
Abstract
The Philadelphia chromosome resulting from t(9;22)(q34;q11.2) or its variants is a defining event in chronic myeloid leukemia. It is also observed in several types of de novo acute leukemia, commonly in B lymphoblastic leukemia, and rarely in acute myeloid leukemia, acute leukemia of ambiguous lineage, and T lymphoblastic leukemia. Acquisition of the Philadelphia chromosome during therapy of acute leukemia and myelodysplastic syndrome is rare. We reported 19 patients, including 11 men and 8 women with a median age of 53 years at initial diagnosis. The diagnoses at initial presentation were acute myeloid leukemia (n = 11), myelodysplastic syndrome (n = 5), B lymphoblastic leukemia (n = 2), and T lymphoblastic leukemia (n = 1); no cases carried the Philadelphia chromosome. The Philadelphia chromosome was detected subsequently at relapse, or at refractory stage of acute leukemia or myelodysplastic syndrome. Of 14 patients evaluated for the BCR-ABL1 transcript subtype, 12 had the e1a2 transcript. In 11 of 14 patients, the diseases before and after emergence of the Philadelphia chromosome were clonally related by karyotype or shared gene mutations. Of 15 patients with treatment information available, 7 received chemotherapy alone, 5 received chemotherapy plus tyrosine kinase inhibitors, 2 received tyrosine kinase inhibitors only, and 1 patient was not treated. Twelve patients had follow-up after acquisition of the Philadelphia chromosome; all had persistent/refractory acute leukemia. Thirteen of 15 patients died a median of 3 months after the emergence of the Philadelphia chromosome. In summary, secondary Philadelphia chromosome acquired during therapy is rare, and is associated with the e1a2 transcript subtype, terminal disease stage, and poor outcome.
Similar content being viewed by others
Introduction
The Philadelphia chromosome resulting from t(9;22)(q34;q11.2) or its variants is the hallmark of chronic myeloid leukemia [1]. It is also observed in several types of de novo acute leukemia, commonly in B lymphoblastic leukemia [2], and rarely in acute myeloid leukemia [3,4,5], acute leukemia of ambiguous lineage [6, 7], and T lymphoblastic leukemia [8, 9]. Different break point sites within the BCR gene result in different BCR-ABL1 fusion transcript subtypes that may affect the disease phenotype. The typical b2a2/b3a2 fusion transcripts, which encode P210, are found in over 95% of patients with chronic myeloid leukemia. The e1a2 fusion transcript, which encodes P190, is seen in most patients with Philadelphia chromosome-positive B lymphoblastic leukemia, and is associated with monocytosis in chronic myeloid leukemia [10]. Patients with chronic myeloid leukemia and the e1a2 transcript have a higher risk of blastic transformation, poor treatment response, and unfavorable survival compared with patients with the typical b2a2/b3a2 transcripts [11]. Cases of chronic myeloid leukemia that carry the rare e19a2 transcript, which encodes P230, often show prominent neutrophilic maturation or thrombocytosis [12, 13].
All subtypes of BCR-ABL1 transcripts encode fusion proteins with constitutive tyrosine kinase activity that is essential for inducing leukemia via increasing tumor cell proliferation and growth, reducing adherence to bone marrow stroma [1], and promoting angiogenesis, metastasis, and a defective apoptotic response [14]. An understanding of the abnormal signaling pathway has led to the development of small molecule tyrosine kinase inhibitors targeting BCR-ABL1. This targeted therapy has dramatically improved the outcome of patients with chronic myeloid leukemia [15], and to a lesser extent, Philadelphia chromosome-positive B lymphoblastic leukemia [16,17,18]. Improved survival in patients with de novo Philadelphia chromosome-positive acute myeloid leukemia has also been reported anecdotally [19].
Myeloid and lymphoid neoplasms in which the Philadelphia chromosome appears as a primary change at disease onset are well-established entities, and have been extensively studied. However, secondary Philadelphia chromosome acquired during therapy of myeloid or lymphoid neoplasms is extremely rare, and only sporadically described in the form of single-case reports in the literature [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. The biological and clinical features of these cases have not been systematically studied, and the prognostic significance of secondary Philadelphia chromosome is poorly understood. Here, we reported 19 patients who acquired the Philadelphia chromosome during therapy of myeloid or lymphoid neoplasms and provided an overview of cases reported in the literature.
