
Abstract
Tubulocystic renal cell carcinoma, a unique tumor, was recently included as a new entity in the World Health Organization classification of renal tumors. It has variably been reported to be related to other renal cell carcinomas, including papillary renal cell carcinoma, fumarate hydratase-deficient carcinoma, and others, likely because many such carcinomas may show variable amounts of tubulocystic architecture. The published data characterizing the molecular features of these tumors are inconsistent. We studied nine “pure” tubulocystic renal cell carcinomas, as defined by International Society of Urologic Pathologists (ISUP) and World Health Organization (WHO), by targeted next-generation sequencing, and fluorescence in situ hybridization for X and Y chromosomes, to investigate if these show any unique characteristics or any overlap with known mutational/molecular profiles or copy number alterations in other subtypes of renal cell carcinoma. All nine tubulocystic carcinomas demonstrated combined losses at chromosome 9 and gains at chromosome 17, as well as, loss of chromosome Y (in 5/5). None of the tumors showed mutational profiles characteristic of other renal neoplasms, including those seen in fumarate hydratase-deficient renal cell carcinoma. Recurrent mutations in chromatin-modifying genes, KMT2C and KDM5C, were detected in two of nine tumors. Thus, tubulocystic renal cell carcinoma, if defined strictly, at the clinical and pathologic level, demonstrates genomic features distinct from other subtypes of renal cell carcinoma. These findings support the contention that tubulocystic renal cell carcinoma should be diagnosed only using strict morphological criteria and only when presenting in a “pure” form; presence of variable papillary, poorly differentiated, or other architectural patterns most likely do not belong to the category of tubulocystic renal cell carcinoma.
Similar content being viewed by others

Introduction
Tubulocystic renal cell carcinoma in the older literature has variably been described as Bellinian epithelioma and low grade collecting duct carcinoma [1, 2]. The term tubulocystic renal cell carcinoma was first used by Amin et al. [3] at the 2004 United States and Canadian Academy of Pathology annual meeting where they described a series of 29 cases defining the characteristics of this tumor; the series was subsequently published in 2009. The tumor is now included as a distinct entity in the WHO 2016 classification of tumors of the kidney [4]. Less than 100 cases of pure tubulocystic renal cell carcinoma have been reported in literature. These tumors are considered to be relatively less aggressive with metastasis reported in ~ 6% of the cases [4].
Both the International Society of Urologic Pathologists (ISUP) and World Health Organization (WHO) recommend that the term tubulocystic renal cell carcinoma be restricted to tumors that display the typical macroscopic and microscopic features [4, 5]. According to the ISUP, the term should not be used in situations in which there is a tubulocystic pattern admixed with the usual elements of papillary renal cell carcinoma or collecting duct carcinoma. Tubulocystic-like architecture may be variably present in many renal tumors, including renal oncocytoma, chromophobe renal cell carcinoma, fumarate hydratase-deficient renal cell carcinoma/hereditary leiomyomatosis renal cell carcinoma-associated renal cell carcinoma, and collecting duct carcinoma, among others [6,7,8]. Many publications on tubulocystic carcinomas have included tumors with areas showing papillary architecture [9, 10]. In recent years, there have been some publications describing tubulocystic renal cell carcinoma with high-grade or poorly differentiated areas exhibiting tubulopapillary and nonglandular (solid/sheet-like, nested, cord-like, rhabdoid) components in addition to areas showing tubulocystic architecture [11,12,13].
Molecular alterations of “pure” tubulocystic renal cell carcinoma (as defined by ISUP and WHO) remain largely unknown. Using Affymetrix X3P oligonucleotide microarray analysis, Amin et al. [3] reported that tubulocystic carcinoma overexpresses genes related to amino-acid metabolism and cell cycle, whereas underexpressing a number of biopolymer metabolism genes. A few studies on molecular/genetic features of tubulocystic renal cell carcinoma have suggested that the tumor is related to papillary renal cell carcinoma [9, 10]. However, these studies also included tumors with areas showing papillary architecture, and would not be considered to be “pure”. One recent study that concentrated on miRNA expression analysis, also reported on mutations detected by targeted next-generation sequencing [14]. Unfortunately, a number of the cases in this study lacked paired constitutional DNA samples for comparison, raising the question of whether the findings truly represent recurrent somatic driver events.
