
Abstract
Patients with neuropathic pain are usually accompanied by depression. Chemokine-mediated neuroinflammation is involved in a variety of diseases, including neurodegenerative diseases, depression, and chronic pain. The nucleus accumbens (NAc) is an important area in mediating pain sensation and depression. Here we report that spinal nerve ligation (SNL) induced upregulation of chemokine CCL2 and its major receptor CCR2 in both dopamine D1 and D2 receptor (D1R and D2R)-containing neurons in the NAc. Inhibition of CCR2 by shRNA lentivirus in the NAc shell attenuated SNL-induced pain hypersensitivity and depressive behaviors. Conversely, intra-NAc injection of CCL2-expressing lentivirus-induced nociceptive and depressive behaviors in naïve mice. Whole-cell patch clamp recording of D1R-positive or D2R-positive medium spiny neurons (MSNs) showed that SNL increased NMDA receptor (NMDAR)-mediated currents that are induced by stimulation of prefrontal cortical afferents to MSNs, which was inhibited by a CCR2 antagonist. Furthermore, Ccr2 shRNA also reduced NMDAR-mediated currents, and this reduction was mainly mediated via NR2B subunit. Consistently, NR2B, colocalized with CCR2 in the NAc, was phosphorylated after SNL and was inhibited by intra-NAc injection of Ccr2 shRNA. Furthermore, SNL or CCL2 induced ERK activation in the NAc. Inhibition of ERK by a MEK inhibitor reduced NR2B phosphorylation induced by SNL or CCL2. Finally, intra-NAc injection of NR2B antagonist or MEK inhibitor attenuated SNL-induced pain hypersensitivity and depressive behaviors. Collectively, these results suggest that CCL2/CCR2 signaling in the NAc shell is important in mediating neuropathic pain and depression via regulating NR2B-mediated NMDAR function in D1R- and D2R-containing neurons following peripheral nerve injury.
Similar content being viewed by others


Introduction
Neuropathic pain resulted from peripheral nerve injury is difficult to treat, and the patients not only suffer from severe pain, but also are accompanied with negative affects, such as anhedonia, decreased motivation, and depression [1, 2]. Understanding the molecular and synaptic mechanisms underlying neuropathic pain and associated depression is important for the treatment of these debilitating pain syndromes.
A number of studies have shown that pain and depression share some common mechanisms. Indeed, the typical antidepressant medications are successful in treating chronic pain [3, 4]. Moreover, inflammation has been recently demonstrated to be associated with pain and depression in the supraspinal structures [5, 6]. For example, neuropathic pain is associated with the increased gene expression of proinflammatory cytokines (TNF and IL-1β) in the prefrontal cortex and brainstem [7, 8]. Spinal injury also induces a delayed increase of chemokines CCL2, CCL3 in the hippocampus [9]. On the other hand, systemic injection of bacterial lipopolysaccharide upregulates TNF, IL-1β, IL-6, and CCL2 level in the hippocampus and hypothalamus [10]. Consistently, increasing the brain cytokines produces persistent pain-like symptoms [11, 12] and depression-like behaviors [13,14,15]. The evidence collectively suggests that brain inflammation is involved in the pathogenesis of both pain and depression.
Chemokine CCL2 and its major receptor CCR2 have been demonstrated to mediate neuroinflammation and increase the synaptic transmission in the spinal cord dorsal horn under neuropathic pain condition [16]. In the brain, CCL2 contributes to Parkinson’s disease in the striatum [17] and also underlies drug addiction in the prefrontal cortex and nucleus accumbens (NAc) [18]. The NAc is a critical brain region in mediating reward and motivation and is also implicated in the pathophysiology of depression [19,20,21]. The NAc is divided into the core and shell. Both regions have been shown to play a role in depression [22, 23]. Recent studies showed that the shell also plays an important role in mediating neuropathic pain [24, 25]. In addition, the glutamate NMDA receptor (NMDAR)-mediated synaptic transmission in the NAc is involved in natural rewards, emotion, and pain regulation [26, 27]. Whether CCL2 and CCR2 modulate NMDAR-mediated synaptic activity in the NAc shell and further contribute to neuropathic pain and associated depressive behaviors is important to be investigated.
The NAc is comprised predominantly of medium spiny neurons (MSNs), which can be divided into two populations: dopamine D1 receptor (D1R)-positive and D2 receptor (D2R)-positive. Previous studies showed that D1R-positive and D2R-positive MSNs contribute to distinct nociceptive or motivational behaviors [23, 25]. In the present study, using spinal nerve ligation (SNL)-induced neuropathic pain model and transgenic mice, we investigated the distribution of CCL2/CCR2 in D1R-expressing and D2R-expressing neurons and explored the role and mechanism of CCL2/CCR2 in SNL-induced pain hypersensitivity and depression.
Materials and methods
Animals, surgery, and behavior tests
Adult ICR male mice, transgenic BAC Drd1a-tdTomato mice, and BAC Drd2-EGFP were used. SNL was performed as previously described [28]. Pain hypersensitivity (von Frey test and Hargreaves test) and depressive behaviors (forced swim test (FST) and sucrose preference test (SPT)) were assessed as previously reported [29,30,31].
Multiplexing protein array
Animals were transcardially perfused with PBS. The NAc was dissected and homogenized. Cytokine and chemokine expression profiles were evaluated using Bio-Plex System according to the manufacturer’s protocols.
Real-time quantitative PCR (qPCR)
The total RNA of the NAc was extracted using Trizol reagent. One microgram of total RNA was reverse transcribed using an oligo (dT) primer. qPCR analysis was performed in the Real-time Detection System by SYBR green I dye detection. The sequences of the primers are shown in Table 1. The PCR amplifications were performed as previously described [32].
Western blot
Protein samples were separated on SDS–PAGE gel and transferred to nitrocellulose blots. The blots were blocked with 5% milk and incubated overnight at 4 °C with antibody against CCR2, pNR2B, NR2B, pERK, ERK, and GAPDH, and further incubated with the HRP-conjugated secondary antibody, developed in ECL solution, and exposed onto Hyperfilm. Specific bands were evaluated by molecular size.
