
Abstract
Due to the efficacy of immune checkpoint inhibitor therapy in tumors with deficient mismatch repair, there has been a surge in demand for mismatch repair deficiency testing in various tumor types. Mismatch repair deficiency is not known to play a significant role in the pathogenesis of sarcomas, and the utility of testing these tumor types is not established. This study aimed to determine the frequency, pattern, and clinicopathologic correlates of mismatch repair deficiency in sarcomas. Three hundred and four sarcomas were profiled using a genomic platform that employs massively parallel sequencing to interrogate 447 cancer-associated genes. Mismatch repair status was evaluated by determining the number of small insertion/deletion events occurring in homopolymer regions per megabase of exonic sequence data across all genes. Molecular characteristics of mismatch repair-deficient sarcomas were compared to mismatch repair-deficient carcinomas (n = 70) also identified using the sequencing panel. Seven sarcomas (2.3%) were classified as mismatch repair-deficient: four unclassified sarcomas, and one each of pleomorphic rhabdomyosarcoma, epithelioid leiomyosarcoma and malignant PEComa. One patient had an established diagnosis of Lynch syndrome. In the remaining patients, the mismatch repair gene mutation was confirmed or suspected to be somatic. Mismatch repair immunohistochemistry confirmed the mismatch repair-deficiency status of all cases with alterations in the tested proteins. As expected, mismatch repair-deficient sarcomas showed a significantly elevated tumor mutation burden relative to mismatch repair-proficient sarcomas (median 16 versus 4.6, p < 0.001). However, in comparison to mismatch repair-deficient carcinomas, mismatch repair-deficient sarcomas showed a lower tumor mutation burden (median 28 versus 16, p = 0.006) and a significantly greater degree of chromosomal instability. Among mismatch repair-deficient sarcomas, PD-L1 was variably expressed on tumor-associated macrophages but not on tumor cells. Three patients received pembrolizumab: two progressed and one has stable disease with five months follow-up. Mismatch repair deficiency in histologically classifiable sarcomas is rare (1%) and is more common in unclassified sarcomas (10%). Additional study is required to determine the predictive role of mismatch repair-deficiency in sarcomas for immunotherapy.
Similar content being viewed by others


Introduction
Sarcomas comprise a diverse group of solid tumors that may arise at any anatomic site, with a predilection for soft tissue and bone primary locations. More than fifty different types of sarcoma have been defined based on specific histologic, immunohistochemical, and/or genomic features, including chromosomal translocations, amplification events, or point mutations [1]. Despite the increasingly well-defined genomic landscape of sarcomas, treatment options remain limited. Surgery is the therapeutic mainstay for limited stage/localized disease. Neoadjuvant or adjuvant radiotherapy offers some benefit in certain histotypes [2,3,4]; however, outcomes are generally poor for patients with advanced disease. Few targeted therapy options are available, as only a very small subset of these tumors have a targetable driver event, such as KIT or PDGFR activation in gastrointestinal stromal tumor or ALK rearrangement in inflammatory myofibroblastic tumor. There is a significant need, therefore, to identify additional treatment approaches for this rare group of tumors.
Immune checkpoint inhibitor therapy has demonstrated efficacy across subsets of patients with solid tumors with deficient mismatch repair/high level microsatellite instability, and the United States Food and Drug Administration has approved PD-1 inhibitor therapy in this context, irrespective of pathologic diagnosis (https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560040.htm). As a result, there has been a surge in demand for mismatch repair and microsatellite instability testing in various solid tumor types. Sarcomas, in particular liposarcoma and leiomyosarcoma, have been recognized within the spectrum of malignancies occurring in Lynch Syndrome patients and demonstrate mismatch repair-deficiency/microsatellite instability in this context [5]. The rate of tumoral mismatch repair-deficiency among unselected cohorts of sarcoma patients has not been established, but appears low (fewer than 2% of cases) [6,7,8,9,10,11].
In a phase 2 study of the PD-1 inhibitor pembrolizumab in sarcoma, 5% of bone sarcomas and 18% of soft tissue sarcomas demonstrated objective responses, including 40% of patients with undifferentiated pleomorphic sarcomas [12]. PD-L1 expression by immunohistochemistry was observed in only 4% of the overall cohort, but notably its expression was restricted to undifferentiated pleomorphic sarcoma, including in two patients with objective response; mismatch repair status was not reported. Separately, one patient with mismatch repair-deficient/microsatellite instability-high sarcoma receiving pembrolizumab within a clinical trial experienced progressive disease (http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf). It is not yet clear which biomarkers predict efficacy of PD-1 inhibitors in sarcoma; however, given the promising responses to PD-1 inhibitors in diverse tumor types with mismatch repair-deficiency and the availability of an approved therapy in this context, it is of critical importance to define the patterns of mismatch repair-deficiency in sarcomas and to determine the role of this mutational signature as a biomarker for response to immunotherapy.