In this study, we aimed to clarify the molecular alterations of “pure” tubulocystic renal cell carcinoma and investigated the somatic mutations and copy number alterations using targeted next-generation sequencing in nine “pure” tubulocystic renal cell carcinomas fulfilling the current diagnostic recommendations of WHO and ISUP. As both these next-generation sequencing methods are not designed to detect numerical alterations in the Y chromosome, fluorescence in situ hybridization (FISH) assays were performed to evaluate such changes in sex chromosomes.
Materials and methods
Nine cases of “pure” tubulocystic renal cell carcinoma were included in this multi-institutional study approved by the respective institutional research boards. Five cases were obtained from the pathology archives of Memorial Sloan Kettering Cancer Center; the other four represented those from the control group of previously published study by Smith et al. [8] on “tubulocystic carcinoma with poorly differentiated areas”. All tumors were well circumscribed and had pure tubulocystic morphological features as defined previously [5], namely, small to intermediate-sized tubules admixed with larger cysts, lined by a single layer of flattened, cuboidal/columnar, and hobnail epithelium with enlarged and irregular nuclei, intermediate to large (WHO/ISUP nucleolar grade 3) nucleoli and fibrotic stroma.
Detailed histomorphologic features were evaluated by pathologists with expertize in genitourinary pathology (SKT, RM, and YBC); other pathological and clinical details, including sex, age, tumor laterality, gross features, focality, tumor size, and stage, were obtained from the pathology reports and patient charts. In all cases, tissues had been fixed in neutral-buffered formalin and embedded in paraffin as part of routine surgical pathology evaluation.
Immunohistochemical evaluation and analysis
Immunohistochemical staining for CK7, cytokeratin 34βE12, alpha methyl acyl co-A racemase (AMACR) and CD10 was performed on 5-micron-thick sections on the Ventana, Benchmark XT (Ventana Medical Systems, Oro Valley, AZ) immunohistochemical stainer, using standard protocols. The sections were deparaffinized and subjected to antigen retrieval before primary incubation with the antibody panel. Visualization of bound antibodies was performed using the peroxidase-labeled streptavidin–biotin system (DAKO, LSAB2 kit) with 3, 3- diaminobenzidine as a chromogen. Positive and negative controls were run concurrently and exhibited appropriate immunostaining.
Immunohistochemical staining for fumarate hydratase (FH) was performed using commercially available anti-FH mouse monoclonal antibody, clone J-13, Santa Cruz Biotechnology (Dallas, TX) as previously described [8]. An absence of FH immunohistochemical expression in the neoplastic cells, in the presence of a positive internal control in non-neoplastic cells, including inflammatory and endothelial cells, was interpreted as true negative expression/staining (loss or FH-deficient status). All other patterns of immunohistochemical expression with cytoplasmic and granular staining were considered as retained positivity/positive. Immunohistochemical staining for S-(2-succino)-cysteine (2SC) was performed using a polyclonal antibody as described previously [6, 15]. The 2SC immunohistochemical expression was assessed for intensity (1 + to 3 + ) and staining pattern (nuclear and cytoplasmic vs. cytoplasmic only); only 3 + intensity, nuclear, and cytoplasmic staining was interpreted as positive.