Methods
Patient selection
Patients with myeloid or lymphoid neoplasms diagnosed in our institution from 1998 through 2016 were reviewed for the presence of the Philadelphia chromosome acquired during therapy. Patients with following Philadelphia chromosome-positive diseases were not included: chronic myeloid leukemia, de novo B- or T lymphoblastic leukemia, de novo acute myeloid leukemia, and de novo acute leukemia of ambiguous lineage. Patient demographics, clinical histories, including treatment and follow-up information, and laboratory data were obtained by electronic chart review. This study was approved by the Institutional Review Board at the University of Texas MD Anderson Cancer Center and was conducted in accordance with the Declaration of Helsinki.
Flow cytometric immunophenotyping
Flow cytometric immunophenotypic analysis was performed on bone marrow aspirates using standard multicolor analysis, which evolved substantially during the study interval. The following antibodies were used in various combinations: CD2, CD3 (surface and cytoplasmic), CD4, CD5, CD7, CD13, CD14, CD15, CD19, CD20, CD22, CD25, CD33, CD34, CD36, CD38, CD41, CD45, CD56, CD64, CD117, CD123, HLA-DR, MPO, and TDT (Becton-Dickinson, Biosciences, San Jose, CA, USA). Data were analyzed using FCS Express (De novo Software, Glendale, CA).
Cytogenetic studies
Conventional chromosomal analysis was performed on G-banded metaphase cells prepared from unstimulated bone marrow aspirate cultures (24 and 48 h) using standard GTG banding. At least 20 metaphases were analyzed. Results were reported using the 2013 International System for Human Cytogenetic Nomenclature [54]. Fluorescence in situ hybridization (FISH) analysis for BCR-ABL1 fusion was performed on freshly harvested aspirate smears or cultured cells with LSI-BCR-ABL1 ES fusion probes (Abbott Molecular/Vysis, Des Plaines, IL) using previously described methods [55]. At least 200 interphase nuclei were analyzed.
Detection of BCR-ABL1 transcripts
Reverse transcription-quantitative polymerase chain reaction (PCR) for detection of BCR-ABL1 transcripts [e1a2, e13a2 (b2a2), and e14a2 (b3a2)] was performed using RNA extracted from bone marrow or peripheral blood samples with sensitivities of 1 in 10,000 and 1 in 100,000, respectively, according to methods described previously [56, 57]. Quantitative results were expressed as the percent ratio of BCR-ABL1 to ABL1. The sizes of fusion transcripts were determined by capillary electrophoresis.
Mutation analysis
Mutation analysis was performed using DNA extracted from bone marrow aspirate samples in a subset of patients using the following techniques. Next-generation sequencing-based analysis was performed to detect somatic mutations in the entire coding sequences of 28 genes (ABL1, EGFR, GATA2, IKZF2, MDM2, NOTCH1, RUNX1, ASXL1, EXH2, HRAS, JAK2, MLL, NPM1, TET2, BRAF, FLT3, IDH1, KIT, MPL, NRAS, TP53, DNMT3A, GATA1, IDH2, KRAS, MYD88, PTPN11, WT1) or in hotspots of 53 genes (ABL1, CSF1R, FGFR1, HRAS, KRAS, PIK3CA, SRC, AKT1, CTNNB1, FGFR2, IDH1, MET, PTEN, STK11, ALK, DNMT3A, FGFR3, IDH2, MLH1, PTPN11, TP53, APC, EGFR, FLT3, JAK2, MPL, RB1, VHL,ATM, ERBB2, GNA11, JAK3, NOTCH1, RET, XPO1,BRAF, ERBB4, GNAQ, KDR, NPM1, SMAD4, CDH1, EZH2, GNAS, KIT, NRAS, SMARCB1, CDKN2A, FBXW7, HNF1A, KLHL6, PDGFRA, SMO1). The next-generation sequencing assays were performed using the Illumina MiSeq sequencer (Illumina, San Diego, CA) with a sensitivity of 5% as described previously [58, 59]. FLT3-ITD, FLT3 D835, and NPM1 (exon 12, codons 956–971) mutations were assessed by PCR followed by capillary electrophoresis on a genetic analyzer (Prism ABI 3130, Applied Biosystems, Foster City, CA) as described previously [60]. NRAS (codons 12, 13, 61), KRAS (codons 12, 13, 61), IDH1 (exon 4, codons 87–138), IDH2 (exon 4), KIT (exons 8 and 17), and JAK2 (codon 617) mutations were assessed by PCR followed by Sanger sequencing with a sensitivity of 20% as described previously [58, 61]. PCR-based cDNA sequencing of BCR-ABL1 was performed to detect mutations in codons 221–500 of the ABL1 kinase domain by using pyrosequencing PSQ96 HS System (Biotage AB, Uppsala, Sweden) per the manufacturer’s instructions. The lower limit of detection of the assay is 5% mutation-bearing cells in the test samples. PCR-based microsatellite polymorphism analysis was performed by using GENESCAN on the pre-transplant, donor, and post-transplant samples.