Using a validated next generation sequencing-based tool for mismatch repair-deficiency detection, this study aimed to determine the frequency, pattern, and clinicopathologic correlates of mismatch repair deficiency in an unselected cohort of adult soft tissue and bone sarcomas.
Materials and methods
Three hundred and four sarcomas or mesenchymal neoplasms of intermediate biologic potential in adults were analyzed as part of a research study or as part of clinical care at a single institution (Table 1). This study was approved by the Dana-Farber Cancer Institute Institutional Review Board. Tumors were profiled using a genomic platform that employs massively parallel sequencing to interrogate 447 cancer-associated genes for detection of single nucleotide and insertion-deletion variants, copy number alterations, and structural variants. Mismatch repair status was evaluated by determining the number of small insertion/deletion events occurring in homopolymer regions per megabase of exonic sequence covered. A threshold of >1.5 such events per Mb was used to classify cases as mismatch repair-deficient, modified from a previously validated method in our laboratory [13]. Separately, tumor mutational burden was calculated as the number of non-synonymous somatic mutations per megabase of exonic sequence data. The number of copy number alterations per tumor was defined as the number of genes per tumor that were assigned a non-neutral copy number status based upon an internally developed copy number assessment algorithm and molecular pathologist review. This count was divided by the total number of genes on the panel to generate a percentage of copy number altered genes per tumor.
For comparison of tumor mutation burden and copy number levels to other tumor types, the Dana-Farber cBioPortal database was queried to identify all cases (n = 2610) from the current version of OncoPanel that had an assigned mismatch repair status of either proficient or deficient based upon the aforementioned algorithm and a tumor content of ≥30%, the minimum validated threshold for copy number analysis by OncoPanel. Of the 304 sarcomas matching these criteria, 286 had adequate tumor content and available copy number data for tumor mutation burden and copy number comparisons. For analysis of TP53 variant effect, all nonsense, frameshift, and canonical splice site alterations (including p.T125T) were classified as deleterious. Additionally, all missense variations annotated as having a non-functional or partially functional transactivation class as defined by the IARC TP53 database (R19) were classified as deleterious [14]. All remaining TP53 variants were considered functional. A two-sample Wilcoxon test was used to evaluate differences in tumor mutation burden and copy number alterations between tumor types. All data analysis was performed in R version 3.4.3 (R Foundation for Statistical Computing, Vienna, Austria).
Mismatch repair immunohistochemistry was performed on all mismatch repair-deficient cases. Immunohistochemistry was conducted after pressure cooker heat induced epitope retrieval (0.01 mol/L citrate buffer, ph 6.0) on 4-mm thick formalin-fixed, paraffin-embedded tissue sections using mouse anti-MLH1 monoclonal antibody (1:100 dilution; clone ES05; Novocastra, Buffalo Grove, IL), mouse anti-MSH2 monoclonal antibody (1:150 dilution; clone FE11; Calbiochem, San Diego, CA), mouse anti-PMS2 monoclonal antibody (1:50 dilution; clone MRQ-28; Cell Marque, Rocklin, CA), and mouse anti-MSH6 monoclonal antibody (1:50 dilution; clone PU29; Novocastra, Buffalo Grove, IL) using the Envision Plus Detection System (Dako, Carpinteria, CA). For mismatch repair proteins, expression was considered lost when there was complete absence of nuclear staining in tumor cells, in the presence of intact staining in non-neoplastic cells, which act as positive internal control. For mismatch repair-deficient cases, immunohistochemistry was performed for PD-L1 (1:100 dilution; clone EIL3N; Cell Signaling), CD3 (rabbit anti-CD3 polyclonal antibody, 1:300 dilution, Dako), CD8 (1:200 dilution; clone CD8/144B; Dako), and p53 (1:500 dilution; clone D07; Dako). PD-L1 was scored according to the percentage of tumor cells and/or infiltrating immune cells showing membranous expression of PD-L1 based on pathologist review [15]. For CD3 and CD8, the percentage of positive cells relative to all tumor-infiltrating immune cells was estimated. For p53, multifocal or scattered weak nuclear staining pattern was considered consistent with a “wild type” pattern, and diffuse strong nuclear staining considered consistent with a “mutant” pattern [16].