Molecular analysis
DNA was extracted from manually macro-dissected tumor and matched normal formalin-fixed paraffin-embedded tissue using QIAamp DNA formalin-fixed paraffin-embedded tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Four tumors were analyzed using the Ion Torrent Comprehensive Cancer Panel, a multiplexed PCR-based next-generation sequencing platform targeting ~ amplicons, including the complete coding sequence of 409 cancer-related genes, as previously reported [16,17,18]. Paired tumor and normal DNA samples of five other cases from Memorial Sloan Kettering Cancer Center were analyzed using the targeted capture-based next-generation sequencing (Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets, MSK-IMPACTTM) to identify somatic mutations and copy number alterations as described previously [19]. Both assays involve deep sequencing of all exons of oncogenes and tumor suppressor genes, whereas MSK-IMPACT TM also captures intergenic and intronic single-nucleotide polymorphisms (tiling probes), interspersed homogenously across the genome, aiding the accurate assessment of genome-wide copy number. The 410 genes included in MSK-IMPACTTM are listed in Supplementary Table 1. The allele-specific copy number analysis of MSK-IMPACTTM data were conducted using open-source (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing) tool [20]. The functional impact of the detected mutations was determined using OncoKB (http://oncokb.org), a precision oncology knowledge base maintained at Memorial Sloan Kettering Cancer Center [21], and separated into two categories: known or likely oncogenic vs. variant of unknown significance.
FISH analysis
FISH assay for X and Y chromosome was performed on five of the nine tumors. In all, 4 µm formalin-fixed paraffin-embedded tissue sections were used following standard protocols. The dual color probes used for the X and Y chromosomes (Abbott Molecular, Des Plaines, IL), labeled in Spectrum Orange and Spectrum Green, respectively, are specific for the centromere region of the X chromosome, and for the Yq12 regions. After applying the FISH probes to the tissue areas, both tissue and probes were co-denatured at 94 °C for 7 min, and then incubated at 37˚C overnight, followed by post-hybridization washing in 2×SSC/0.3% NP-40 at 77 °C for 2 min. Tissue sections were counterstained with 4′,6-diamidino-2-phenylindole. Signal analysis was performed in combination with morphology correlation within the marked tumor areas.
Results
The clinicopathologic features of the nine cases are summarized in Table 1. All nine patients were males. Seven of the tumors were staged as pT1a, 1 as pT2b, and 1 as pT3a. None of them had metastasis at the time of presentation.
The pathological features of all tumors are tabulated (Table 2). On gross examination, all tumors had spongy to grossly microcystic cut surface. All except one tumor were well circumscribed, but none showed encapsulation; the one non-circumscribed tumor showed renal sinus fat invasion.
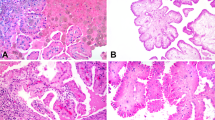
On microscopic examination, all the tumors were entirely tubulocystic without any solid, papillary, or cribriform architecture (Fig. 1). They were composed of small-to-intermediate-sized tubules admixed with cystically dilated tubules, with intervening hypocellular and fibrotic stroma. The lining cells consisted of a single layer of flat to cuboidal to hobnailed cells, with mostly abundant pink cytoplasm and large nuclei with prominent nucleoli (equivalent to WHO/ISUP nucleolar grade 3). Perinucleolar halos were not identified in any case.

Tubulocystic carcinoma showing a well circumscribed but unencapsulated tumor composed entirely of tubules and cysts. No solid areas are allowed, per WHO & ISUP classification a. At higher magnification, this tumor shows the characteristic tubulocystic appearance with the cysts lined by cuboidal to hobnail cells separated by hypocellular and hyalinized stroma b. The cyst and tubular lining often has cells with abundant pink cytoplasm with prominent nucleoli. Notice no perinucleolar halos are present c
Immunohistochemical findings are listed in Table 3. All nine tumors showed retained FH immunohistochemical staining (Fig. 2a). No nuclear or cytoplasmic positivity for 2SC stain was present in any case (Fig. 2b). CK7 and CD10 showed patchy positivity in all six cases tested, whereas AMACR was diffusely positive. Cytokeratin 34ßE12 showed patchy positivity in four of these six tumors, but in general was less diffuse than either CK7 or CD10.

Immunohistochemical stain with fumarate hydratase (FH) showed cytoplasmic positivity in all the nine tumors a, whereas S-(2-succino)-cysteine (2SC) stain was negative in all b
Molecular findings
Copy number analysis of all nine tumors showed loss of chromosome 9 and gain of chromosome 17 (Fig. 3) [8]. Other chromosomal gains or losses were also identified, including chromosome 16 gain in six cases, chromosome 2 and 8 gains in two cases, chromosome 6 gain in one case, and loss of chromosome 19 in one case. Overall, only chromosome 9 loss and chromosome 17 gain were consistently identified across all tumors.

