
Abstract
Emerging data continues to point towards a relationship between neuroinflammation and neuropsychiatric disorders. ATP-induced activation of P2X7 results in IL-1β release causing neuroinflammation and microglial activation. This study describes the in-vitro and in-vivo neuropharmacology of a novel brain-penetrant P2X7 antagonist, JNJ-55308942, currently in clinical development. JNJ-55308942 is a high-affinity, selective, brain-penetrant (brain/plasma of 1) P2X7 functional antagonist. In human blood and in mouse blood and microglia, JNJ-55308942 attenuated IL-1β release in a potent and concentration-dependent manner. After oral dosing, the compound exhibited both dose and concentration-dependent occupancy of rat brain P2X7 with an ED50 of 0.07 mg/kg. The P2X7 antagonist (3 mg/kg, oral) blocked Bz-ATP-induced brain IL-1β release in conscious rats, demonstrating functional effects of target engagement in the brain. JNJ-55308942 (30 mg/kg, oral) attenuated LPS-induced microglial activation in mice, assessed at day 2 after a single systemic LPS injection (0.8 mg/kg, i.p.), suggesting a role for P2X7 in microglial activation. In a model of BCG-induced depression, JNJ-55308942 dosed orally (30 mg/kg), reversed the BCG-induced deficits of sucrose preference and social interaction, indicating for the first time a role of P2X7 in the BCG model of depression, probably due to the neuroinflammatory component induced by BCG inoculation. Finally, in a rat model of chronic stress induced sucrose intake deficit, JNJ-55308942 reversed the deficit with concurrent high P2X7 brain occupancy as measured by autoradiography. This body of data demonstrates that JNJ-55308942 is a potent P2X7 antagonist, engages the target in brain, modulates IL-1β release and microglial activation leading to efficacy in two models of anhedonia in rodents.
Similar content being viewed by others
Introduction
Inflammatory disorders are often co-morbid with mood disorders; emerging science suggests a connectivity between the imbalance of the immune system and the central nervous system (CNS), leading to the hypothesis that mental health may be modulated by systemic inflammatory tone. As such, subsets of patients suffering from depression and bipolar disorder, especially the treatment-resistant population, may benefit if targeted treatments are offered to patients with higher neuroinflammatory burden. Microglial cells in the CNS drive neuroinflammation by release of pro-inflammatory cytokines and chemokines, and cell-surface receptors modulating such pro-inflammatory signals ought to be attractive neuroimmune targets in psychiatry [1]. There is a growing body of clinical literature demonstrating microglial activation in neuropsychiatric disorders and dampening glia derived inflammatory signaling may offer benefit towards the unmet need in psychiatry [2].
Microglia abundantly express the ATP-gated P2X7 ion channel [3,4,5]. Activation of P2X7 leads to non-selective cation flux followed by NLRP3-driven release of IL-1β and IL-18 [6], initiating the cascade of neuroinflammation in the CNS [7, 8]. The NLRP3 inflammasome pathway leading to IL-1β and IL-18 release has been linked to the pathogenesis of depression [9]; likewise, increased levels of IL-1β in plasma and CSF have been associated with depression and mania [10, 11]. In addition, IL-1β has been linked with both geriatric depression and postpartum depression [12, 13]. P2X7 SNPs and gain-of-function haplotypes have also been associated with both depression [14,15,16] and bipolar disorder [17], highlighting the P2X7-NLRP3-IL1β signaling cascade as a potential pathway of therapeutic interest in neuropsychiatry.
Supporting the clinical literature is an emerging body of preclinical data linking the role of the P2X7-NLRP3-IL1β pathway in chronic models of stress-induced anhedonia-like behaviors [18,19,20,21,22,23]. Likewise, several laboratories have demonstrated that P2X7 knockout mice manifest a protective phenotype in models of despair and mania [22, 24,25,26], strengthening the hypothesis that P2X7 antagonism may be therapeutically beneficial in mood disorders. Consistent with the antidepressant phenotype observed in P2X7 knockout mice, additional data suggest that P2X7 antagonists can reverse despair-like behaviors in rodents, although it remains to be understood the mechanism by which ATP can build-up under “acute” stress to generate efficacy by P2X7 antagonism [27]. “Chronic” stress, on the other hand has been shown to cause ATP release [28], inflammasome activation [20, 29], and microglial activation [30, 31]; it is plausible that P2X7 may play a role in models of chronic stress [20, 32], which is known to be causal to clinical depression [33]. In addition to depression, preclinical data also suggest that P2X7 antagonism may produce anti-manic or mood stabilizing effects in bipolar disorder [34]. More recently, the role of P2X7 in the PCP model of schizophrenia has come to light [35]. The current body of data suggests that a selective and brain-penetrant P2X7 antagonist may be therapeutically beneficial in several neuropsychiatric disorders [36].
This study describes the neuropharmacology of JNJ-55308942, a P2X7 antagonist [37], which is currently a clinical compound in development (https://clinicaltrials.gov/ct2/show/NCT03151486). The manuscript represents a systematic and thorough evaluation of the molecule from in-vitro to in-vivo test systems, with an emphasis on brain target engagement and biomarker endpoints. Finally, the molecule was used in rodent models to probe the role of P2X7 in a lipopolysaccharide (LPS) model of microglial activation, in a Bacillus Calmette-Guerin (BCG) model of neuroinflammatory depression and in a chronic model of stress-induced anhedonia. The study provides insight into the mechanism of P2X7 in neuroinflammation and adds preclinical rationale for designing proof-of-concept studies in patients with anhedonia, such as seen in neuropsychiatric disorders.
Materials & methods
Calcium influx, radioligand binding, and IL-1β assays
These assays have been described in details elsewhere [4, 38, 39].
Mouse primary microglia isolation, IL-1β release, and cell death assay
Mouse (C57BL/6) primary microglia were harvested from cortices (post-natal day 0–3) of mouse pups, as described [3]. For IL-1β release, microglia were incubated with different concentrations of JNJ-55308942 for 1 h before 100 ng/ml LPS challenge for 22 h in the presence or absence of 380 μM Bz-ATP for an additional 2 h. Mouse IL-1β ELISA kit (R&D systems) were used for quantification of IL-1β in cell culture media following the manufacturer’s instruction. For cell death assay, microglia were pre-treated with different concentrations of the P2X7 antagonist JNJ-55308942 for 1 h before exposure to 380 μM Bz-ATP for an additional 4.5 h. Cell viability was determined by cell counting kit-8 (CCK-8, Dojindo), which measures mitochondrial dehydrogenase activity of cells. Briefly, 10 μl of CCK-8 solution was added to 100 μl of media in each well of the plate. After incubating the plate for 4 h at 37 °C, the absorbance at 450 nm was measured using the Bio-Rad microplate reader. The ratio of microglial cell death was normalized to the condition of 380 μM Bz-ATP alone.