Microsatellite instability polymerase chain reaction (PCR) was performed on tumor-derived DNA using a 5 marker microsatellite panel as previously reported [13].
Histologic evaluation and detailed clinical review to include family history, treatment history, and outcome data was performed for cases identified as mismatch repair-deficient.
Results
Molecular screening for mismatch repair deficiency
Three hundred and four mesenchymal neoplasms (including 40 unclassified sarcomas or unclassified malignant neoplasms most consistent with sarcoma) were analyzed, from 145 females and 159 males and with a median patient age of 57.5 (range 18–90 yrs). Seven tumors (2.3%) were classified as mismatch repair-deficient based on the number of small insertion/deletion events occurring in homopolymer regions per megabase of exonic sequence data, as described in the methods section. The tumor mutational burden of the seven cases ranged from 9.1–25.856 (Table 2 and Fig. 1). Affected mismatch repair genes included MSH2 (including via EPCAM deletion), MSH6, PMS2, and MLH3; specific mutations are detailed in Table 2.

Comparison of tumor mutational burden between mismatch repair-proficient and deficient sarcomas and all mismatch repair-proficient and deficient tumors (a). Mismatch repair-deficient sarcomas have a significantly elevated tumor mutational burden relative to mismatch repair-proficient sarcomas and other mismatch repair-proficient tumor types (Pan-Cancer), although they have a lower mutational burden compared to other mismatch repair-deficient tumor types (predominantly tumors of gastrointestinal and endometrial origin). Comparison of percentage of targeted genes with copy number alterations between mismatch repair-proficient and deficient sarcomas and all mismatch repair-proficient and deficient tumors (b). Mismatch repair-deficient sarcomas have a significantly higher percentage of copy number alterations in targeted genes compared to other mismatch repair-deficient tumor types. In both panels, center bar within each box plot represents the median, upper and low box boundaries represent boundaries of the second and third quartiles, and whiskers denote values ±1.5 times the inter-quartile range. Box plots are superimposed on violin plots representing the overall distribution density of values
One patient was known to have Lynch syndrome, and germline testing had confirmed the presence of an MSH2 mutation. For the 6 non-Lynch syndrome patients, the allele fraction of the identified mismatch repair gene mutation was consistent with somatic origin. Dedicated germline testing was performed in an additional two patients and no mutations were detected, confirming the somatic status.
Mismatch repair immunohistochemistry and/or microsatellite instability polymerase chain reaction confirmed the mismatch repair-deficient status of all cases and showed protein expression patterns corresponding to the molecular abnormalities (see below).
Clinical demographics and presentation of patients with mismatch repair-deficient sarcomas
Four mismatch repair-deficient cases occurred in male and three in female patients (Table 2). The median age of patients with mismatch repair-deficient tumors was 64 years (range 36–79 yrs). The patient with Lynch syndrome was the youngest. The anatomic locations of primary tumors were as follows: uterus (n = 2), deep soft tissues of the extremities or trunk (n = 3), anterior mediastinum (n = 1), and renal/perirenal (n = 1). The patient with known Lynch syndrome had a family history of several first-degree relatives with colon cancer and thyroid cancer. One patient had a daughter with Cowden syndrome and breast cancer, however the patient himself underwent germline testing and did not have a PTEN mutation detected. The remaining five patients did not have a family history suggestive of a specific inherited tumor syndrome; one patient had first-degree family relatives with throat cancer, thyroid cancer, and non-Hodgkin lymphoma; one had first-degree family members with prostate cancer and lung cancer (with smoking histories). For all patients, this was their first diagnosed malignancy.
Molecular, histologic and immunohistochemical correlations
The tumor mutational burden in mismatch repair-deficient sarcomas was significantly higher than that observed in mismatch repair-proficient sarcomas (median mutational burden 16 versus 4.6, p < 0.001) (Fig. 1a). Notably, however, the mutational burden for mismatch repair-deficient sarcomas was significantly lower than the mutational burden for other mismatch repair-deficient tumor types (median tumor mutational burden 16 versus 28, p = 0.006). The mutational burden for mismatch repair-proficient sarcomas was similar to that of all mismatch repair-proficient tumor types (median tumor mutational burden 4.56 versus 5.32). In contrast to other mismatch repair-deficient tumor types, sarcomas with mismatch repair-deficiency had a high level of copy number alterations (median 24.6% altered genes per tumor), consistent with chromosomal instability (Fig. 1b). This is not significantly different from mismatch repair-proficient sarcomas, many of which show chromosomal instability as a dominant feature [6], but is significantly different from the pattern seen in other mismatch repair-deficient carcinomas (median 4.9% altered genes per tumor, p = 0.002). The three mismatch repair-proficient tumors with the highest mutation burdens (angiosarcoma; unclassified dermal sarcoma; unclassified radiation-associated sarcoma) were all cutaneous in origin and displayed a predominance of C/G > T/A and CC/GG > TT/AA substitutions most characteristic of ultraviolet light exposure.