Allele-specific copy number analyses of the next-generation sequencing data revealed loss of chromosome 9 and gain of chromosome 17 in all nine cases. Mutational analysis did not reveal FH mutation in any of the tumors. This figure shows loss of chromosome 9 and gain of chromosome 17 in all five cases analyzed by Memorial Sloan Kettering Cancer Center compared with the normal diploid chromosomes
Table 4 details the mutations identified in these nine cases. None of the cases exhibited mutations in genes well known to occur in other renal cell carcinomas subtypes including FH, VHL, TSC1/2, MET, and SDHB. Recurrent mutations in two chromatin modifying genes lysine N-methyltransferase (KMT2C), a histone methyltransferase, and lysine-specific demethylase 5 C (KDM5C), a histone demethylase, were each identified in two of nine (22%) cases. Other known or likely oncogenic mutations in PMS2, RAD21, TP53, FBXW7, and KEAP1 were also detected. Case TC2 also showed amplification of MDM2 and CDK4 genes. Other genes listed in the table harbored mutations considered as variant of unknown significance.
FISH results
In all five cases, no Y chromosome (green) signal was observed in tumor cells, whereas both X (orange) and Y signals were observed in cells in the stroma between tumor tubules and cysts, as well as the non-neoplastic tissues (Fig. 4).

FISH analysis using the probes for the X chromosome (orange), and Y chromosome (green) reveals loss of the Y chromosome in the lining epithelial cells of cysts. All lining cells display one red signal for the X chromosome, with no green signal (Y chromosome), whereas those cells in the septal region have signals for both X and Y chromosomes
Discussion
As its description as a distinct entity by Amin et al., there have been multiple studies detailing the clinicopathologic features of tubulocystic renal cell carcinoma. Some of these studies have also reported molecular findings based on FISH, comparative genomic hybridization, gene expression profiling, etc [3, 9, 10, 22, 23]. However, until recently, no detailed molecular characterization of tubulocystic renal cell carcinoma using next-generation sequencing had been reported in the literature [14]. Tubulocystic renal cell carcinoma, although more frequently reported to be associated with papillary renal cell carcinoma, has also been reported to occur in association with other subtypes of renal cell carcinoma including clear cell renal cell carcinoma, renal oncocytomas, hybrid tumors, and chromophobe renal cell carcinoma [7, 24, 25].
A few studies have reported that tubulocystic renal cell carcinoma is related to papillary renal cell carcinoma. This is primarily based on their finding of papillary areas in some tumors and presence of other papillary renal cell carcinoma in other areas of the same kidney. Hence, it is not surprising that at least some of previously described tubulocystic renal cell carcinoma have shown genomic features overlapping with papillary renal cell carcinoma.
Zhou et al. performed multicolor FISH assay for chromosomes 7, 17, and Y, on 12 cases of tubulocystic renal cell carcinoma and 20 papillary renal cell carcinoma. Ten of the tubulocystic renal cell carcinoma had associated papillary renal cell carcinoma or papillary adenoma within the same kidney. Ten of 12 tubulocystic renal cell carcinomas demonstrated chromosome 7 gain, eight of 12 cases had a chromosome 17 gain, and eight of nine cases had a loss of Y chromosome [9]. Chen et al. [10] also performed FISH analysis on two cases, one pure and one mixed case of tubulocystic renal cell carcinoma and papillary renal cell carcinoma and showed gains of chromosomes 7 and 17 in the pure as well as in the mixed tubulocystic and papillary renal cell carcinoma components. Such studies have suggested some genomic overlap between these two entities.