Rat brain ex-vivo autoradiography
Male Sprague-Dawley rats approximately 300–400 g in body weight were used. Animals received a bolus dose via a 16-gauge intragastric gavage. JNJ-55308942 was formulated at a concentration or 0.002, 0.006, 0.02, 0.06, 0.2, 0.6, or 2 mg/ml in 20% (w/v) HP-β-CD and was delivered in a volume of 5 ml/kg as a solution. Blood and tissue sampling were conducted at 2 h post dose. Blood and tissue samples were taken immediately after euthanasia by CO2 asphyxiation. Brains from each animal were removed and frozen on dry ice for autoradiography and determination of brain concentration by LC/MS-MS. Brain tissue was cryosectioned at the level of the hippocampus. Three 20 μm sections (two for total binding and one for non-specific binding) were thaw mounted on superfrost microscope slides for each animal. Occupancy was assessed in rat brain with 10 nM [3H]JNJ-54232334 in 50 mM Trizma hydrochloride (Tris) buffer with 0.1% bovine serum albumin (BSA). Non-specific binding was assessed in the same buffer with the addition of 10 μM A-740003. Tissue sections were incubated for 1 min at room temperature followed by 4 × 5 min washes at 4 °C in 50 mM Tris with 0.1% BSA buffer followed by 2 dips in deionized water. Quantitative autoradiography was performed after 3-hour acquisition using a β Imager, Biospace (Paris, France). The digitized computer images generated by the β Imager were visualized and quantified using M3 Vision software (Biospace Paris, France).
IL-1β microdialysis (rat brain)
A guide cannula was surgically placed in the brain (male, Sprague-Dawley, 280–350 g) to facilitate the insertion of the dialysis probe in the hippocampus, as described previously [38]. Animals were allowed at least 4 days to recover from surgery prior to experimentation. JNJ-55308942 was formulated in 20% HP-β-CD. The compound was formulated at 0.6 mg/ml and orally dosed in a volume of 5 ml/kg. Bz-ATP was formulated at a concentration of 100 mM in 1 × aCSF (147 mM NaCl, 4 mM KCl, 0.85 mM MgCl2, 2.3 mM CaCl2) with 0.15% BSA and was delivered via reverse dialysis over 2 h.
LPS-induced brain microglial activation in mice
Sixteen male C57/BL6J mice were purchased from Jackson Laboratory (Sacramento, CA, USA) at an age of 6–7 weeks, housed in groups of four under controlled conditions with a 12:12 light/dark schedule (lights on at 6 AM) and temperature maintained at 22 ± 2 °C. This dosing was achieved by weighing the compound and creating a 3 mg/ml stock solution of JNJ-55308942 in 0.5% hydroxypropyl methylcellulose. Each animal was allocated to one of four groups in a 2 × 2 design with one group receiving three oral doses of JNJ-55308942 (30 mg/kg at t = 0, 24, and 31 h) in combination with an injection of LPS (0.8 mg/kg i.p. at t = 1 h), one group receiving only the compound combined with a vehicle (PBS) injection, one group receiving only LPS combined with vehicle (0.5% HPMC) instead of drug, and one control group only receiving vehicle for both treatments. Animals were sacrificed by CO2 asphyxiation followed by cardiac puncture for blood collection and transcardial perfusion with PBS supplemented with Heparin (5 Units/ml). Brains were collected in ice-cold DPBS containing sodium pyruvate and glucose (ThermoFisher). Brains were dissociated and cleared from myeloid debris and red blood cells using a commercial kit (Miltenyi Biotec). All samples were washed and re-suspended in cold PBS and filtered through a 70 µm cell strainer. For each sample 100 µl of cell suspension at a concentration of approximately 107 live cells/ml was incubated with 3 µl of Zombie Violet (BioLegend 423113) or 1 µl Ghost Dye Red 780 (Tonbo biosciences) on ice for 20 min in order to stain dead cells. Cells were washed and re-suspended in cell staining buffer (BioLegend 420201). A 10 µl FcR blocking reagent (Miltenyi Biotec 130-092-575) was added to each 100 µl sample and incubated for 10 min. The following fluorescence conjugated antibodies were added as a cocktail, respectively. Brain panel 1 (microglia surface characterization): BV605 anti-CD45 (2 µg/ml, clone 30-F11, BioLegend 103155), FITC anti-TREM2 (2 µg/ml, clone 78.18, abcam ab119852), PE anti-CD206 (2 µg/ml, BioLegend 141705), PE-Cy7 anti-CD11b (1 µg/ml, clone M1/70, BD Biosciences 552850), APC anti-P2X7R (2 µg/ml, clone 1F11, BioLegend 148705), APC-Cy7 anti-I-A/I-E (MHC-II) (2 µg/ml, clone M5/114.15.2, BioLegend 107627). Brain panel 2 (proliferation): BV605 anti-CD45 (2 µg/ml, clone 30-F11, BioLegend 103155), AF488 anti-CD31 (5 µg/ml, clone MEC13.3, BioLegend 102513), PE anti-ACSA-2 (1 ug/ml, clone IH3-18A3, Miltenyi Biotec 130-102-365), PerCP-Cy5.5 anti-CD11b (2 µg/ml, clone M1/70, BioLegend 101227). Samples and controls were acquired on a 10-color BD FACSCanto-II cytometer and analyzed using Cytobank Enterprise (Cytobank, Inc.) and Graphpad Prism 7. All data sets were analyzed by 2-way ANOVA with Tukey-corrected post hoc tests for multiple comparisons using Graphpad Prism 7. Corrected p values < 0.05 were considered significant.