Immunohistochemistry
Animals were deeply anesthetized with isoflurane and perfused with PBS followed by 4% paraformaldehyde as we described previously [33]. The brain sections were cut in a cryostat and were first blocked with 5% donkey serum, then incubated with the following primary antibodies: CCL2, CCR2, NR2B, and NeuN, GFAP, and IBA-1. The sections were then incubated with Cy3-conjugated or FITC-conjugated secondary antibodies. The stained sections were examined with a Leica fluorescence microscope.
Lentiviral vectors production and intra-NAc injection
The recombinant lentivirus shRNA expressing vectors containing Ccr2 shRNA (LV-Ccr2 shRNA) or NC shRNA (LV-NC) were constructed using pGCSIL-GFP vector. Recombinant lentivirus expressing CCL2 vector was constructed using the pGV365 lentiviral expression vector to produce pLV-Ubi-Ccl2-3FLAG-CMV-EGFP (LV-ccl2). A lentiviral vector that expresses GFP alone (LV-CON) was generated as a control. For the lentivirus injection, mice were anesthetized with isoflurane. The head was fixed by stereotaxic apparatus, and two small holes were drilled on each side. A 32 G syringe was directly lowered into the NAc. A volume of 0.5 μl lentivirus solution was injected over a period of 10 min.
Brain cannula implantation and drug injection
The animals were anesthetized with isoflurane. A guided cannula (26 G) was implanted above the bilateral NAc. An injection needle (32 G) was inserted through the guide cannula, and drug solution or vehicle (0.5 μl) was slowly injected over 5 min using a Hamilton syringe.
Brain slices preparation
NAc slices preparation and whole cell patch-clamp recordings were conducted as described previously [34]. The NAc shell and prefrontal cortex were cut using a vibratome. The slices were stored with oxygenated normal aCSF for 30 min at 34 °C and subsequently incubated in room temperature at least 1-h prior to experiment recording.
Whole-cell patch-clamp recording in NAc shell slices
MSNs in NAc shell were identified by their morphology and hyperpolarized resting membrane potential. Afferents were stimulated at 0.1 Hz by a concentric bipolar electrode [35]. The GABAA receptor antagonist picrotoxin and AMPA receptor antagonist CNQX were present in the aCSF throughout the experiment. Data were filtered at 2 kHz and digitized at 10 kHz. The NMDAR current–voltage (I–V) relationship experiments were performed via clamping the neuron at −70, −40, −20, 0, +20 and + 40 mV, respectively.
Quantification and statistics
All data were expressed as mean ± SEM. The behavioral data were analyzed by two-way RM ANOVA followed by Bonferroni test. For western blot, the density of specific bands was measured with Image J. Differences between groups were compared using one-way ANOVA followed by Bonferroni test. Two-tailed Student’s t-test was used if only two groups were applied. The criterion for statistical significance was P < 0.05.
Additional experimental procedures are described in Supplemental Materials.
Results
SNL induces CCL2 upregulation in NAc neurons
To investigate whether inflammatory mediators in the NAc were changed after SNL, we examined the expression of three proinflammatory cytokines and five chemokines in the NAc, 7 days after SNL or sham operation using a multiplexing protein array system. The level of CCL2, CCL4, and CXCL1 was significantly increased (Fig. 1a). Given the important role of CCL2 in the spinal cord in mediating chronic pain [16], we focused on CCL2 in this study.

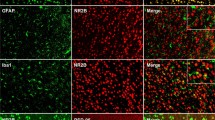
SNL increases CCL2 expression in the neurons of NAc. a A multiplexing protein array shows the expression of three cytokines and five chemokines in the NAc, 7 days after SNL or sham operation. *P < 0.05, Student’s t-test. n = 5 mice/group. b Quantitative RT-PCR shows the increased Ccl2 mRNA expression in the NAc at days 3, 7, and 21 after SNL. *P < 0.05, vs. sham. One-way ANOVA followed by Bonferroni’s test. c Schematic drawing indicating the site of images taken in (d–f). d–f CCL2 had low expression in naïve (d) and sham-treated animals (e) and was markedly increased in SNL-treated animals (f). g–i Double staining of CCL2 with neuronal marker NeuN (g), astrocytic marker GFAP (h), and microglial marker IBA-1 (i) in the NAc 7 days after SNL. j–k Expression of NeuN in the NAc of Drd1a-tdTomato (j) and Drd2-EGFP (k) mice. l–m Expression of CCL2 in the NAc of Drd1a-tdTomato (l) and Drd2-EGFP (m) mice 7 days after SNL
qPCR showed that SNL increased Ccl2 mRNA expression at days 3, 7, and 21. Immunofluorescence staining showed that, in the medial shell of the NAc (Fig. 1c), CCL2 has low expression in naïve (Fig. 1d) and sham-treated mice (Fig. 1e), and was remarkably increased after SNL (Fig. 1f). In addition, most of the CCL2-positive cells (97%) were colocalized with neuronal marker NeuN (Fig. 1g), rarely with astrocytic marker GFAP (1%, Fig. 1h), or microglial marker IBA-1 (1%, Fig. 1i). Furthermore, of NeuN-positive neurons in the NAc of Drd1a-tdTomato mice, 67% expressed Tomato (Fig. 1j), whereas 46% of NeuN-positive neurons in the NAc of Drd2-EGFP mice expressed EGFP (Fig. 1k), suggesting that 13% neurons express both D1R and D2R. Immunostaining of CCL2 in these mice showed that 73% D1R-positive (Fig. 1l) and 68% D2R-positive neurons express CCL2 (Fig. 1m), suggesting that CCL2 is expressed in both D1R-containing and D2R-containing neurons in the NAc shell.
SNL increases CCR2 expression in the neurons of the NAc shell
CCR2 is the major receptor of CCL2 [36]. CCR2 mRNA was increased at days 3, 7, and 21 after SNL (Fig. 2a). CCR2 protein was also increased in the ipsilateral and contralateral NAc 7 days after SNL (Fig. 2b). Immunostaining showed that CCR2 was expressed in naïve (Fig. 2c) and sham-operated mice (Fig. 2d), and increased in SNL mice (Fig. 2e). Furthermore, CCR2 was highly coexpressed with NeuN (Fig. 2f), but not with GFAP (Fig. 2g) or IBA-1 (Fig. 2h). In addition, 84% D1R-positive and 58% D2R-positive neurons express CCR2 7 days after SNL (Fig. 2i, j).