Of classified sarcoma types, three (1.1%) cases were mismatch repair-deficient, whereas among unclassified sarcomas four (10%) were mismatch repair-deficient. The patient with Lynch syndrome had a pleomorphic rhabdomyosarcoma arising in deep soft tissue of the lower extremity. Other histologic diagnoses included four undifferentiated sarcomas/malignant neoplasms: pleomorphic malignant neoplasm most suggestive of undifferentiated sarcoma arising in the anterior mediastinum (Fig. 2); malignant epithelioid and spindle cell neoplasm involving the kidney, undifferentiated sarcoma with epithelioid features occurring in deltoid muscle, and undifferentiated pleomorphic sarcoma of deep soft tissue of the back. For one of these cases (case 3), after review of a subsequent resection it was felt that the tumor may have represented a spindle cell angiosarcoma due to the presence of nuclear ERG expression. For the other three unclassified tumors, the histologic appearances, immunohistochemical profiles (to evaluate for hematolymphoid neoplasm, carcinoma, or melanoma where relevant), anatomic location and clinical histories all favored diagnoses of undifferentiated/unclassified sarcoma. The two female patients in this study both had uterine primary tumors; malignant PEComa (Fig. 3) and epithelioid leiomyosarcoma, both of which showed classic histologic features of each tumor type.

Pleomorphic malignant neoplasm arising in the mediastinum of a 79-year old man. The tumor was composed of atypical cells admixed with a dense lymphoplasmacytic infiltrate with scattered eosinophils that in many areas obscured the neoplastic cells (a). In areas of less inflammation it is easier to identify the large pleomorphic atypical cells (b). The tumor had an MSH2 nonsense mutation and showed loss of expression of MSH2 (c) and MSH6 (d) and intact staining for MLH1 (e) and PMS2 (f)

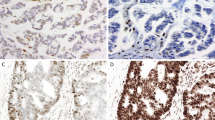
Malignant PEComa of the uterus composed of large pleomorphic atypical epithelioid cells with granular and eosinophilic cytoplasm (a). There was two copy deletion of MSH2, and loss of expression of both MSH2 (b) and MSH6 (c)
For all cases, the immunohistochemical profile reflected the underlying genetic change: complete loss of MSH2 and MSH6 expression in tumor cells in the four tumors with MSH2 mutations or copy loss (Figs. 2 and 3); complete loss of MSH6 expression (only) in the tumor with MSH6 mutation (Fig. 4); complete loss of PMS2 expression (only) in the tumor with a PMS2 mutation. Notably, there was intact staining for MLH1, PMS2, MSH2 and MSH6 in the tumor with MLH3 mutation and PMS2 deep deletion, despite microsatellite instability confirmed by PCR.

Unclassified sarcoma arising within the deltoid muscle of a 64-year old man (a) with MSH6 mutation and loss of MSH6 expression (b), showing PD-L1 expression in macrophages at the tumor-stroma interface (c). Immunohistochemistry for P53 showed aberrant diffuse strong nuclear positivity (d), consistent with the presence of a TP53 c.818 G > A nonsense mutation
Three tumors (cases 1–3) had a prominent inflammatory cell infiltrate primarily composed of lymphocytes and plasma cells present within pre-treatment specimens (Fig. 2). One case had a prominent histiocytic infiltrate with smaller numbers of lymphocytes in the post-neoadjuvant therapy resection (case 7). The remaining three cases did not have a significant intratumoral inflammatory infiltrate.
Five of seven cases showed PD-L1 expression in tumor-infiltrating macrophages, ranging from 2–50% of immune cells (Table 2). No staining was seen in tumor cells in any case. Intratumoral CD3 + T lymphocytes ranged from <5 to 30% of tumor-infiltrating immune cells and CD8 + T lymphocytes from <2 to 40%. The number of CD3 and CD8 positive T-lymphocytes increased corresponding to increasing PD-L1 expression. In one case the inflammatory cell infiltrate showed a striking tumor-stromal interface distribution (Fig. 4c).