BCG model of neuroinflammation (mouse)
All experimental procedures were approved by the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee. For all studies, 10–12-week-old male C57BL6/J mice (Jackson Laboratory) were used. BCG stocks were prepared from a seed vial of BCG Live TheraCys® (Sanofi, lot# C3705AA) cultured in Middlebrook 7H9 supplemented whith Tween® 80 and 10% ADC enrichment at 5% CO2. Stocks were aliquoted to an approximate concentration of 1 × 108 CFU and stored at −80 °C. Mice then received either 3 × 107 CFU of BCG intraperitoneally or an equivalent volume of physiological saline. Mice were observed for unexpected signs of distress and weighed daily by research staff between 9:00 am and 11:00 am. JNJ-55308942 was weighed (10 mg) and diluted with vehicle to a 3 mg/ml solution each night prior to use. The suspension was continuously vortexed at medium speed overnight at 4 degrees. Compound or vehicle were then administered each morning at a dose of 30 mg/kg (test compound) or equivalent volume (vehicle was 0.5% hydroxypropyl methylcellulose). Sucrose preference was measured using a continuous two-bottle paradigm over a 48 h period. Mice were first trained to the two-bottle paradigm for three days prior to BCG/vehicle injections. During this time, the mice have ad libitum access to both bottles, which are placed on outside of the cage lid with food in the middle. One bottle contains water, and the other contains a 1% sucrose solution. Bottles are weighed daily to measure intake. Stable preference [preference = sucrose intake/(sucrose + water intake)*100] of 80–90% is reached by the end of training. Mice were tested on days 11–13 post BCG using the same two-bottle test to determine sucrose preference during the 48 h period 2 weeks post-BCG. Initial bottle placement (right vs. left) was determined randomly and changed each day to avoid location bias. General locomotor activity and anxiety-like behavior were measured in the open field. Immediately following the open field assay, a novel conspecific adult mouse was placed into one corner of the open field arena. The novel “guest” mouse was contained under a small cage with vertical bars to restrict its ability to move about the arena but enable both visual and olfactory interaction to occur. The latency of the test mouse to approach the stranger and the duration of time that the test mouse spent in a virtual “interaction zone” was determined using Noldus XT tracking software. Data were initially analyzed by two-factor ANOVA (saline/BCG x treatment group), and where interactions were significant (<0.05) bonferroni post hoc testing was conducted. Data were also analyzed for simple treatment effects of BCG using t-test.
Chronic mid stress (CMS) in rats
Male Wistar rats (Charles River, Germany) were brought into the laboratory one month before the start of the experiment. Except as described below, the animals were singly housed with food and water freely available, and were maintained on a 12 h light/dark cycle and in a constant temperature (22 ± 2 °C) and humidity (50 ± 5%) conditions. All procedures used in this study have conformed to the rules and principles of the 86/609/EEC Directive, and have been approved by the Local Bioethical Committee at the Institute of Pharmacology, Polish Academy of Sciences, Krakow, Poland. After a period of 3 weeks of adaptation to laboratory and housing conditions, the animals were trained to consume a 1% sucrose solution; training consisted of nine 1 h baseline tests in which sucrose was presented, in the home cage, followed by 14 h of food and water deprivation. The sucrose intake was measured by weighing pre-weighed bottles containing the sucrose solution, at the end of the test. Subsequently, sucrose consumption was monitored, under similar conditions, at weekly intervals throughout the whole experiment. Based on their sucrose intakes following 2 weeks of initial stress, both stressed and control groups were each divided further into matched subgroups (n = 8), and for subsequent 5 weeks they received once daily administration of vehicle (1% (w/v) hydroxyethylcellulose, 1 ml/kg, PO), test compounds (each at 1 mg/kg, PO) or imipramine HCl (10 mg/kg, i.p., dissolved in saline) as the reference treatment. The volume of all injections was 1 ml/kg. The drugs were administered at approx. 10.00 and the weekly sucrose tests were carried out 24 h following the last drug injections. Stress was continued throughout the entire period of treatment. Each week of stress regime consisted of: two periods of food or water deprivation, two periods of 45 degree cage tilt, two periods of intermittent illumination (lights on and off every 2 h), two periods of soiled cage (250 ml water in sawdust bedding), one period of paired housing, two periods of low intensity stroboscopic illumination (150 flashes/min), and three periods of no stress. All stressors were 10–14 h of duration and were applied individually and continuously, day and night. Control animals were housed in separate rooms and had no contact with the stressed animals. They were deprived of food and water for 14 h preceding each sucrose test, but otherwise food and water were freely available in the home cage. All results obtained in this study were analysed (Statistica for Windows v. 6.0) by multiple analyses of variance with three between-subject’s factors (stress/control, drug treatments, and successive sucrose tests). The Fisher’s LSD test was used for post-hoc comparisons of means.
Results
In-vitro pharmacology of JNJ-55308942
The chemical structure of JNJ-55308942 is shown in Fig. 1a. The molecule was discovered by Janssen and went through rigorous medicinal chemistry optimization for drug-like properties [37]. In this manuscript, we describe the neuropharmacology of JNJ-55308942. The in-vitro pharmacology of this molecule is summarized in Table 1. In an assay probing for the ability of JNJ-55308942 to block calcium flux through the P2X7 ion channel, the compound was a potent antagonist of P2X7 across a host of species, including the human isoform (Table 1a). Since potency (pIC50) is generally dependent on agonist concentration and other experimental conditions, we also tested the equilibrium binding affinity of JNJ-55308942 at both the recombinant and native P2X7. The affinity (pKi) of JNJ-55308942 for recombinant human and rat P2X7 channels, as assessed by radioligand binding, was 8.1 ± 0.08 and 8.5 ± 0.04, respectively (Table 1b). The binding affinity of JNJ-55308942 was similar in rat cortex (8.2 ± 0.06) to the recombinant system. Since P2X7 activation is linked to IL-1β release (see “Introduction”), and blood IL-1β can be used as surrogate of target engagement [40], we also assessed the in-vitro pharmacology of JNJ-55308942 in both human blood and human blood derived peripheral blood mononuclear cell (PBMCs). As summarized in Table 1c, the compound is also a potent antagonist of P2X7 dependent IL-1β release. Finally, the selectivity of JNJ-55308942 was assessed across P2X subtypes, as well as a broader panel of receptors, transporters, and enzymes. The compound did not block P2X1, P2X2, P2X3, P2X4 homomers, and P2X2/3 heteromers (all human P2X) at concentrations as high as 10 μM. Likewise, JNJ-55308942 (1 μM) did not hit any of the known receptors and enzymes used in our screening panel at CEREP (receptors) or Millipore (enzymes). In summary, JNJ-55308942 is a potent and selective P2X7 antagonist.

P2X7 antagonism attenuates IL-1β release and cell death of microglia. a Chemical structure of the P2X7 antagonist JNJ-55308942. b Secretion of IL-1β of microglia after pre-treatment with different concentrations of JNJ-55308942 in the presence of 100 ng/ml LPS followed by 380 μM Bz-ATP for additional 2 h and quantified by ELISA. c Cell number of microglia after pre-treatment with different concentrations of JNJ-55308942 in the presence of 380 μM Bz-ATP quantified by CCK-8 kit. The proportion of microglial cell death was obtained by normalizing to the condition of Bz-ATP treatment only. Data shown is mean ± standard deviation. Experiments were done in six replicates and repeated twice independently
Since microglia are the predominant cell type that release IL-1β in the CNS, the next step was to characterize the efficacy of JNJ-55308942 in primary microglial cultures. As shown in Fig. 1b, JNJ-55308942 blocked microglial IL-1β release in a concentration-dependent manner. Since activation of P2X7 is also known to cause cell death [3], the effect of P2X7 antagonism in rescuing cell death was also tested (Fig. 1c). Consistent with P2X7 antagonism, the compound attenuated ATP-induced microglial cell death in a concentration-dependent manner, although the potency is right shifted for these end-points (cell death & IL-1β) in neonatal mouse microglia, as compared to the pharmacology noted in Table 1 for the recombinant isoform.