CCR2 is involved in SNL-induced neuropathic pain and depression. a Quantitative PCR shows that Ccr2 mRNA expression was increased at days 3, 7, and 21 in the NAc after SNL. *P < 0.05, vs. sham, one-way ANOVA followed by Bonferroni’s test. b Western blot shows increased CCR2 protein 7 days in the ipsilateral (Ipsi) and contralateral (Contra) NAc after SNL. *P < 0.05, one-way ANOVA followed by Bonferroni’s test. n = 3 mice/group. c–e Fluorescence immunostaining of CCR2 in the NAc of naïve (c), sham (d), and SNL (e) mice. f–h Double immunostaining of CCR2 with NeuN (f), GFAP (g), and IBA-1 (h). i, j Expression of CCR2 in the NAc of Drd1a-tdTomato (i) and Drd2-EGFP (j) mice 7 days after SNL. k, l SNL decreased paw withdrawal threshold (k) and paw withdrawal latency (l) at days 7, 14, and 21. n = 5 mice/group. m, n SNL increased the immobility time of mice in forced swim test (M) and decreased the preference to sucrose in sucrose preference test (n) 14 days after SNL. n = 6 mice/group. *P < 0.05, SNL vs. sham, two-way RM ANOVA followed by Bonferroni’s test. o, p Pretreatment with LV-Ccr2 shRNA dramatically attenuated SNL-induced mechanical allodynia (o) and blocked SNL-induced heat hyperalgesia (p). *P < 0.05, vs. LV-NC, two-way RM ANOVA followed by Bonferroni’s test. n = 9–12 mice/group. q, r LV-Ccr2 shRNA decreased the immobility time (q) and prevented the decrease of the preference for sucrose (r) induced by SNL at day 14. *P < 0.05, vs. LV-NC, two-way RM ANOVA followed by Bonferroni’s test. n = 8 mice/group. s, t Intra-NAc injection of CCL2-expressing lentivirus (LV-Ccl2) decreased PWT (s) and PWL (t) in naïve mice from 7 days to 21 days after injection. u Intra-NAc injection of LV-Ccl2 increased the immobility time of mice. v LV-Ccl2 decreased the preference to sucrose at 14 days after injection. *P < 0.05, LV-CON, two-way RM ANOVA followed by Bonferroni’s test. n = 5–6 mice/group
Inhibition of CCR2 attenuated SNL-induced neuropathic pain and depression
To investigate whether CCL2/CCR2 signaling is involved in neuropathic pain and/or depression, pain hypersensitivity via Hargreaves test and von Frey filament test, and depressive behaviors via FST and SPT were tested. Consistent with previous studies [32, 37], nerve injury produced persistent mechanical allodynia (Fig. 2k) and heat hyperalgesia (Fig. 2l), also increased the immobility time in forced swim (Fig. 2m), and decreased the preference to sucrose (Fig. 2n), suggesting that SNL induces nociceptive and depressive behaviors.
The recombinant lentivirus shRNA can effectively and persistently knock down the expression of a target gene [38]. To reduce the expression of CCR2, we delivered Ccr2 shRNA lentivirus (LV-Ccr2 shRNA) to bilateral NAc shell. The timeline of surgeries, tissue collection, and behavioral tests is shown in Supplementary Figure 1. GFP fluorescence was shown 7 days after injection, and was mainly distributed in NeuN-expressing neurons, with a few in GFAP-positive or IBA-1-positive glial cells. Pretreatment with LV-Ccr2 shRNA significantly reduced CCR2 protein level 7 days after SNL (Supplementary Figure 2).
Further behavioral tests showed that pretreatment with LV-ccr2 shRNA in the bilateral NAc, 7 days before SNL, did not affect the motor function (Supplementary Figure 3) or the basal pain responses, but increased paw withdrawal threshold (PWT, Fig. 2o) and paw withdrawal latency (PWL, Fig. 2p) on days 7, 14, and 21 post-SNL. LV-Ccr2 shRNA also inhibited the increase of immobility time in FST (Fig. 2q), and restored the preference to sucrose at day 14 (Fig. 2r), suggesting that CCR2 in the NAc shell is necessary for both nociceptive and depressive behaviors induced by SNL.
Increasing CCL2 induces nociceptive and depressive behaviors in naïve mice
We then checked whether CCL2 is sufficient to induce pain hypersensitivity and depression-like behaviors. The timeline of surgery, behavioral tests, and tissue collection is shown in Supplementary Figure 4. Intra-NAc injection of Ccl2-expressing lentivirus (LV-Ccl2) did not affect the motor function (Supplementary Figure 5), but decreased PWT and PWL at days 7, 14, and 21 after the injection (Fig. 2s, t). Moreover, LV-Ccl2 increased the immobility time in the FST (Fig. 2u), and reduced the preference to sucrose at day 14 (Fig. 2v). RT-PCR and ELISA confirmed that intra-NAc injection of LV-Ccl2 effectively increased Ccl2 mRNA level by 2.79 ± 0.72-fold and protein level by 1.5 ± 0.72-fold in the NAc 7 days after injection (P < 0.05, vs. LV-CON, Student’s t-test). These data suggest that genetic increase of CCL2 in the NAc shell is sufficient to induce both nociceptive and depressive behaviors.
SNL increases NMDAR currents via CCR2 in both D1R-MSNs and D2R-MSNs in the NAc shell
To explore the possible mechanisms, we did electrophysiological experiments. As more than 90% neurons are MSNs in the NAc and NMDARs are densely expressed in striatal MSNs [39, 40], we asked if CCL2/CCR2 regulates NMDAR-mediated synaptic transmission in the MSNs. The EPSCs were evoked by stimulating the boarder of the prefrontal cortex (PFC) and the NAc area, which may primarily activate the synaptic input from the PFC [35]. The NMDAR-mediated EPSCs on both D1R-MSNs and D2R-MSNs were significantly increased in SNL mice (Fig. 3a, b). We further constructed I–V relationship for NMDAR-mediated EPSCs via a series of holding potentials (−70 to +40 mV). The NMDAR-mediated EPSCs on D1R-MSNs or D2R-MSNs were significantly increased in SNL mice (Fig. 3C1 and D1). Furthermore, SNL resulted in a similar level of increase of the conductance in both D1R-MSNs and D2R-MSNs in NAc shell (Fig. 3C2 and D2), indicating that the enhanced currents might be due to the change in the biophysical properties of NMDAR NR2 subunits at the synapse [41, 42].