Yang et al. [22] studied 13 cases of tubulocystic renal cell carcinoma, 5 of which demonstrated associated papillary renal cell carcinoma in close spatial proximity. By immunohistochemistry, all tubulocystic renal cell carcinoma showed diffuse and strong AMACR positivity, and CK7 immunoreactivity was found in the majority, present either focally or as weak and diffuse. By gene expression profiling, they found the molecular signature of tubulocystic carcinoma to closely approximate that of papillary renal cell carcinoma; by comparative genomic microarray analysis, they found gains of chromosome 17p and 17q (representing trisomy 17), but no chromosome 7p and 7q gains (representing trisomy 7). Hence, they concluded tubulocystic renal cell carcinoma to have a close relationship to papillary renal cell carcinoma based on a high frequency (23%) of co-occurrence of these two entities, a close clustering of the two tumors on the basis of the molecular signatures and last, positive immunoreactivity for AMACR and weak CK7 immunoreactivity in tubulocystic carcinoma.
On the other hand, other groups have reported dissimilarities in molecular profiles of tubulocystic and papillary renal cell carcinoma. Tran et al. [23] investigated 12 cases of pure tubulocystic renal cell carcinoma. All 12 cases showed strong and diffuse labeling for AMACR, and five tumors showed focal reactivity for CK7. All 12 tumors were negative for gains of chromosomes 7 or 17, and were negative for TFE3 translocation by FISH analysis. By virtual karyotyping, one study on a single case of tubulocystic renal cell carcinoma showed gains of chromosomes 8 and 17 and loss of chromosome 9 [24]. Similarly, using Affimetrix 250 K Nsp genotyping array essays, Quiroga-Garza et al. [26], reported (in abstract form) recurrent loss of chromosome 9 and gain of 17 in tubulocystic renal cell carcinoma. Our findings of loss of chromosome 9 and gain of chromosome 17 in all nine cases of “pure” tubulocystic renal cell carcinoma by copy number analysis support this claim. Loss of Y chromosome was also observed in all five tested tumors by FISH. Other chromosomal gains or losses were also present in individual cases by next-generation sequencing in our study, however the only consistent finding, across all nine cases, was the loss of chromosome 9 and gain of chromosome 17, as well as the loss of chromosome Y.
Tubulocystic-like architecture has been described in many different subtypes of kidney tumors. In a study of 15 cases of renal cell carcinoma with tubulocystic pattern, including pure tubulocystic renal cell carcinoma, tubulocystic renal cell carcinoma with poorly differentiated areas, and hereditary leiomyomatosis renal cell carcinoma-associated renal cell carcinoma studied for status of chromosomes 7, 17, and Y by Ulamec et al. [27], none of the six cases of pure tubulocystic renal cell carcinoma showed combined polysomies of chromosomes 7 and 17, (similar to our observations) as were seen in papillary renal cell carcinoma. One of their pure tubulocystic carcinomas showed loss of Y chromosome. We could not test the status of Y chromosome by next-generation sequencing, as both MSK-IMPACTTM and Ion Torrent Comprehensive Cancer Panel do not include any genes from Y chromosome. Therefore, we additionally used FISH assay for the sex chromosomes, which showed consistent loss of Y chromosome, as well.
One of the contentious issues about tubulocystic carcinoma has been its relationship with papillary renal cell carcinoma. The consistent gains of chromosome 17 and losses of Y chromosome, similar to that seen in papillary renal cell carcinoma, in theory would be in favor of such a close relationship. However, multiple factors point against any such connection: (1) Pure tubulocystic carcinoma as described here and as defined by WHO, has a distinct morphology with complete lack of any papillary features. (2) Most tubulocystic carcinomas that were described to show papillary areas in the past, with the current understanding now, most likely represent examples of fumarate hydratase-deficient renal cell carcinoma [8]. (3) Gains of chromosome 7, an almost universal feature in type 1 papillary renal cell carcinoma [28], are not seen in tubulocystic carcinoma. (4) Unlike tubulocystic carcinoma, consistent chromosome 9 losses are not a feature of papillary renal cell carcinoma.