To investigate the mechanism driving chromosomal instability in these mismatch repair-deficient sarcomas, we evaluated the mutational status of TP53, RB1, and ATRX—all genes highly implicated in genomic instability in sarcoma and other tumor types—as well as p53 protein status [6]. Three cases had hotspot TP53 mutations (p.R175H, p.R273H, and one case with both TP53 p.R273H and p.T125T) and showed diffuse strong staining in tumor cells consistent with a mutant pattern of p53 protein expression (Fig. 4d). Two TP53-mutated cases had ATRX co-mutations (p.R1022* and p.R1504*). The remaining cases showed a wild-type staining pattern, and lacked TP53 mutations. One case showed a RB1 mutation (p.Q436*). Although TP53-mutant mismatch repair-deficient sarcomas did not exhibit a significantly higher percentage of genes with copy alterations as compared to TP53 functional mismatch repair-deficient sarcomas, this comparison was likely limited by the small overall number of mismatch repair-deficient sarcomas and may be confounded by the presence of other DNA damage-repair pathway alterations detected in all but one of the examined cases (Table 2) [17]. To investigate whether TP53 status can, in general, contribute to aneuploidy even in a mismatch repair-deficient state, we analyzed the effect of TP53 mutations in our pan-cancer cohort of mismatch repair-deficient tumors. Notably, the presence of a deleterious TP53 mutation within a mismatch repair-deficient tumor is significantly correlated with an increased percentage of genes with copy number alterations as compared to a mismatch repair-deficient tumor with a functional TP53 status (11.8% versus 3.8%, p = 0.01, Fig. 5), suggesting contributions from both mismatch repair deficiency and chromosomal aneuploidy to overall genomic instability in mismatch repair-deficient TP53-mutant tumors regardless of type.

Comparison of percentage of genes with copy number alterations between TP53 functional and mutant tumors stratified by mismatch repair status. Within mismatch repair-proficient tumors of any type (n = 2540), the presence of a deleterious TP53 mutation is associated with a significant increase in the percentage of genes with copy number alterations (12.3% versus 23.0%, p < 0.001). Similarly, within mismatch repair-deficient tumors of any type (n = 70), the presence of a deleterious TP53 mutation is also associated with a significant increase in the percentage of genes with copy number alterations (3.8% versus 11.8%, p = 0.01). Center bar within each box plot represents the median, upper, and low box boundaries represent boundaries of the second and third quartiles, and whiskers denote values ±1.5 times the inter-quartile range. Box plots are superimposed on violin plots representing the overall distribution density of values
Treatment data and clinical outcomes
All patients underwent surgical resection of the primary tumor (Table 3). Two patients (cases 4 & 7) received neoadjuvant radiation therapy, and one patient (case 1) received neoadjuvant chemoradiation therapy. Two patients (cases 2 & 3) received adjuvant chemotherapy or radiation therapy alone. Both patients with uterine primaries (cases 5&6) have had the longest clinical courses, and underwent surgical resections, multiple agent chemotherapy, and pelvic radiation prior to commencement of pembrolizumab.
The follow-up duration ranged from 9 to 60 months. Three patients remain free of disease 13, 14, and 25 months after initial diagnoses. Two patients alive with extensive disease received pembrolizumab (both patients with uterine primaries). There has been no measurable response to treatment thus far in one patient, but this patient has had stable disease for 5 months, while another progressed after 4 months of pembrolizumab and is now receiving other chemotherapy. Two patients developed distant soft tissue and lung metastases and died of disease at 9 and 15 months after the initial diagnoses—one (patient 3) received pembrolizumab for persistent/progressive disease after surgical resection but had rapid progression, the other patient did not receive pembrolizumab (patient 7).
Discussion
Significant advances in understanding the molecular genetics of different sarcoma types have been made in the last decade, resulting in improved tumor classification and improved prognostication. However, there is still a significant gap between our knowledge of the molecular pathogenesis of different mesenchymal tumor types and targeted treatment approaches [18]. Recent additions to the chemotherapeutic approach to sarcoma include olaratumab, pazopanib, trabectedin, and eribulin, which are usually administered with or following doxorubicin, and while these have resulted in some responses, the overall improvements in survival have been at best modest [2, 19,20,21,22]. For metastatic or unresectable disease there are still very limited effective treatment options, and many sarcomas, particularly unclassified sarcomas, are relatively resistant to standard sarcoma chemotherapeutic regimes [3].