SNL increases NMDA receptor-mediated currents in the D1R-MSNs and D2R-MSNs of the NAc shell. a Sample traces of NMDAR-mediated EPSCs (A1) in the D1R-MSNs of the NAc shell evoked by stimulation intensity from 25 to 150 μA from sham and SNL mice. Each trace is an average of 6–10 EPSCs at each holding potential. EPSCs were measured at the peak of the responses. (A2) SNL enhanced the input–output response of NMDAR-mediated EPSCs in D1R-MSN (10 neurons from three mice for each group). P < 0.05, Two-way RM ANOVA. b Sample traces of NMDAR-mediated EPSCs (B1) from D2R-MSNs in the NAc shell. (B2) SNL enhanced the input–output response of NMDAR-mediated EPSCs in D2R-MSN (Sham, seven neurons from three mice, SNL, eight neurons from three mice). P < 0.05, two-way RM ANOVA. c Sample traces (left) and the I–V relationship (right) of NMDAR-mediated EPSCs (C1) in sham (n = 10 neurons from three mice) and SNL mice (10 neurons from three mice). The amplitudes EPSCs were normalized to mean current amplitudes from sham at +40 mV. (C2) Mean conductance of NMDAR-mediated EPSCs was increased in SNL mice (10 neurons from three mice for each group). P < 0.05, SNL vs. sham, Student’s t-test. d Sample traces (left) and the I–V relationship (right) of NMDAR-mediated EPSCs (D1) in sham (12 neurons from five mice) and SNL mice (11 neurons from four mice). (D2) Mean conductance of NMDAR-mediated EPSCs was increased in SNL mice (Sham, 12 neurons from five mice; SNL, 11 neurons from four mice). P < 0.05, SNL vs. sham, Student’s t-test. e Average NMDAR-mediated EPSCs traces (E1) in D1R-MSNs from sham and SNL mice (left panel). Time course of the peak NMDAR-mediated EPSCs treated by RS504393 in sham or SNL mice (eight neurons from three mice for each group). The peaks of NMDAR-mediated EPSCs were normalized to that of sham at +40 mV (right panel). (E2) Summary graph of RS504393-caused depression on NMDAR-mediated EPSCs in D1R-MSNs. *P < 0.05, vs. baseline. Student’s t-test. f Average NMDAR-mediated EPSCs traces (F1) in D2R-MSNs from sham or SNL mice (left panel). Time course of the peak NMDAR-mediated EPSCs treated by RS504393 in sham or SNL mice (sham, seven neurons from three mice; SNL, six neurons from three mice). The peaks of NMDAR-mediated EPSCs were normalized to that of sham at +40 mV (right panel). (F2) Summary graph of RS504393-caused depression on NMDAR-mediated EPSCs in D2R-MSNs. *P < 0.05, vs. baseline. Student’s t-test
To examine the effect of CCR2 activity on NMDAR-mediated responses at synapses of D1R-MSNs and D2R-MSNs, we incubated the NAc slices with CCR2 antagonist RS504393. RS504393 (10 μM) caused a significant depression of NMDAR-mediated EPSCs in both D1R-MSNs (Fig. 3e) and D2R-MSNs (Fig. 3f) from either sham control or SNL mice (Fig. 3E2 and F2), suggesting that CCR2 is involved in the modulation of NMDAR activity under both sham and SNL condition in the MSNs in the NAc shell.
CCR2 contributes to altered NMDAR current via the increase of NR2B component function
To further confirm if CCR2 contributes to the altered synaptic transmission after SNL, we injected LV-Ccr2 shRNA into the NAc shell 7 days before SNL and recorded NMDAR-mediated EPSCs in the MSNs 7–10 days after SNL in ICR mice (Fig. 4a). Knockdown of CCR2 by shRNA significantly reduced the increase in NMDAR-mediated EPSCs compared to LV-NC treatment (Fig. 4b, c). In addition, the voltage-dependence of NMDAR EPSCs (Fig. 4d) and the conductance (Fig. 4e) were also markedly reduced, confirming that CCR2 in the NAc shell contributes to the enhancement of NMDAR-current after SNL.

Inhibition of CCR2 decreases the enhanced NMDAR-mediated currents in the MSNs of the NAc shell. a Timeline of experiments in (b–g). b Samples of NMDAR-mediated EPSCs recorded in SNL mice pretreated with LV-NC or LV-Ccr2 shRNA. c Intra-NAc injection of LV-Ccr2 shRNA significantly reduced the I-O response of NMDAR-mediated EPSCs (LV-NC, 11 neurons from four mice; LV-Ccr2 shRNA, 10 neurons from four mice). *P < 0.05, two-way RM ANOVA. d Sample traces (left) and the I–V relationship (right) of NMDAR-mediated EPSCs in LV-NC-pretreated (11 neurons from five mice) or LV-Ccr2 shRNA-pretreated mice (10 neurons from four mice). P < 0.05, two-way RM ANOVA. e Mean conductance of NMDA receptor was reduced by LV-Ccr2 shRNA injection (LV-NC, 11 neurons from five mice; LV-Ccr2 shRNA, 10 neurons from four mice). *P < 0.05, Student’s t-test. f Sample traces of NMDAR-mediated EPSCs triggered at +40 mV in the presence of picrotoxin and CNQX from neurons of sham or SNL mice, and fitted with double exponential curves (green line). Stimulus artifacts were truncated for clarity. g Mean decay weighted time of the NMDAR-mediated EPSCs was slower in SNL mice (Sham, eight neurons from four mice; SNL, 10 neurons from five mice). * P < 0.05, SNL vs. sham, Student’s t-test. h Representative NMDAR-mediated EPSCs from SNL mice treated with LV-NC or LV-Ccr2 shRNA, and fitted with double exponential curves (green line). i Mean decay weights of the NMDAR-mediated EPSCs in SNL mice pretreated with LV-NC (11 neurons from five mice) or LV-Ccr2 shRNA (10 neurons from four mice). * P < 0.05, Student’s t-test. j Sample traces of NMDAR-mediated EPSCs from TCN-201-pretreated or Ro 25-6981-pretreated slices. Currents are normalized to the peak of NMDAR-mediated EPSCs. k Time course of the peak of NMDAR-mediated EPSCs in conditions of TCN-201 + RS504393 or Ro25-6981 + RS504393. l Summary of the relative inhibition of the peak amplitudes of NMDAR-mediated EPSCs by pretreatment with TCN-201 (6 neurons from three mice) or Ro25-6981 (6 neurons from two mice). * P < 0.05, Student’s t-test
As the time constant of decay can reflect the change of the proportion of NR2B subunits [23, 43], we fitted the NMDAR EPSC decay time course with a double exponential function. The decay of the NMDAR-mediated EPSC was significantly slowed in NAc slice after SNL (Fig. 4f, g), indicating the increase in the proportion of synaptic NR2B subunit [23, 43]. Consistently, the NMDAR decay kinetics in LV-Ccr2 shRNA-injected SNL mice returned to normal (Fig. 4h, i), indicating that CCR2 may mediate the change of NMDARs subunit composition in SNL mice.