Shared chromosomal losses/gains, based on 1 or 2 chromosomes alone, cannot be justifiable as a proof of relatedness of different tumors. For example, loss of chromosome Y is not uncommon in a large number of non-renal tumors, as well as in many renal cell tumors, including clear cell and chromophobe renal cell carcinoma, and renal oncocytoma [29,30,31,32,33]. Similarly, gain of chromosome 17 is particularly common in hereditary leiomyomatosis renal cell carcinoma/fumarate hydratase-deficient tumors [34]. In brief, a combination of loss of chromosome 9 and gain of chromosome 17, with loss of Y chromosome appears to be a consistent feature in tubulocystic renal cell carcinoma, and to date has not been reported in any other renal cell tumor.
A majority of the reported tubulocystic renal cell carcinomas have shown indolent behavior, mostly presenting as pT1 disease, and with metastasis in < 10%. In some of the larger series, totaling a combined 89 cases, only three developed metastasis [3, 9, 22, 23, 35]. Two of the cases had pure tubulocystic architecture [3], whereas one case had metastasis of the high-grade papillary carcinoma component [22].
In more recent years, there have been some publications describing tubulocystic renal cell carcinoma with high-grade areas [11,12,13]. More recently, another more comprehensive multi-institutional study about tubulocystic renal cell carcinoma with poorly differentiated foci based on 29 cases was published [8]. However, 24 of these 29 cases (83%) had immunohistochemical staining patterns suggestive of fumarate hydratase-deficiency (FH-/2SC + n = 16; FH ± /2SC + n = 8); and 8 of 11 cases with FH-deficiency (72%) on sequencing showed FH gene mutations.
Using targeted next-generation sequencing, we found recurrent alterations of KMT2C and p53/Rb tumor suppressor pathway genes in two tumors each. None of the nine cases showed mutations in genes that are commonly altered in other renal cell carcinomas including VHL, TSC1/2, MET, or SDHB. More importantly, FH gene mutations were not seen in any of these pure tubulocystic carcinomas, in contrast to that recently reported in tubulocystic carcinoma with poorly differentiated foci, which is a frequent morphologic pattern of FH-deficient renal cell carcinoma [8]. A recent study on tubulocystic renal cell carcinoma by Lawrie et al. [14] reported recurrent mutations in ABL1 and PDFGRA genes. However, as constitutional DNA samples were not available for sequencing analysis in some patients in this study, whether these reported mutations truly represent recurrent somatic mutations may need further validation. ABL1 and PDGFRA are indeed included in both our targeted next-generation sequencing panels, but we did not identify any somatic mutations in these two genes in any of our nine tumors. Our study identified recurrent oncogenic mutations in chromatin modifying genes KMT2C and KDM5C, as well as other oncogenic mutations such as those in involving DNA repair and DNA damage response genes such as PMS2, RAD21, and TP53.
In this study, we used targeted next-generation sequencing methodology compared with the more unbiased approaches of whole-exome or whole-genome sequencing. The reasons that we chose the targeted next-generation sequencing approach are mainly its proven performance in formalin-fixed paraffin-embedded tissue and its much higher depth of coverage for detecting low-frequency variants. Cost and availability of tissue (e.g., frozen tissue for whole-genome sequencing) are other limiting factors we had to consider when planning the study. Other strengths of targeted next-generation sequencing include requirement of much less DNA than is required for whole-exome sequencing or whole-genome sequencing, as well as a much faster turnaround time [36]. The targeted next-generation sequencing panels used in our study, MSK-IMPACTTM and Ion Torrent Comprehensive Cancer Panel, had 410 or 409 cancer-related genes, respectively, including the vast majority of genes with recurrent mutations that have been identified in large-scale molecular characterizations of renal cell carcinoma.