Sarcomas are classified based on morphologic, immunohistochemical and molecular genetic characteristics, and most will show a line of differentiation that may resemble a normal cell type, e.g., smooth muscle differentiation in leiomyosarcoma or vascular differentiation in angiosarcoma [1]. However, even after careful histologic examination and ancillary testing, a significant subset of sarcomas will remain unclassified. Undifferentiated pleomorphic/spindle cell sarcomas represent a significant subset of all sarcomas, particularly in adults over 50 years of age, and most have complex karyotypes with loss of tumor suppressor genes such as TP53 and RB1. Less is known about the molecular pathology of this group when compared to sarcomas with simple karyotypes, which include tumors characterized by recurrent translocations, amplifications or single point mutations that result in activation of specific genes/pathways, several of which have effective therapeutic targets.
The efficacy of immune checkpoint inhibitor therapy and recent approval of pembrolizumab across all solid tumors with demonstrable mismatch repair deficiency has led to a surge in demand for mismatch repair testing and has changed current approaches to the evaluation and management of many different tumor types. The utility of global mismatch repair testing in sarcoma has not been established, despite a previous report that some sarcomas respond to pembrolizumab [12]. The identification of mismatch repair deficiency in even a small subset of sarcomas offers a window of opportunity for these otherwise relatively chemoresistant tumors, yet mismatch repair status has been studied in only small numbers of sarcomas, and usually by immunohistochemistry or PCR analysis [5,5,7]. There is, therefore, a need to characterize the frequency and pattern of mismatch repair deficiency in sarcomas—a task more readily undertaken by employing comprehensive sequencing approaches.
Mismatch repair deficiency was detected by next generation sequencing in 2.3% of our total cohort of sarcomas, and was uncommon in classified sarcoma types at 1% but enriched to 10% when evaluating undifferentiated pleomorphic/spindle cell sarcomas and unclassified malignant neoplasms thought best to represent sarcoma. Among classifiable sarcoma types in this study, mismatch repair deficiency was limited to one male patient with pleomorphic rhabdomyosarcoma in the context of Lynch syndrome and two female patients with uterine sarcomas—leiomyosarcoma and malignant PEComa, the latter of which contained conventional leiomyosarcoma-like areas. Loss of mismatch repair protein expression has previously been described in uterine leiomyosarcomas in two patients, neither of whom had a family history suggestive of a hereditary cancer syndrome [10]. In contrast, pleomorphic rhabdomyosarcoma has previously been associated with Lynch syndrome [23, 24]. Of the four mismatch repair-deficient undifferentiated sarcomas, two were initially diagnosed as malignant neoplasms most likely representing sarcomas based on anatomic location, histologic appearances and ancillary studies, and clinical history. The frequency of mismatch repair deficiency among unclassified sarcomas was higher than one might have anticipated given that mismatch repair defects are not known to play a significant role in the pathogenesis of sarcomas. This finding raises the possibility that mismatch repair deficiency plays a role in the pathogenesis of this tumor type and may offer a window of opportunity for treatment of this relatively chemoresistant group of sarcomas. The results are particularly notable when taken in context with findings from a recent study that showed the greatest benefit from pembrolizumab was seen in unclassified sarcomas (partial or complete response in 4 of 10 patients), but mismatch repair status was not reported [12]. In the current study, no sarcomas characterized by simple karyotypes. e.g., translocation associated sarcomas, were found to be mismatch repair-deficient. Mismatch repair deficiency was rare among leiomyosarcoma and not detected among the other common mesenchymal neoplasms, including well/dedifferentiated liposarcoma and GIST.
Somatic sequencing from all seven cases demonstrated a clear genetic cause for mismatch repair deficiency, including nonsense and frameshift alterations, and two copy deletion of either EPCAM, MSH2, or PMS2. Mismatch repair immunohistochemistry correlated well with the genomic findings, and immunohistochemistry may be a reasonable surrogate for sequencing, but in rare cases, mismatch protein expression may be intact despite evidence of mismatch repair-deficiency at the genomic level. Interestingly, no MLH1 methylation abnormalities were detected, and two patients had PMS2 alterations. Instead, the mutated genes were more typical of those seen in Lynch syndrome. As expected, the mismatch repair-deficient sarcomas displayed an elevated tumor mutational burden compared to mismatch repair-proficient cases, consistent with a DNA repair defect. Notably, the three mismatch repair-proficient tumors with the highest mutation burdens all displayed a mutational signature characteristic of ultraviolet light exposure, underscoring the fact that not all hypermutated sarcomas are mismatch repair-deficient.