Target engagement of JNJ-55308942
Ex-vivo autoradiography
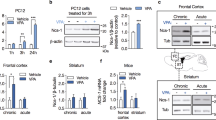
The next step in the characterization of JNJ-55308942 was to test in-vivo pharmacodynamic properties of the molecule. To determine dose-occupancy relationship for JNJ-55308942, the compound was orally administered to rats at various dose levels (0.01, 0.03, 0.1, 0.3, 1, 3, or 10 mg/kg), followed by an assessment of ex-vivo brain P2X7 occupancy at 2 h post dose. As depicted in Fig. 2a, the compound exhibited a robust dose dependency, with P2X7 brain occupancy saturating near 1–3 mg/kg. JNJ-55308942 demonstrated an ED50 of 0.07 mg/kg. Corresponding plasma and brain exposures of the molecule are shown in Fig. 1a as an inset. Dose-dependent exposure of JNJ-55308942 was observed in both plasma and brain tissue with compound partitioning into the brain efficiently with a brain/plasma ratio close to unity. The plasma and brain EC50 of JNJ-55308942 was approximately 15 and 12 ng/ml, respectively. Since the Hill slope of the plasma exposure-effect curve was not significantly different from unity, EC80 and EC90 plasma exposures of the compound were estimated to be around 60 ng/ml and 135 ng/ml, respectively. As such, plasma exposures around 100 to 150 ng/ml will be sufficient for close to maximal brain P2X7 occupancy. To better understand the duration of P2X7 occupancy, and to assess elimination of target engagement as a function of the compound’s pharmacokinetics, a thorough autoradiography study was undertaken along with assessing plasma and brain exposures over three days (Fig. 2b). As shown in the Fig. 2b, the compound occupied brain P2X7 rapidly (88% occupancy after 0.25 h) after a 10 mg/kg (p.o.) dose. The level of receptor occupancy was maintained over time (>80% occupancy through 6 h) and JNJ-55308942 slowly dissociated from the receptor (70% at 24 h, 14% at 48 h and 0% at 72 h; Fig. 2b). Plasma and brain concentrations of the compound as a function of time are shown in the inset. This data provided a high degree of confidence that JNJ-55308942 when dosed orally, engaged the target (P2X7) in brain.

Demonstration of target engagement of JNJ-55308942 in the brain and blood. a Dose vs. brain occupancy curve at 2 h post a single oral administration of JNJ-55308942 in the rat. Plasma and brain exposure of the compound for each dose is shown as an inset table. b Time course of rat brain P2X7 occupancy after a single 10 mg/kg dose (oral) of JNJ-55308942. Plasma and brain was harvested at various time points (X-axis) to monitor changes in occupancy and exposure (inset). c Bz-ATP induced increases in brain IL-1β levels as measured by microdialysis in freely moving rats. The increase in IL-1β was observed during the time of Bz-ATP infusion, as indicated by the shaded bar/line. The compound was administered at a dose of 3 mg/kg (oral), 4 h prior to Bz-ATP infusion. d Effect of a single 30 mg/kg (oral) dose of JNJ-55308942 on ex-vivo IL-1β release in mouse blood. Mice were sacrificed at various time points to assess exposure in the brain/plasma and to study the effect of the compound in the blood on LPS primed Bz-ATP stimulated IL-1β release (black closed circle). At least three animals were used to generate data for each symbol in the figure. p < 0.01 at the 5 h time point, Wilcoxon test
Effects on IL-1β in the brain and blood
To test whether target engagement, as assessed by autoradiography, results in a functional effect in rat brain, we tested the effect of JNJ-55308942 in blocking brain IL-1β release from freely moving rats. As shown clearly in Fig. 2c, a 3 mg/kg oral dose of JNJ-55308942 attenuated Bz-ATP-induced IL-1β release in the rat brain. This set of data unequivocally demonstrated that dosed orally, JNJ-55308942 had access to brain P2X7 where it attenuated IL-1β release, demonstrating robust central target engagement of the molecule. Since peripheral blood cells express P2X7 ion channels, and when primed with LPS and challenged with ATP these cells release IL-1β, we next sought to understand the relationship of JNJ-55308942’s exposure in the plasma and functional effect of attenuation of IL-1β in mice. As depicted in Fig. 2d, a single dose of JNJ-55308942 led to high exposure of the compound in both mouse plasma and brain, with an acceptable pharmacokinetic profile and brain to plasma ratio of unity (similar to rat). At the time points indicated in the figure, mouse blood was collected, primed with LPS and challenged with Bz-ATP to induce IL-1β release ex-vivo. As noted in Fig. 2d, full attenuation of IL-1β release was observed up to 6 h post dose. The block started to wash off, as the exposure of JNJ-55308942 declined with time, with moderate effects observed at 1 day post dose. Thus, JNJ-55308942 causes functional effects in rats and mice on IL-1β release, providing confidence to start evaluating efficacy readouts in models of disease.
Effect of JNJ-55308942 in LPS-induced microglial activation in mice
Mice were dosed with JNJ-55308942 1 h before as well as 7 and 24 h after an i.p. injection of LPS (0.8 mg/kg, i.p.) or vehicle and were sacrificed 48 h after LPS injection. The experimental scheme is shown in Fig. 3a. Brains were collected, dissociated, and stained with conjugated antibodies for FACS. Gating strategy is shown in Fig. 3b. LPS causes microglial activation as determined by significant increases in forward scatter (FSC, correlating with cell size), side-scatter (SSC, correlating with cell complexity), and increases in expression of CD45 and CD11b (3C). JNJ-55308942 significantly attenuated the effect of LPS on FSC, CD45 surface expression and CD11b surface expression, whereas in vehicle-treated controls the compound shows no effect. A second aliquot of the dissociated brains was stained with the intranuclear proliferation marker Ki-67, since microglial proliferation is also a marker of activation profile. After a series of gates to exclude debris, doublets etc. (not shown), the population encompassing microglia and other brain-resident macrophages was identified by their intermediate CD45 signal and positive CD11b staining. The percentage of cells positive for Ki-67 was evaluated in Fig. 3e. Injection of LPS dramatically increased the percentage of proliferating (Ki-67-positive) cells within brain microglia. This effect was significantly blocked in the mice treated with JNJ-55308942. These data show conclusive evidence that P2X7 antagonism modulates microglial activation in the brain strengthening the hypothesis that P2X7 activation plays a role in microglial activation. The measured brain concentration of JNJ-55308942 2 h after dosing was ~ 7000 ng/ml. Terminal exposure of the compound in the brain was below the limit of detection, suggesting that the compound cleared the system as expected by the pharmacokinetic properties of the molecule.