To examine whether NMDAR NR2 subunits (NR2A and NR2B) contribute to this change, we pre-incubated the slices with selective NR2A antagonist TCN-201 (3 μM) or selective NR2B antagonist Ro 25-6981 (1 μM) before RS504393 treatment (Fig. 4j). In slices pretreated with TCN-201, RS504393 still reduced NMDAR-mediated EPSCs (Fig. 4k, l). However, in slices treated with Ro 25-6981, RS504393 resulted in a mild change for NMDAR-mediated EPSCs (Fig. 4k, l). These data suggest that the alteration of NMDAR currents by CCR2 might be mainly due to the increase of NR2B component function at NAc synapse.

CCR2/pNR2B/pERK pathway mediates SNL-induced neuropathic pain and depression. a Western blot shows the increase of pNR2B, but not NR2B at days 7 and 21 after SNL. *P < 0.05, vs. sham. One-way ANOVA followed by Bonferroni’s test. b Double staining shows the colocalization of NR2B and CCR2 in the NAc. c Pretreatment with LV-Ccr2 shRNA markedly decreased pNR2B expression in the NAc 7 days after SNL. *P < 0.05, vs. LV-NC. Student’s t-test. d Intra-NAc infusion of LV-Ccl2 increased pNR2B expression in the NAc 7 days after injection. * P < 0.05, vs. LV-CON. Student’s t-test. e Incubation of NAc slices with CCL2 increased pNR2B expression, which was reduced by pretreatment with Ro25-6981 (Ro). *P < 0.05, one-way ANOVA followed by Bonferroni’s test. f–h Intra-NAc injection of Ro25-6981 attenuated SNL-induced mechanical allodynia (f) and heat hyperalgesia (g), and reduced the immobility time of FST (h). A guided cannula was implanted after baseline 1 (BL1) testing. Baseline 2 (BL2) was tested 7 days after cannula implantation and 1 day before SNL. Seven days after SNL, Ro25-6981 was microinjected. The PWT, PWL, and FST were tested 1–2 h after Ro 25–6981 injection. *P < 0.05, one-way ANOVA followed by Bonferroni’s test. n = 6–7 mice/group. i SNL increased pERK expression in the NAc. *P < 0.05. One-way ANOVA. n = 3–4 mice/group. j, k Expression of pERK in the NAc shell of Drd1a-tdTomato (j) and Drd2-EGFP mice (k). l Intra-NAc injection of LV-Ccr2 shRNA reduced SNL-induced pERK expression. *P < 0.05, Student’s t-test. m Intra-NAc injection of Ccl2-expressing lentivirus-induced pERK expression in naïve mice. *P < 0.05, Student’s t-test. n Intra-NAc injection of MEK inhibitor PD98059 markedly reduced SNL-induced pNR2B expression. *P < 0.05, Student’s t-test. o–q Intra-NAc injection of PD98059 7 days after SNL attenuated SNL-induced mechanical allodynia (o) and heat hyperalgesia (p), and reduced the immobility time of FST (q). *P < 0.05, two-way ANOVA followed by Bonferroni’s test. n = 5–6 mice/group
SNL increases pNR2B expression via CCR2 in the NAc
To further test whether NR2B expression or phosphorylation is regulated by CCL2/CCR2 signaling, we examined the protein level of NR2B and pNR2B (phosphorylated NR2B) after SNL. pNR2B, but not NR2B was markedly increased at both 7 and 21 days (Fig. 5a). In addition, NR2B was highly coexpressed with CCR2 (Fig. 5b), supporting the possible interaction between NR2B and CCR2 in NAc neurons.
We then asked if CCR2 is involved in the increase of pNR2B after SNL. LV-Ccr2 shRNA or LV-NC shRNA was injected into the NAc shell 7 days before SNL, and NAc was harvested 7 days after SNL. Compared to LV-NC treatment, LV-Ccr2 shRNA significantly reduced the expression of pNR2B (Fig. 5c). Furthermore, intra-NAc injection of LV-Ccl2 remarkably increased pNR2B expression in the NAc (Fig. 5d). Incubation of NAc slices with CCL2 (100 ng/ml, 10 min) also significantly increased pNR2B expression, which was inhibited by preincubation with 1 μM Ro 25-6981 (Fig. 5e). These data suggest that CCL2, increased by SNL, phosphorylates NR2B via CCR2 in the NAc.
We then checked the role of NR2B antagonist on SNL-induced nociceptive behaviors and depressive behaviors. The timeline of surgeries and behavioral tests is shown in Supplementary Figure 6. NR2B antagonist Ro 25-6981 (1 μg/μl) or vehicle was microinjected into the NAc shell 7 days after SNL. Ro 25-6981 attenuated SNL-induced mechanical allodynia (Fig. 5f) and heat hyperalgesia (Fig. 5g) 1–2 h after microinjection, also reduced the immobility time of mice 2 h after injection (Fig. 5h). These behavioral data suggest that inhibition of NR2B in the NAc attenuates SNL-induced nociceptive and depressive behaviors.
