, et al. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J. 2017;38:1402–12.
Schneider T, Hung LH, Schreiner S, Starke S, Eckhof H, Rossbach O, et al. CircRNA-protein complexes: IMP3 protein component defines subfamily of circRNPs. Sci Rep. 2016;6:31313.
Qiu W, Wang X, Buchanan M, He K, Sharma R, Zhang L, et al. ADAR1 is essential for intestinal homeostasis and stem cell maintenance. Cell Death Dis. 2013;4:e599.
Samuel CE. ADARs: viruses and innate immunity. Curr Top Microbiol Immunol. 2012;353:163–95.
de Bruin RG, Shiue L, Prins J, de Boer HC, Singh A, Fagg WS, et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat Commun. 2016;7:10846.
Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. 2014;19:486–94.
Widagdo J, Zhao QY, Kempen MJ, Tan MC, Ratnu VS, Wei W, et al. Experience-dependent accumulation of N6-methyladenosine in the prefrontal cortex is associated with memory processes in mice. J Neurosci. 2016;36:6771–7.
Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20:2262–76.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27:626–41.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mahmoudi, E., Fitzsimmons, C., Geaghan, M.P. et al. Circular RNA biogenesis is decreased in postmortem cortical gray matter in schizophrenia and may alter the bioavailability of associated miRNA. Neuropsychopharmacol. 44, 1043–1054 (2019). https://doi.org/10.1038/s41386-019-0348-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0348-1
This article is cited by
-
Circular RNAs arising from synaptic host genes during human neuronal differentiation are modulated by SFPQ RNA-binding protein
BMC Biology (2023)
-
Integrative Analysis of Morphine-Induced Differential Circular RNAs and ceRNA Networks in the Medial Prefrontal Cortex
Molecular Neurobiology (2023)
-
Circular RNAs: Diagnostic and Therapeutic Perspectives in CNS Diseases
Current Medical Science (2023)
-
Circ-PKD2 promotes Atg13-mediated autophagy by inhibiting miR-646 to increase the sensitivity of cisplatin in oral squamous cell carcinomas
Cell Death & Disease (2022)
-
Circular RNA: A novel and potential regulator in pathophysiology of schizophrenia
Metabolic Brain Disease (2022)