The genomic correlates of microsatellite instability in sarcomas appear different from other well-characterized microsatellite instability-high tumor types, particularly colon and endometrial carcinomas. For one, the overall mutational burden of mismatch repair-deficient sarcomas, while significantly higher than mismatch repair-proficient sarcomas, is still lower than that of other mismatch repair-deficient tumors. In general, microsatellite instability-high colon and endometrial carcinomas—the most numerous and therefore best studied of the mismatch repair-deficient tumors—have few numeric chromosomal changes [25, 26]. In contrast, the number of copy alterations seen in mismatch repair-deficient sarcomas is strikingly higher than that of other mismatch repair-deficient tumors (predominantly gastrointestinal and endometrial carcinomas) sequenced in our institutional cohort. Four of the seven mismatch repair-deficient sarcomas identified here had mutations in TP53, RB1, and ATRX genes—recognized drivers of chromosomal instability in this tumor type.
A prominent inflammatory infiltrate was seen in three of seven cases prior to any treatment, and in one case after neoadjuvant chemoradiation. The remaining three cases lacked a conspicuous inflammatory infiltrate. The presence of inflammation alone is therefore unlikely to be helpful in identifying cases that may be mismatch repair-deficient, and further studies will be needed to establish the role of inflammatory infiltration in sarcoma as an independent biomarker for response to checkpoint blockade. PD-L1 staining in sarcomas has previously been shown to be uncommon and heterogenous across similar sarcoma types and alone does not predict response to pembrolizumab [12, 15, 27]. In other tumor types, an association between mismatch repair deficiency by immunohistochemistry and PD-L1 expression has been reported [11], however none of the mismatch repair-deficient sarcomas in this cohort showed tumor PD-L1 expression. Rather, PD-L1 expression was restricted to infiltrating immune cells. Interestingly, in a trial of metronomic pembrolizumab and cyclophosphamide in 57 patients with advanced soft tissue sarcomas, only one showed a partial response. This patient’s tumor (a solitary fibrous tumor) was notable for being the only case with >10% PD-L1 staining in infiltrating immune cells [27]. In the current cohort, of five mismatch repair-deficient tumors with PD-L1 expression, four had staining in ≥10% of immune cells.
Among mismatch repair-deficient tumors, mismatch repair-deficient sarcomas may have some distinctive features, including high levels of chromosomal instability. Chromosomal instability predicts poor outcomes in patients with non-small cell lung carcinoma [28], but the implications of the combined presence of mismatch repair deficiency and chromosomal instability for response to immunotherapies remains to be determined. Mismatch repair deficiency is thought to contribute to the evolution of potent mutation-associated neoantigens that can trigger an immune response, with implications for improved prognosis via immune surveillance and for efficacy of checkpoint blockade therapy [29]. There is emerging evidence that tumors may escape immune recognition—and therefore become resistant to immune checkpoint blockade—via elimination of clonal neoantigens through chromosomal deletion events and loss of heterozygosity, or as a result of mutations that interfere with HLA presentation [30]. One may speculate that the lack of response in the pembrolizumab-treated patients in this series could reflect in part a genomic instability-mediated mechanism of immune escape via elimination of neoantigens. Further studies employing whole exome sequencing will be required to determine the implications of genomic instability for neoantigen detection in sarcomas.
In light of the unique status of mismatch repair deficiency as a tumor-type agnostic FDA-approved biomarker for pembrolizumab therapy, knowledge of the expected frequency of mismatch repair deficiency and clinicopathologic correlates is critical, especially for aggressive tumors with limited other treatment options. Our series suggests that focused screening of undifferentiated sarcomas, where the rate of mismatch repair deficiency is 10%, may be a cost-effective strategy for identifying patients who may be eligible for on-label pembrolizumab therapy. Given the good concordance between mismatch repair immunohistochemistry, microsatellite instability, or broad genomic profiling, any of these approaches may be acceptable, depending on the practice setting. Of pembrolizumab-treated mismatch repair-deficient sarcoma patients in our series, one died of disease, one is alive with disease following progression, and one has stable disease on therapy. Given the small study size, it is difficult to draw conclusions about mismatch repair deficiency as a prognostic or predictive biomarker. Ultimately, the best biomarker of response to immunotherapy in sarcomas, and the contexts in which mismatch repair deficiency may be predictive, remain to be determined.
References
Gronchi A and Fletcher CDM. Tumors of Soft Tissue. In: Fletcher CBJ, Hogendoorn P, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2013, p. 13–8.
Gronchi A, Ferrari S, Quagliuolo V, Broto JM, Pousa AL, Grignani G, et al. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): an international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet Oncol. 2017;18:812–22.
Savina M, Le Cesne A, Blay JY, Ray-Coquard I, Mir O, Toulmonde M, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15:78.
von Mehren M, Randall RL, Benjamin RS, Boles S, Bui MM, Ganjoo KN, et al. Soft Tissue Sarcoma, version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:536–63.