Systemic P2X7 antagonism alleviates LPS-induced microglial activation. a Mice (N = 4/group) received the compound (30 mg/kg, oral) 1 h before as well as 7 and 24 h after an i.p. injection of LPS (0.8 mg/kg, i.p.) or vehicle and were sacrificed 48 h after LPS injection. Brains were collected, dissociated and stained with conjugated antibodies for FACS. b Gating strategy for (c): after a series of gates to exclude events with inconsistent flow rate, debris, doublets, and cell clumps (not shown), remaining events were gated for live cells by their relative size (forward scatter, FSC) and negative Zombie dye signal. Microglia were identified as CD45 int, CD11b+, CD206- cells within the live cell gate, and the median of their relative signal for CD11b and other markers (not shown) was assessed. c Relative fluorescent signal of microglia cells for FSC (correlating with cell size), side-scatter (SSC, correlating with cell complexity), CD45-BV605, and CD11b-PE-Cy7. LPS treatment causes significant increases in all four parameters. JNJ-55308942 (JNJ) significantly attenuates the effect of LPS on FSC, CD45 surface expression and CD11b surface expression, whereas in vehicle-treated controls the compound shows no effect. d A second aliquot of the dissociated brains was stained with the intranuclear proliferation marker Ki-67. After a series of gates to exclude debris, doublets etc. (not shown) the population encompassing microglia and other brain-resident macrophages was identified by their intermediate CD45 signal and positive CD11b staining. The percentage of cells positive for Ki-67-BV421 was evaluated in (e) e Injection of LPS dramatically increases the percentage of proliferating (Ki-67-positive) cells within brain-resident macrophages. This effect is significantly alleviated by systemic P2X7 blockade (JNJ). Results are shown as % of the non-treated control group. Boxes show mean with upper and lower quartiles with whiskers depicting the total range of the data. ##p < 0.01, ###p < 0.001 LPS vs. VEH, * p < 0.05 CPD vs. CTRL (two-way ANOVA followed by Tukey-corrected multiple comparisons)
Effects of JNJ-55308942 in a neuroinflammatory model of depression in mice
A robust model of neuroinflammation that is known to cause depressogenic behavior in mice is induced by Bacillus Calmette-Guerin (BCG) [41, 42]. Since JNJ-55308942 demonstrated efficacy in the LPS model of microglial activation, and since the BCG model causes microglial activation [43], we wanted to test the efficacy of the P2X7 antagonist in the BCG model. The compound was dosed prior to BCG inoculation to test the hypothesis under maximal target coverage by JNJ-55308942 (Fig. 4a). Sucrose preference was measured as an index of anhedonia-like behavior (Fig. 4b). In vehicle-treated control mice, BCG precipitated a significant reduction in sucrose preference during a 48 h probe at 2 weeks post challenge. Two-factor ANOVA revealed a significant drug x BCG interaction, and post hoc analysis confirmed that drug-treated mice did not exhibit BCG-induced reduction in sucrose preference. BCG also disrupted social interaction in a P2X7 receptor dependent manner (Fig. 4c). BCG reduced the amount of time that vehicle-treated mice spent in the interaction zone proximal to a novel conspecific. Drug treatment significantly attenuated the reduction in social interaction induced by BCG challenge. These data collectively suggest that JNJ-55308942 produced efficacy in two behavioral endpoints akin to depression; more importantly, to our knowledge, this is the first demonstration of a P2X7 mechanism in this model of neuroinflammation-mediated depression.

JNJ-55308942 is efficacious in a BCG-model of depression. a The experimental schematic is shown; JNJ-55308942 (JNJ, 30 mg/kg, oral) was dosed a day prior to a single administration of BCG and every day thereafter. Behavior was measured on days indicated. b Sucrose preference was measured as an index of anhedonia-like behavior. In vehicle-treated control mice, BCG precipitated a significant 10% reduction in sucrose preference during a 48 h probe at day 11 post challenge (p < 0.05). Two-factor ANOVA revealed a significant drug x BCG interaction, and post hoc analysis confirmed that drug-treated mice did not exhibit BCG-induced reduction in sucrose preference. Data represent mean ± SEM, n = 8–11 mice/group. c BCG disrupts social interaction in a P2X7 receptor dependent manner. BCG reduced the amount of time that vehicle-treated mice spent in the interaction zone proximal to a novel stranger mouse (p < 0.05). Drug treatment significantly attenuated the reduction in social investigation precipitated by BCG challenge (drug × BCG interaction, p < 0.02) Data represent mean + /- SEM, n = 8–11 mice/group
Effect of JNJ-55308942 in a chronic model of stress-induced depression in rats
The final objective in this study was to investigate the neuropharmacology of JNJ-55308942 in chronic models of stress. It is well accepted that chronic stress precipitates clinical depression; and our hypothesis has been that P2X7 antagonists may be beneficial in a sub-population of patients suffering from stress-induced depression, perhaps with a heightened inflammatory tone. The chronic mild stress model was chosen due to the chronicity of the stress paradigm and robustness of anhedonic behavior (deficit of sucrose intake) [44]. Moreover, it has been demonstrated that P2X7-IL-1β pathway is engaged in chronic stress models [20], and as such we wanted to test whether JNJ-55308942 and other chemically and pharmacokinetically distinct P2X7 antagonists produced an efficacy signal in the model. Rats were subjected to a chronic mild stress (CMS) procedure for a period of seven consecutive weeks. The effect of stress was evaluated by measurement of the consumption of a 1% sucrose solution (see “Methods”). On the basis of their sucrose intake following 2 weeks of initial stress, both stressed and control groups were each divided further into matched subgroups, and for the following 5 weeks they received once daily administration of vehicle, imipramine (10 mg/kg, i.p.) or P2X7 antagonists including JNJ-55308942. Effects of chronic treatment with vehicle or imipramine on the consumption of 1% sucrose solution in controls (open symbols), and in animals exposed to CMS (closed symbols), are depicted in Fig. 5a. Compared to vehicle administration, imipramine had no significant effect on sucrose intake in control animals (data not shown) and gradually increased the sucrose consumption in stressed animals only. Thus, the reversal of sucrose intake deficit in the continued presence of imipramine was slow, and complete reversal took approximately 5 weeks.