ERK, activated by CCR2, contributes to NR2B phosphorylation and SNL-induced neuropathic pain and depressive behaviors
To further examine how NR2B is phosphorylated by CCL2/CCR2, we checked the activation of extracellular signal-regulated kinase (ERK) in the NAc after SNL. SNL increased phosphorylated ERK (pERK) expression in the NAc 7 days after SNL (Fig. 5i). Immunostaining further showed the distribution of pERK in both D1R-positive and D2R-positive neurons (Fig. 5j, k).
Furthermore, inhibition of CCR2 expression by LV-Ccr2 shRNA reduced SNL-induced pERK upregulation (Fig. 5l). Conversely, intra-NAc injection of LV-Ccl2 lentivirus increased pERK expression 7 days after injection in naïve mice (Fig. 5m). These data suggest that SNL induces CCR2-dependent ERK activation in the NAc.
To check if ERK is involved in SNL-induced NR2B phosphorylation, MEK inhibitor PD98059 (0.1 μg each site) [44] was injected into bilateral NAc shell at day 7 after SNL. pNR2B expression was significantly reduced by PD98059 1 h after injection (Fig. 5n). In addition, PD98059 also attenuated SNL-induced mechanical allodynia (Fig. 5o) and heat hyperalgesia (Fig. 5p), and decreased the immobility time in FST (Fig. 5q). These data indicate that ERK, as a downstream kinase of CCL2/CCR2, mediates SNL-induced NR2B phosphorylation in the NAc.
Discussion
Previous studies have shown that depression is present in more than 50% of individuals suffering from chronic pain [1, 2], and comorbid depression and chronic pain are highly prevalent in individuals suffering from physical illness [5, 45]. Here we provide the first demonstration that chemokine CCL2/CCR2 signaling in the NAc shell plays a pivotal role in mediating peripheral nerve injury-induced nociceptive behaviors and associated depressive behaviors. Our study also showed that CCL2/CCR2 signaling increased NMDA-mediated synaptic transmission in both D1R-expressing and D2R-expressing MSNs, which may be caused by ERK-dependent phosphorylation of NMDAR subunit 2B. Our results demonstrated a novel role of CCL2/CCR2 in mediating both neuropathic pain and depression in the NAc shell.
Supraspinal inflammation has been demonstrated to be responsible for the transition from sickness to depression and acute to chronic pain [5, 46]. Chemokines are increasingly recognized as important inflammatory mediators in the nervous system. CCL2 contributes to Parkinson’s disease in the striatum [17] and also underlies drug addiction in the PFC and the NAc [18]. We for the first time showed that CCL2 and CCR2 are increased in the NAc neurons after SNL. In addition, inhibition of CCR2 largely attenuated SNL-induced nociceptive and depressive behaviors, and genetic increase of CCL2-induced pain hypersensitivity and depressive behaviors in naïve mice. These data suggest that CCL2/CCR2 in the NAc is necessary and sufficient in the pathogenesis of nerve injury-induced neuropathic pain and depression.
The MSNs in the core and shell of the NAc receive glutamatergic inputs from the PFC, hippocampus, and amygdala. In the brain, glutamatergic synaptic transmission is mainly mediated by the AMPAR and NMDAR. A recent study showed that spared nerve injury increased the calcium-permeable AMPA receptors function in the NAc core, and an AMPA receptor potentiator delivered into the NAc decreases pain-induced depression [37]. Differently, Schwartz et al. reported that D2R-containing MSNs showed a decreased ratio of AMPA-mediated EPSCs to NMDAR-mediated EPSCs after spinal nerve injury, and the change may be in part due to a decrease in the number and/or function of AMPARs. In addition, by examining the time constant of decay, they reported that chronic pain may affect NMDAR-mediated synaptic transmission [23]. Here, we showed that the amplitude of NMDAR current was remarkably increased in both D1R-MSNs and D2R-MSNs after SNL. Interestingly, CCR2 antagonist or shRNA reduced the NMDAR-mediated currents in these neurons, indicating that NMDAR is involved in the change of synaptic transmission in the NAc shell, and CCR2 may regulate the function of NMDAR. Different from Schwartz et al.’ results, in which NMDAR EPSCs were not changed in D1R-MSNs in the NAc core under chronic pain condition, our data showed a similar increase of NMDAR-mediated currents in both D2R-MSNs and D1R-MSNs after SNL. The discrepancy may be due to different regions of the recorded MSNs. Additionally, recent studies implicated that D1-MSNs and D2-MSNs may play different roles in mediating depression and pain hypersensitivity. For example, chronic social defeat stress or restraint stress decreased the strength of excitatory synapses on D1R-MSNs, and manipulation of D1R-MSNs activity changed stress-induced depression behaviors [21, 47,48,49]. Ren et al. reported that spared nerve injury increased excitability of D2R MSNs in the NAc, and inhibiting D2-MSNs reversed injury-induced tactile allodynia [25]. As our results showed that inhibition of CCR2 in D1R-MSNs and D2R-MSNs attenuated both SNL-induced neuropathic pain and depression, further studies using D1R/D2R-cre mice to specifically manipulate CCR2 in D1R/D2R-MSNs may help to differentiate the cell type-specific role of CCR2 in mediating neuropathic pain and associated depression.
NMDARs consist of heterotetrameric assemblies of different subunits within a repertoire of three subtypes, NR1, NR2, and NR3. Functional NMDARs in mammalian cells require the heteromeric combination of at least one NR1 and one NR2 subtype (from NR2A to NR2D). NR2B in the NAc plays a key role in the remodeling of excitatory synapses [50]. Our recording showed a prolonged weighted decay time for NMDAR-mediated EPSCs after SNL, which was normally due to an increase in the proportion of synaptic NMDARs containing NR2B subunit [43]. Similarly, recent works demonstrated that spared nerve injury selectively increased NR2B containing NMDAR in the NAc [23, 25]. We further showed that the prolonged weighted time was depressed in SNL mice pretreated with Ccr2 shRNA. CCR2 antagonist also decreased the isolated NMDAR-mediated EPSCs, and the effect was blocked by pretreatment with the NR2B antagonist. Therefore, the upregulation of CCR2 expression after SNL may promote the function of NR2B-containing NMDAR at synapses on MSNs in NAc shell after SNL. However, we cannot exclude the possibility that CCL2/CCR2 might modulate trafficking of NMDARs [51] and ionic conductance of individual NMDARs [42], which are worthy to be investigated in the future.