Nilbert M, Therkildsen C, Nissen A, Akerman M, Bernstein I. Sarcomas associated with hereditary nonpolyposis colorectal cancer: broad anatomical and morphological spectrum. Fam Cancer. 2009;8:209–13.
Cancer Genome Atlas Research Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell . 2017;171:950–65 e28.
Cote GM, He J, Choy E. Next-generation sequencing for patients with sarcoma: a single center experience. Oncologist. 2018;23:234–42.
Davis JL, Grenert JP, Horvai AE. Loss of heterozygosity and microsatellite instability are rare in sporadic dedifferentiated liposarcoma: a study of 43 well-characterized cases. Arch Pathol Lab Med. 2014;138:823–7.
Ericson K, Engellau J, Persson A, Lindblom A, Domanski H, Akerman M, et al. Immunohistochemical loss of the DNA mismatch repair proteins MSH2 and MSH6 in malignant fibrous histiocytomas. Sarcoma. 2004;8:123–7.
Hoang LN, Ali RH, Lau S, Gilks CB, Lee CH. Immunohistochemical survey of mismatch repair protein expression in uterine sarcomas and carcinosarcomas. Int J Gynecol Pathol. 2014;33:483–91.
Kim ST, Klempner SJ, Park SH, Park JO, Park YS, Lim HY, et al. Correlating programmed death ligand 1 (PD-L1) expression, mismatch repair deficiency, and outcomes across tumor types: implications for immunotherapy. Oncotarget. 2017;8:77415–23.
Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18:1493–501.
Nowak JA, Yurgelun MB, Bruce JL, Rojas-Rudilla V, Hall DL, Shivdasani P, et al. Detection of mismatch repair deficiency and microsatellite instability in colorectal adenocarcinoma by targeted next-generation sequencing. J Mol Diagn. 2017;19:84–91.
Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–76.
D’Angelo SP, Shoushtari AN, Agaram NP, Kuk D, Qin LX, Carvajal RD, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2015;46:357–65.
Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih Ie M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011;24:1248–53.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 2018;23:239–254 e6.
George S. Evolving treatment of soft tissue sarcoma. J Natl Compr Canc Netw. 2017;15:733–6.
Demetri GD, von Mehren M, Jones RL, Hensley ML, Schuetze SM, Staddon A, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34:786–93.
Schoffski P, Chawla S, Maki RG, Italiano A, Gelderblom H, Choy E, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387:1629–37.
Tap WD, Jones RL, Van Tine BA, Chmielowski B, Elias AD, Adkins D, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388:488–97.
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–86.
Cranmer LD, Chen CC, Morgan S, Martino G, Ray J. Pleomorphic rhabdomyosarcoma in a patient with hereditary nonpolyposis colorectal cancer. J Clin Oncol. 2013;31:e108–10.
den Bakker MA, Seynaeve C, Kliffen M, Dinjens WN. Microsatellite instability in a pleomorphic rhabdomyosarcoma in a patient with hereditary non-polyposis colorectal cancer. Histopathology. 2003;43:297–9.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: a phase 2 clinical trial. JAMA Oncol. 2018;4:93–7.
Richard C, Fumet JD, Chevrier S, Derangere V, Ledys F, Lagrange A, et al. Exome analysis reveals genomic markers associated with better efficacy of nivolumab in lung cancer patients. Clin Cancer Res. 2018. 30154227. Epub ahead of print.
Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–20.
Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung. Cancer Cancer Discov. 2017;7:264–76.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
LMS: Consultant for Foghorn Therapeutics, LOXO Oncology, AstraZeneca Pharmaceuticals. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Doyle, L.A., Nowak, J.A., Nathenson, M.J. et al. Characteristics of mismatch repair deficiency in sarcomas. Mod Pathol 32, 977–987 (2019). https://doi.org/10.1038/s41379-019-0202-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0202-3
This article is cited by
-
Biomarkers for immune checkpoint inhibition in sarcomas – are we close to clinical implementation?
Biomarker Research (2023)
-
Pleomorphic rhabdomyosarcoma in a young adult harboring a novel germline MSH2 variant
Human Genome Variation (2022)
-
Uterine PEComas: correlation between melanocytic marker expression and TSC alterations/TFE3 fusions
Modern Pathology (2022)
-
Molecular and clinicopathological analysis revealed an immuno-checkpoint inhibitor as a potential therapeutic target in a subset of high-grade myxofibrosarcoma
Virchows Archiv (2022)
-
Myxofibrosarcoma harboring an MLH1 pathogenic germline variant associated with Muir-Torre syndrome: a case report
Hereditary Cancer in Clinical Practice (2021)