JNJ-55308942 is efficacious in a chronic stress model of depression. Chronic stress resulted in a deficit of sucrose intake as demonstrated by open (non-stressed) and closed (stressed) circles in all panels a, b. The deficit of sucrose intake in the stressed rats was maintained during the 5 weeks of vehicle treatment. The effect of imipramine (10 mg/kg, i.p.) is shown in panel a. Efficacy of JNJ-55308942 (0.1 and 1 mg/kg, oral) is shown in panel b. Drug treatment had no effect on non-stressed rats (data not shown). c Relationship between sucrose intake (Y axis), P2X7 antagonists dosed at 1 mg/kg (oral) for 5 weeks (JNJ-47965567: white bar; JNJ-42253432: brown bar; JNJ-55471300: orange bar; JNJ-55308942: green bar) and corresponding mean brain P2X7 occupancy shown as numbers (±SEM) within the bar. Occupancy (as shown by the numbers within the bar graphs) represents binding of the compound at 3 h post dose. Sucrose intake was measured weekly from N = 8 rats, divided across various treatment groups. Individual analysis of each doses revealed that in stressed animals the two doses of JNJ-55308942 caused significant treatment effect [0.1 mg: F(1,84) = 28.749; p < 0.001, 1 mg: F(1,84) = 38.766; p < 0.001]. When compared to the week zero scores, the increase of intakes caused by 0.1 mg/kg was apparent after already first week of treatment, but it reached significance at two time points (i.e., week 3 & 5) only. The dose of 1 mg/kg increased intakes also already after first week of treatment (p = 0.014) and, except for week 3 & 4, this effect was further enhanced in the following tests. *p < 0.05, **p < 0.01, ***p < 0.001; relative to vehicle-treated or drug-treated control groups. #p < 0.05, ## p < 0.01; relative to drug-treated stressed animals at week 0
The effect of chronic treatment with vehicle or the P2X7 antagonist JNJ-55308942 (0.1, & 1.0 mg/kg, p.o.) on the consumption of 1% sucrose solution in animals exposed to CMS (closed symbols) is shown in Fig. 5b. The two doses were picked to address the important question of occupancy needed for efficacy, based on autoradiography dose response presented in Fig. 2, even though in this CMS study rats were exposed to multiple doses (once daily for 5 weeks) of the compound. As compared to vehicle administration, JNJ-55308942 had no effect on sucrose intake in control animals. This demonstrated that the compound had no baseline effect on normal drinking in non-stressed rats. Individual analysis of each dose revealed that in stressed animals both doses had a significant effect. When compared to the week zero scores, the increased intake caused by 0.1 mg/kg was apparent after 1 week of treatment but it reached significance at two time points (i.e., week 3 & 5) only. The 1 mg/kg dose increased intake after the first week of treatment and, except for week 3 and 4, this effect was further enhanced in the subsequent tests. To get an estimate of brain P2X7 occupancy needed for efficacy in the chronic stress model, terminal brain samples were subjected to ex-vivo occupancy as described in the previous sections. In addition to JNJ-55308942, three distinct P2X7 antagonists were also tested for efficacy in the stress model. While JNJ-55471300 produced efficacy (Fig. 5c), the other two P2X7 antagonists (JNJ-47965567 and JNJ-42253432) did not produce efficacy on week 5; when these compounds were assessed for P2X7 brain occupancy, we noticed that efficacy and occupancy correlated extremely well. For example, both JNJ-55308942 and JNJ-55471300 demonstrated high terminal occupancy and were efficacious in the stress model; contrary to this, the two compounds without efficacy had low brain P2X7 occupancy. This data illustrates that high sustained brain P2X7 occupancy is essential to maintain efficacy. In summary, JNJ-55308942 reversed the sucrose intake deficit and appears to have the right mix of pharmacology and pharmacokinetic properties to maintain efficacy after 5 weeks of daily dosing.
Discussion
The ATP-gated ion channel P2X7 is expressed abundantly in microglia and is hypothesized to play a role in neuroinflammatory disorders of the CNS, including psychiatric diseases. In this manuscript, we describe the neuropharmacology of JNJ-55308942, a P2X7 antagonist currently in clinical evaluation. The molecule is orally bioavailable, binds to brain P2X7 and blocks IL-1β release from adult rodent brain. As shown in Fig. 1 and Table 1, JNJ-55308942 is a P2X7 antagonist across multiple species, including rats and mice. This is an important attribute as prior P2X7 antagonists tested in clinical trials for rheumatoid arthritis were devoid of rodent activity and were peripherally restricted. Activity of JNJ-55308942 at the rat & mouse P2X7 helped us test key hypotheses in rodent models of target engagement and efficacy. This was a pre-requisite in our discovery approach as we wanted to optimize molecules for development that had excellent human drug-likeliness and possessed good rodent pharmacology and pharmacokinetic properties. JNJ-55308942 attenuated IL-1β release in human blood, mouse blood, and mouse microglia isolated from pup brains, as expected for this mechanism. The molecule was efficacious in preventing Bz-ATP-induced cell death in a concentration-dependent manner (Fig. 1c). This is important as intracellular ATP, released as a consequence of cell necrosis and pyroptosis, can cause sustained P2X7 activation causing amplification of further ATP release; as such the ability of JNJ-55308942 to block cell death is important and can have differentiating features in-vivo from other IL-1β attenuating mechanisms (such as NLRP3 inhibition). JNJ-55308942 prevented LPS-induced microglial activation (Fig. 3). Whether modulation of microglial activation will result in benefit for patients remains to be seen in the clinic. Several reports have suggested a positive relationship between clinical depression, bipolar, schizophrenia, and microglial activation as assessed by enhanced PET signal using TSPO ligands. The efficacy data reported here brings hope to patients suffering from CNS neuroinflammatory disorders. For example, we have shown efficacy of JNJ-55308942 in a BCG model of neuroinflammation-induced anhedonia (Fig. 4). The mouse BCG model is a neuroinflammatory model causing microglial activation, inducing deficits of sucrose preference and social interaction. Likewise, in a 5-week chronic model of stress, JNJ-55308942 reversed the sucrose intake deficits. This was particularly exciting and of therapeutic relevance as in this case (Fig. 5), the compound was administered after the animals had been stressed for 2 weeks with deficits in sucrose intake. More importantly, as shown in Fig. 5d, there seems to be striking relationship between brain P2X7 occupancy and efficacy. In addition to JNJ-55308942, three distinct P2X7 antagonists were also tested for efficacy in the stress model. Pharmacology, including selectivity profiles and pharmacokinetic & brain/plasma ratios of these three P2X7 antagonists has been published elsewhere [37,38,39]. While JNJ-55471300 produced efficacy (Fig. 5d), the other two P2X7 antagonists (JNJ-47965567 and JNJ-42253432) did not produce any efficacy; on assessing terminal brain P2X7 occupancy (numbers in the bar graph), we were encouraged to find a nice and robust relationship between high central P2X7 occupancy and efficacy.