Phosphorylation of NR2B subunit is critical for the neural plasticity underlying allodynia/hyperalgesia [52]. Nerve injury or tissue inflammation enhances NR2B phosphorylation in the dorsal horn [52, 53], and inhibition of spinal NR2B activation attenuated cancer pain or neuropathic pain [52, 54, 55]. Here SNL increased NR2B phosphorylation, which was inhibited by CCR2 shRNA. Intra-NAc injection of CCL2-induced NR2B phosphorylation in naïve mice, suggesting the involvement of CCL2/CCR2 in SNL-induced phosphorylation of NR2B.
CCR2 belongs to GPCR family, and activation of GPCR can regulate various signaling pathways. Mitogen-activated protein kinases (MAPKs), which consists of three members (ERK, JNK, and p38), are key components in GPCR-induced intracellular signaling and have been implicated in mediating chronic pain in the spinal cord [56]. CCL2-induced ERK activation in the spinal cord [16]. ERK is involved in the induction of activity-dependent neuronal plasticity [57]. In the NAc, CCL2 directly increased pERK expression in D1R-positive and D2R-positive neurons. SNL-induced ERK activation was blocked by CCR2 shRNA, suggesting that ERK is downstream of CCL2/CCR2. In addition, MEK inhibitor reduced SNL-induced NR2B phosphorylation, suggesting that ERK contributes to CCL2/CCR2-mediated NR2B phosphorylation. Besides, ERK activation in nociceptive pathways increases various kinds of immediate early gene transcription, which causes a transition from short-term adaptive processes to long-term hyperexcitability [57, 58]. In addition, intra-NAc injection of NR2B inhibitor or MEK inhibitor attenuated SNL-induced pain hypersensitivity and depressive behaviors. Our data suggest the pivotal role of CCL2/CCR2/ERK/NR2B in the NAc in mediating neuropathic pain and associated depression.
Taken together, we identify that CCL2/CCR2 in the NAc shell contribute to neuropathic pain and associated depression. Our results also reveal that CCL2/CCR2 regulates NMDAR-mediated synaptic transmission in the MSNs of NAc shell after SNL, which may be through ERK activation and NR2B phosphorylation. These findings provide new insights into the molecular mechanisms underlying neuropathic pain and associated depression. Combining with the important role of CCL2/CCR2 in the spinal cord and DRG regulating pain hypersensitivity [59], targeting CCL2/CCR2 signaling may provide an effective approach for the treatment of neuropathic pain and its comorbidity depression.
References
Miller LR, Cano A. Comorbid chronic pain and depression: who is at risk? J Pain. 2009;10:619–27.
Dworkin RH, Gitlin MJ. Clinical aspects of depression in chronic pain patients. Clin J Pain. 1991;7:79–94.
Finnerup NB, Otto M, McQuay HJ, Jensen TS, Sindrup SH. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118:289–305.
Dharmshaktu P, Tayal V, Kalra BS. Efficacy of antidepressants as analgesics: a review. J Clin Pharmacol. 2012;52:6–17.
Walker AK, Kavelaars A, Heijnen CJ, Dantzer R. Neuroinflammation and comorbidity of pain and depression. Pharmacol Rev. 2014;66:80–101.
Lees JG, Fivelman B, Duffy SS, Makker PG, Perera CJ, Moalem-Taylor G. Cytokines in neuropathic pain and associated depression. Mod Trends Pharm. 2015;30:51–66.
Apkarian AV, Lavarello S, Randolf A, Berra HH, Chialvo DR, Besedovsky HO, et al. Expression of IL-1beta in supraspinal brain regions in rats with neuropathic pain. Neurosci Lett. 2006;407:176–81.
Norman GJ, Karelina K, Zhang N, Walton JC, Morris JS, Devries AC. Stress and IL-1beta contribute to the development of depressive-like behavior following peripheral nerve injury. Mol Psychiatry. 2010;15:404–14.
Knerlich-Lukoschus F, Noack M, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J. Spinal cord injuries induce changes in CB1 cannabinoid receptor and C-C chemokine expression in brain areas underlying circuitry of chronic pain conditions. J Neurotrauma. 2011;28:619–34.
Cazareth J, Guyon A, Heurteaux C, Chabry J, Petit-Paitel A. Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. J Neuroinflamm. 2014;11:132.
Martuscello RT, Spengler RN, Bonoiu AC, Davidson BA, Helinski J, Ding H, et al. Increasing TNF levels solely in the rat hippocampus produces persistent pain-like symptoms. Pain. 2012;153:1871–82.
Minami M, Katayama T, Satoh M. Brain cytokines and chemokines: roles in ischemic injury and pain. J Pharmacol Sci. 2006;100:461–70.
Connor TJ, Song C, Leonard BE, Merali Z, Anisman H. An assessment of the effects of central interleukin-1beta, -2, -6, and tumor necrosis factor-alpha administration on some behavioural, neurochemical, endocrine and immune parameters in the rat. Neuroscience. 1998;84:923–33.
O’Connor JC, Andre C, Wang Y, Lawson MA, Szegedi SS, Lestage J, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. 2009;29:4200–9.
Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW. Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures. J Neuroinflamm. 2010;7:43.
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–108.
Guyon A, Skrzydelski D, De Giry I, Rovere C, Conductier G, Trocello JM, et al. Long term exposure to the chemokine CCL2 activates the nigrostriatal dopamine system: a novel mechanism for the control of dopamine release. Neuroscience. 2009;162:1072–80.
Wakida N, Kiguchi N, Saika F, Nishiue H, Kobayashi Y, Kishioka S. CC-chemokine ligand 2 facilitates conditioned place preference to methamphetamine through the activation of dopamine systems. J Pharmacol Sci. 2014;125:68–73.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8.
Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–9.
Chen YW, Rada PV, Butzler BP, Leibowitz SF, Hoebel BG. Corticotropin-releasing factor in the nucleus accumbens shell induces swim depression, anxiety, and anhedonia along with changes in local dopamine/acetylcholine balance. Neuroscience. 2012;206:155–66.
Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, et al. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science. 2014;345:535–42.
Chang PC, Pollema-Mays SL, Centeno MV, Procissi D, Contini M, Baria AT, et al. Role of nucleus accumbens in neuropathic pain: linked multi-scale evidence in the rat transitioning to neuropathic pain. Pain. 2014;155:1128–39.
Ren W, Centeno MV, Berger S, Wu Y, Na X, Liu X, et al. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nat Neurosci. 2016;19:220–2.
Hikida T, Yawata S, Yamaguchi T, Danjo T, Sasaoka T, Wang Y, Pathway-specific modulation of nucleus accumbens in reward and aversive behavior via selective transmitter receptors. Proc Natl Acad Sci USA. 2013;110:342–7.
Loureiro M, Kramar C, Renard J, Rosen LG, Laviolette SR. Cannabinoid transmission in the hippocampus activates nucleus accumbens neurons and modulates reward and aversion-related emotional salience. Biol Psychiatry. 2016;80:216-25.
Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126:745–61.
Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–2.
Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–62.
Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88.
Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–97.
Jing PB, Cao DL, Li SS, Zhu M, Bai XQ, Wu XB, et al. Chemokine receptor CXCR3 in the spinal cord contributes to chronic itch in mice. Neurosci Bull. 2018;34:54–63.
Wu X, Shi M, Wei C, Yang M, Liu Y, Liu Z, et al. Potentiation of synaptic strength and intrinsic excitability in the nucleus accumbens after 10 days of morphine withdrawal. J Neurosci Res. 2012;90:1270–83.
Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–23.
Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, et al. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. 2009;29:8051–62.
Goffer Y, Xu D, Eberle SE, D’Amour J, Lee M, Tukey D, et al. Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. J Neurosci. 2013;33:19034–44.
Jiang BC, He LN, Wu XB, Shi H, Zhang WW, Zhang ZJ, et al. Promoted interaction of C/EBPalpha with demethylated Cxcr3 gene promoter contributes to neuropathic pain in mice. J Neurosci. 2017;37:685–700.
Standaert DG, Friberg IK, Landwehrmeyer GB, Young AB, Penney JB Jr.. Expression of NMDA glutamate receptor subunit mRNAs in neurochemically identified projection and interneurons in the striatum of the rat. Brain Res Mol Brain Res. 1999;64:11–23.
Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB, Jr. .Young AB, NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–307.
Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Science’s STKE: Signal Transduct Knowl Environ. 2004;2004:re16.
Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400.
Sanz-Clemente A, Nicoll RA, Roche KW. Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist. 2013;19:62–75.
Yang YL, Su YW, Ng MC, Chao PK, Tung LC, Lu KT. Extract of Ginkgo biloba EGb761 facilitates extinction of conditioned fear measured by fear-potentiated startle. Neuropsychopharmacology. 2007;32:332–42.
Kroenke K, Bair MJ, Damush TM, Wu J, Hoke S, Sutherland J, et al. Optimized antidepressant therapy and pain self-management in primary care patients with depression and musculoskeletal pain: a randomized controlled trial. JAMA. 2009;301:2099–110.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56.
Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, Evans B, et al. Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol Psychiatry. 2017;22:1512–9.
Francis TC, Lobo MK. Emerging role for nucleus accumbens medium spiny neuron subtypes in depression. Biol Psychiatry. 2017;81:645–53.
Muir J, Lorsch ZS, Ramakrishnan C, Deisseroth K, Nestler EJ, Calipari ES, et al. In vivo fiber photometry reveals signature of future stress susceptibility in nucleus accumbens. Neuropsychopharmacology. 2018;43:255–63.
Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, et al. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12:602–10.
Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, et al. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–49.
Kim Y, Cho HY, Ahn YJ, Kim J, Yoon YW. Effect of NMDA NR2B antagonist on neuropathic pain in two spinal cord injury models. Pain. 2012;153:1022–9.
Slack S, Battaglia A, Cibert-Goton V, Gavazzi I. EphrinB2 induces tyrosine phosphorylation of NR2B via Src-family kinases during inflammatory hyperalgesia. Neuroscience. 2008;156:175–83.
Swartjes M, Morariu A, Niesters M, Aarts L, Dahan A. Nonselective and NR2B-selective N-methyl-D-aspartic acid receptor antagonists produce antinociception and long-term relief of allodynia in acute and neuropathic pain. Anesthesiology. 2011;115:165–74.
Gu X, Zhang J, Ma Z, Wang J, Zhou X, Jin Y, et al. The role of N-methyl-D-aspartate receptor subunit NR2B in spinal cord in cancer pain. Eur J Pain (Lond, Engl). 2010;14:496–502.
Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–48.
Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705.
Woolf CJ, Costigan M, Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–7730.
Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol life Sci. 2017;74:3275–91.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (NSFC 31671091, 81571070, 81771197, and 31700899), the Natural Science Foundation of Jiangsu Province (BK20171255), and the Qing-Lan Project of Jiangsu Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wu, XB., Jing, PB., Zhang, ZJ. et al. Chemokine receptor CCR2 contributes to neuropathic pain and the associated depression via increasing NR2B-mediated currents in both D1 and D2 dopamine receptor-containing medium spiny neurons in the nucleus accumbens shell. Neuropsychopharmacol 43, 2320–2330 (2018). https://doi.org/10.1038/s41386-018-0115-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0115-8
This article is cited by
-
Spinal MCP-1 Contributes to Central Post-stroke Pain by Inducing Central Sensitization in Rats
Molecular Neurobiology (2023)
-
Progranulin from different gliocytes in the nucleus accumbens exerts distinct roles in FTD- and neuroinflammation-induced depression-like behaviors
Journal of Neuroinflammation (2022)
-
Mechanistic insights into the role of the chemokine CCL2/CCR2 axis in dorsal root ganglia to peripheral inflammation and pain hypersensitivity
Journal of Neuroinflammation (2021)
-
Targeting the CCL2-CCR2 axis in depressive disorders
Pharmacological Reports (2021)
-
CCL2/CCR2 Contributes to the Altered Excitatory-inhibitory Synaptic Balance in the Nucleus Accumbens Shell Following Peripheral Nerve Injury-induced Neuropathic Pain
Neuroscience Bulletin (2021)