In conclusion, tubulocystic renal cell carcinoma demonstrates genomic features distinct from papillary renal cell carcinoma or any of the other usual subtypes of renal cell carcinoma. Although a tubulocystic growth pattern may be encountered in a number of renal neoplasms, our current study provides comprehensive molecular evidence supporting the ISUP and WHO definitional contentions/criteria associated with “pure” tubulocystic renal cell carcinoma, criteria, which are not strictly adhered to in some studies in the literature. Thus, the entity tubulocystic renal cell carcinoma should be diagnosed only when present as a “pure” form, using well defined strict morphological criteria; presence of papillary, poorly differentiated, or other architectural patterns should exclude a tumor from diagnostic assignment of tubulocystic renal cell carcinoma.
References
Masson P. Tumeurs humaines 1955, in Kobernick S (traduction): Human tumors, histology, diagnosis and technique. 2nd edn. Detroit, MI: Wayne State University Press; 1970. p. 181.
Murphy WM, Beckwith JB, Farrow GM. Tumors of the kidney. In: Murphy, Beckwith, Farrow, eds. Atlas of tumor pathology. Tumors of the kidney, bladder, and related urinary structures. 3rd series. Washington, D.C: Armed Forces Institute of Pathology, 1994. p.118–124.
Amin MB, MacLennan GT, Gupta R, al. Tubulocystic carcinoma of the kidney: clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am J Surg Pathol. 2009;33:384–92.
Comperat R, Cheville J, Rioux-Leclercq N. Tubulocystic renal cell carcinoma. In: Moch H, Humphrey PA, Ulbright TM, Reuter VE eds. WHO classifications of tumours of the urinary system and male genital organs. 4th ed. Lyon: International Agency for Research on Cancer; 2016. p. 38.
Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–89.
Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627–37.
Skenderi F, Ulamec M, Vranic S, et al. Cystic renal oncocytoma and tubulocystic renal cell carcinoma: morphologic and immunohistochemical comparative study. Appl Immunohistochem Mol Morphol. 2016;24:112–9.
Smith SC, Trpkov K, Chen YB, et al. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a frequent morphologic pattern of fumarate hydratase-deficient renal cell carcinoma. Am J Surg Pathol. 2016;40:1457–72.
Zhou M, Yang XJ, Lopez JI, et al. Renal tubulocystic carcinoma is closely related to papillary renal cell carcinoma: implications for pathologic classification. Am J Surg Pathol. 2009;33:1840–9.
Chen N, Nie L, Gong J, et al. Gains of chromosomes 7 and 17 in tubulocystic carcinoma of kidney: two cases with fluorescence in situ hybridisation analysis. J Clin Pathol. 2014;67:1006–9.
Bhullar JS, Thamboo T, Esuvaranathan K. Unique case of tubulocystic carcinoma of the kidney with sarcomatoid features: a new entity. Urology. 2011;78:1071–2.
Al-Hussain TO, Cheng L, Zhang S, et al. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a series of 3 cases with fluorescence in situ hybridization analysis. Hum Pathol. 2013;44:1406–11.
Zhao M, Teng X, Ru G, et al. Tubulocystic renal cell carcinoma with poorly differentiated foci is indicative of aggressive behavior: clinicopathologic study of two cases and review of the literature. Int J Clin Exp Pathol. 2015;8:11124–31.
Lawrie CH, Armesto M, Fernandez-Mercado M, et al. Noncoding RNA expression and targeted next-generation sequencing distinguish tubulocystic renal cell carcinoma (TC-RCC) from other renal neoplasms. J Mol Diagn. 2018;20:34–45.
Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131.
Grasso C, Butler T, Rhodes K, et al. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 2015;17:53–63.
Warrick JI, Hovelson DH, Amin A, et al. Tumor evolution and progression in multifocal and paired non-invasive/invasive urothelial carcinoma. Virchows Arch. 2015;466:297–311.
Kadakia KC, Tomlins SA, Sanghvi SK, et al. Comprehensive serial molecular profiling of an “N of 1” exceptional non-responder with metastatic prostate cancer progressing to small cell carcinoma on treatment. J Hematol Oncol. 2015;8:109.
Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–64.
Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131.
Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017; 2017. https://doi.org/10.1200/PO.17.00011.
Yang XJ, Zhou M, Hes O, et al. Tubulocystic carcinoma of the kidney: clinicopathologic and molecular characterization. Am J Surg Pathol. 2008;32:177–87.