While the preclinical data is promising, the fact of the matter remains that new mechanisms must be tested in the clinical setting as efficacy in animal models of anhedonia do not always translate into clinical efficacy. It must be noted here, that although P2X7 antagonists have been shown by us and others to produce efficacy in chronic models of stress, there is equivocal data on the efficacy of P2X7 antagonists in acute behaviors such as despair and anxiety. We believe the mechanism of P2X7 activation is probably more engaged during chronic bouts of stress rather than in acute despair behaviors. Due to these factors, it is critical to test proof of concept studies in patients that are most likely to respond to the P2X7 antagonism. In other words, it is plausible that patients with a higher IL-1β or inflammatory burden may benefit more from a mechanism such as P2X7 antagonism. Since human blood IL-1β can be used as a biomarker, it may be possible to stratify patients based on how much they respond to a ATP challenge ex-vivo. The use of human blood IL-1β as a biomarker will add value to the clinical program of JNJ-55308942, both to assess drug effect (like mouse blood in Fig. 2d) and patient stratification. For example, since P2X7 is highly polymorphic, it is plausible that patients carrying the loss of function allele, will not respond to ATP challenge ex-vivo; contrary to that, gain-of function variants may be more susceptible to mood disorders (or neuroinflammation) and may respond to ATP with robust IL-1β release from blood lymphocytes. This biomarker can be used to stratify patients in the clinical trial; if this strategy is used in conjunction with P2X7 SNP (pharmacogenomics) analysis, and a correlation emerges, it may even provide impetus for a case of companion diagnostic or personalized medicine.
The recent advances made in P2X7 PET ligands is another area of excitement in this field [36]. With the discovery of P2X7 PET ligands that access brain P2X7 [45], a molecule such as JNJ-55308942 is well poised to generate human PK-PD data to support dose selection for efficacy in patients. Demonstration of brain occupancy in CNS early development programs is critical and the preclinical data presented here (Fig. 2) underscores the translational value of such an approach. Moreover, a P2X7 PET ligand has the potential to be a central biomarker to detect microglial activation and astrogliosis. The neuroinflammatory burden of patients can be assessed by PET scanning, which is probably more robust in severe cases of neurodegenerative disorders including epilepsy, stoke, and multiple sclerosis. Mood disorders often is a sequalae of the primary neurodegenerative pathology and as such P2X7 antagonism may offer additional benefit to these patient sub-populations.
In summary, data shown here provides impetus to develop JNJ-55308942 for depression, perhaps a treatment-resistant population, either as monotherapy or maintenance to prevent relapse to existing medications. Given the genetic association of P2X7 SNPs to bipolar, and efficacy reported in amphetamine-induced models of mania, it is also plausible that P2X7 antagonists may indeed benefit as mood stabilizers. At the time of writing this manuscript, P2X7 remains a clinically unvalidated target for mood disorders; nonetheless, data described in this study, if translated in proof-of-concept studies will provide hope to patients in need for alternate avenues of therapy. P2X7 antagonism hopefully provides this avenue to address the clinical unmet need in treatment-resistant depression with an inflammatory tone, and we remain optimistic that JNJ-55308942 has adequate properties to be tested in the clinic for proof-of-concept.
References
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflamm. 2013;10:43.
Reus GZ, Fries GR, Stertz L, Badawy M, Passos IC, Barichello T, et al. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience. 2015;300:141–54.
He Y, Taylor N, Fourgeaud L, Bhattacharya A. The role of microglial P2X7: modulation of cell death and cytokine release. J Neuroinflamm. 2017;14:135.
Lord B, Ameriks MK, Wang Q, Fourgeaud L, Vliegen M, Verluyten W, et al. A novel radioligand for the ATP-gated ion channel P2X7: [3H] JNJ-54232334. Eur J Pharmacol. 2015;765:551–9.
Bhattacharya A, Biber K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia. 2016;64:1772–87.
Giuliani AL, Sarti AC, Falzoni S, Di Virgilio F. The P2X7 receptor-interleukin-1 liaison. Front Pharmacol. 2017;8:123.
Fantoni ER, Dal Ben D, Falzoni S, Di Virgilio F, Lovestone S, Gee A. Design, synthesis and evaluation in an LPS rodent model of neuroinflammation of a novel (18)F-labelled PET tracer targeting P2X7. EJNMMI Res. 2017;7:31.
Territo PR, Meyer JA, Peters JS, Riley AA, McCarthy BP, Gao M, et al. Characterization of (11)C-GSK1482160 for targeting the P2X7 receptor as a biomarker for neuroinflammation. J Nucl Med: Off Publ, Soc Nucl Med. 2017;58:458–65.
Kaufmann FN, Costa AP, Ghisleni G, Diaz AP, Rodrigues AL, Peluffo H et al. (2017). NLRP3 inflammasome-driven pathways in depression: clinical and preclinical findings. Brain, behavior, and immunity 64:367–383.
Jones KA, Thomsen C. The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci. 2013;53:52–62.
Soderlund J, Olsson SK, Samuelsson M, Walther-Jallow L, Johansson C, Erhardt S, et al. Elevation of cerebrospinal fluid interleukin-1ss in bipolar disorder. J Psychiatry Neurosci. 2011;36:114–8.
Corwin EJ, Johnston N, Pugh L. Symptoms of postpartum depression associated with elevated levels of interleukin-1 beta during the first month postpartum. Biol Res Nurs. 2008;10:128–33.
Diniz BS, Teixeira AL, Talib L, Gattaz WF, Forlenza OV. Interleukin-1beta serum levels is increased in antidepressant-free elderly depressed patients. Am J Geriatr Psychiatry: Off J Am Assoc Geriatr Psychiatry. 2010;18:172–6.
Czamara D, Muller-Myhsok B, Lucae S (2017). The P2RX7 polymorphism rs2230912 is associated with depression: a meta-analysis. Progress in neuro-psychopharmacology & biological psychiatry 82:272–277.
Lucae S, Salyakina D, Barden N, Harvey M, Gagne B, Labbe M, et al. P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum Mol Genet. 2006;15:2438–45.
McQuillin A, Bass NJ, Choudhury K, Puri V, Kosmin M, Lawrence J, et al. Case-control studies show that a non-conservative amino-acid change from a glutamine to arginine in the P2RX7 purinergic receptor protein is associated with both bipolar- and unipolar-affective disorders. Mol Psychiatry. 2009;14:614–20.
Backlund L, Nikamo P, Hukic DS, Ek IR, Traskman-Bendz L, Landen M, et al. Cognitive manic symptoms associated with the P2RX7 gene in bipolar disorder. Bipolar Disord. 2011;13:500–8.