Tran T, Jones CL, Williamson SR, et al. Tubulocystic renal cell carcinoma is an entity that is immunohistochemically and genetically distinct from papillary renal cell carcinoma. Histopathology. 2016;68:850–7.
Quiroga-Garza G, Pina-Oviedo S, Cuevas-Ocampo K, et al. Synchronous clear cell renal cell carcinoma and tubulocystic carcinoma: genetic evidence of independent ontogenesis and implications of chromosomal imbalances in tumor progression. Diagn Pathol. 2012;7:21.
Brennan C, Srigley JR, Whelan C, et al. Type 2 and clear cell papillary renal cell carcinoma, and tubulocystic carcinoma: a unifying concept. Anticancer Res. 2010;30:641–4.
Quiroga-Garza G, Ayala AG, Truong L, et al. Cytogenomic molecular profiles of tubulocystic carcinoma of the kidney. Mod Pathol. 2012;25:235A.
Ulamec M, Skenderi F, Zhou M, et al. Molecular genetic alterations in renal cell carcinomas with tubulocystic pattern: tubulocystic renal cell carcinoma, tubulocystic renal cell carcinoma with heterogenous component and familial leiomyomatosis-associated renal cell carcinoma. clinicopathologic and molecular genetic analysis of 15 Cases. Appl Immunohistochem Mol Morphol. 2016;24:521–30.
Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–45.
Kuroda N, Tamura M, Shiotsu T, et al. Chromosomal abnormalities of clear cell renal cell carcinoma: frequent gain of chromosome 7. Pathol Int. 2010;60:9–13.
Speicher MR, Schoell B, du Manoir S, et al. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol. 1994;145:356–64.
Lindgren V, Paner GP, Omeroglu A, et al. Cytogenetic analysis of a series of 13 renal oncocytomas. J Urol. 2004;171:602–4.
Arseneault M, Monlong J, Vasudev NS, et al. Loss of chromosome Y leads to down regulation of KDM5D and KDM6C epigenetic modifiers in clear cell renal cell carcinoma. Sci Rep. 2017;7:44876.
Klatte T, Rao PN, de Martino M, et al. Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma. J Clin Oncol. 2009;27:746–53.
Koski TA, Lehtonen HJ, Jee KJ, et al. Array comparative genomic hybridization identifies a distinct DNA copy number profile in renal cell cancer associated with hereditary leiomyomatosis and renal cell cancer. Genes Chromosomes Cancer. 2009;48:544–51.
Azoulay S, Vieillefond A, Paraf F, et al. Tubulocystic carcinoma of the kidney: a new entity among renal tumors. Virchows Arch. 2007;451:905–9.
Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46.
Acknowledgements
We acknowledge the members of the members of the Molecular Diagnostics Service in the Department of Pathology, the Integrated Genomics Operation and Bioinformatics Core, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology of Memorial Sloan Kettering Cancer Center. The study is supported by Memorial Sloan Kettering Cancer Center Core Grant (P30 CA008748) and The Society of Memorial Sloan Kettering Research Grant (YBC). S.A.T. was supported as the A. Alfred Taubman Emerging Scholar by the A. Alfred Taubman Medical Research Institute. S.A.T. is a co-founder of and consultant for Strata Oncology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest for the current study. Hikmat Al-Ahmadie: Consultant - Bristol-Myers-Squibb, EMD Serono and AstraZeneca.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sarungbam, J., Mehra, R., Tomlins, S.A. et al. Tubulocystic renal cell carcinoma: a distinct clinicopathologic entity with a characteristic genomic profile. Mod Pathol 32, 701–709 (2019). https://doi.org/10.1038/s41379-018-0185-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0185-5
This article is cited by
-
Recurrent KRAS mutations are early events in the development of papillary renal neoplasm with reverse polarity
Modern Pathology (2022)
-
New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)
-
Charakterisierung der unterschiedlichen Entitäten beim Nierenzellkarzinom
Der Urologe (2020)