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. 2008;13:717–28.
Iwata M, Ota KT, Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 2013;31:105–14.
Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, et al. Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol Psychiatry. 2016;80:12–22.
Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA. 2008;105:751–6.
Yue N, Huang H, Zhu X, Han Q, Wang Y, Li B, et al. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflamm. 2017;14:102.
Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, et al. NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int J Neuropsychopharmacol. 2015;18:1–8.
Csolle C, Ando RD, Kittel A, Goloncser F, Baranyi M, Soproni K, et al. The absence of P2X7 receptors (P2rx7) on non-haematopoietic cells leads to selective alteration in mood-related behaviour with dysregulated gene expression and stress reactivity in mice. Int J Neuropsychopharmacol. 2013;16:213–33.
Basso AM, Bratcher NA, Harris RR, Jarvis MF, Decker MW, Rueter LE. Behavioral profile of P2X7 receptor knockout mice in animal models of depression and anxiety: relevance for neuropsychiatric disorders. Behav Brain Res. 2009;198:83–90.
Boucher AA, Arnold JC, Hunt GE, Spiro A, Spencer J, Brown C, et al. Resilience and reduced c-Fos expression in P2X7 receptor knockout mice exposed to repeated forced swim test. Neuroscience. 2011;189:170–7.
Wilkinson SM, Gunosewoyo H, Barron ML, Boucher A, McDonnell M, Turner P, et al. The first CNS-active carborane: a novel P2X7 receptor antagonist with antidepressant activity. ACS Chem Neurosci. 2014;5:335–9.
Carta S, Penco F, Lavieri R, Martini A, Dinarello CA, Gattorno M, et al. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci USA. 2015;112:2835–40.
Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M. The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain Behav Immun. 2013;28:54–62.
Tan S, Wang Y, Chen K, Long Z, Zou J. Ketamine alleviates depressive-like behaviors via down-regulating inflammatory cytokines induced by chronic restraint stress in mice. Biol Pharm Bull. 2017;40:1260–7.
Choi HB, Ryu JK, Kim SU, McLarnon JG. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci. 2007;27:4957–68.
O’Brien-Brown J, Jackson A, Reekie TA, Barron ML, Werry EL, Schiavini P, et al. Discovery and pharmacological evaluation of a novel series of adamantyl cyanoguanidines as P2X7 receptor antagonists. Eur J Med Chem. 2017;130:433–9.
Seo JS, Wei J, Qin L, Kim Y, Yan Z, Greengard P (2016). Cellular and molecular basis for stress-induced depression. Molecular Psychiatry 22:1440–1447.
Gubert C, Fries GR, Pfaffenseller B, Ferrari P, Coutinho-Silva R, Morrone FB et al. (2014). Role of P2X7 receptor in an animal model of mania induced by D-amphetamine. Mol Neurobiol. 53:611–620
Kovanyi B, Csolle C, Calovi S, Hanuska A, Kato E, Koles L, et al. The role of P2X7 receptors in a rodent PCP-induced schizophrenia model. Sci Rep. 2016;6:36680.
Bhattacharya A (2018). Recent Advances in CNS P2X7 Physiology and Pharmacology: Focus on Neuropsychiatric Disorders. Frontiers in Pharmacology. 9:1–7 doi: 10.3389/fphar.2018.00030
Chrovian CC, Soyode-Johnson A, Peterson AA, Gelin CF, Deng X, Dvorak CA, et al. A dipolar cycloaddition reaction To access 6-methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridines enables the discovery synthesis and preclinical profiling of a P2X7 antagonist clinical candidate. J Med Chem. 2018;61:207–23.
Lord B, Aluisio L, Shoblock JR, Neff RA, Varlinskaya EI, Ceusters M, et al. Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-42253432. J Pharmacol Exp Ther. 2014;351:628–41.
Bhattacharya A, Wang Q, Ao H, Shoblock JR, Lord B, Aluisio L, et al. Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br J Pharmacol. 2013;170:624–40.
Letavic MA, Savall BM, Allison BD, Aluisio L, Andres JI, De Angelis M, et al. 4-Methyl-6,7-dihydro-4H-triazolo[4,5-c]pyridine-Based P2X7 receptor antagonists: optimization of pharmacokinetic properties leading to the identification of a clinical candidate. J Med Chem. 2017;60:4559–72.
Gonzalez-Pena D, Nixon SE, O’Connor JC, Southey BR, Lawson MA, McCusker RH, et al. Microglia transcriptome changes in a model of depressive behavior after immune challenge. PLoS ONE. 2016;11:e0150858.
Moreau M, Andre C, O’Connor JC, Dumich SA, Woods JA, Kelley KW, et al. Inoculation of Bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behavior. Brain Behav Immun. 2008;22:1087–95.
Solano Fonseca R, Mahesula S, Apple DM, Raghunathan R, Dugan A, Cardona A, et al. Neurogenic niche microglia undergo positional remodeling and progressive activation contributing to age-associated reductions in neurogenesis. Stem Cells Dev. 2016;25:542–55.
Willner P. The chronic mild stress (CMS) model of depression: history, evaluation and usage. Neurobiol Stress. 2017;6:78–93.
Ory D, Celen S, Gijsbers R, Van Den Haute C, Postnov A, Koole M, et al. Preclinical evaluation of a P2X7 receptor-selective radiotracer: PET studies in a rat model with local overexpression of the human P2X7 receptor and in nonhuman primates. J Nucl Med: Off Publ, Soc Nucl Med. 2016;57:1436–41.
Acknowledgements
Funding
The study was funded by Janssen R&D. JNJ-55308942 and other P2X7 antagonists described in this study were synthesized at Janssen R&D.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Except Drs. Jason O’Connor and Mariusz Papp, all authors are employees of Johnson & Johnson. The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bhattacharya, A., Lord, B., Grigoleit, JS. et al. Neuropsychopharmacology of JNJ-55308942: evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia. Neuropsychopharmacol 43, 2586–2596 (2018). https://doi.org/10.1038/s41386-018-0141-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0141-6
This article is cited by
-
P2X7 receptors: a bibliometric review from 2002 to 2023
Purinergic Signalling (2024)
-
Role of the P2 × 7 receptor in neurodegenerative diseases and its pharmacological properties
Cell & Bioscience (2023)
-
Chronic cough relief by allosteric modulation of P2X3 without taste disturbance
Nature Communications (2023)
-
Variation along P2RX7 interacts with early traumas on severity of anxiety suggesting a role for neuroinflammation
Scientific Reports (2023)
-
Triggering of Major Brain Disorders by Protons and ATP: The Role of ASICs and P2X Receptors
Neuroscience Bulletin (2023)