Results
Timing of emergence of secondary Philadelphia chromosome
A total of 19 patients were identified to have secondary t(9;22)(q34;q11.2)/BCR-ABL1 detected by chromosomal banding analysis during therapy of acute leukemia or myelodysplastic syndrome. The diagnoses at initial presentation were acute myeloid leukemia (n = 11), myelodysplastic syndrome (n = 5), B lymphoblastic leukemia (n = 2), or T lymphoblastic leukemia (n = 1) (Table 1). No variant BCR-ABL1 rearrangements were identified. There were 11 men and 8 women with a median age of 53 years (range, 18–78 years) at initial diagnosis, and 56 years (range, 19–79 years) at time of emergence of secondary Philadelphia chromosome.
Of the 11 patients with an initial diagnosis of acute myeloid leukemia, 6 patients acquired secondary Philadelphia chromosome at time of first to 3rd relapse, 2 acquired secondary Philadelphia chromosome during progression of acute myeloid leukemia after 1–3 relapses, and 3 acquired secondary Philadelphia chromosome during treatment of refractory disease (patients never achieved remission before the emergence of secondary Philadelphia chromosome) (Table 1). Of the five patients with an initial diagnosis of myelodysplastic syndrome, all developed secondary acute myeloid leukemia, and acquired the Philadelphia chromosome when secondary acute myeloid leukemia developed (n = 1), relapsed (n = 1), relapsed and became refractory to treatment (n = 2), or after relapse with persistent myelodysplastic syndrome (1% blasts in bone marrow) (n = 1). The two patients with an initial diagnosis of B lymphoblastic leukemia acquired the Philadelphia chromosome at relapse or at a refractory stage after relapse, respectively. In the patient with T lymphoblastic leukemia, the Philadelphia chromosome was acquired during treatment of refractory disease after two relapses. Among the patients with an initial diagnosis of acute myeloid leukemia or acute lymphoblastic leukemia, the median interval from the initial diagnosis of acute leukemia to the acquisition of secondary Philadelphia chromosome was 16 months (range, 6–40 months). Among the patients with an initial diagnosis of myelodysplastic syndrome, the median interval from the initial diagnosis of myelodysplastic syndrome to the emergence of secondary Philadelphia chromosome was 38 months (range, 14–72 months), and the median interval from the development of secondary acute myeloid leukemia to the emergence of secondary Philadelphia chromosome was 9 months (range, 0–37 months).
Complete blood count data and bone marrow morphology after acquisition of Philadelphia chromosome
At emergence of secondary Philadelphia chromosome, 18 patients had anemia (range, 8.4–11.5 g/dl), 16 had thrombocytopenia (range, 8–104 × 103/μ), and 8 had leukopenia (range, 1.0–3.8 × 103/μl) (Supplemental Table 1). Circulating blasts (range, 2–96%) were seen in 15 patients. In eight patients who had leukocytosis (range, 13.9–89.2 × 103/μl), blasts were the predominant cell population in five patients. Six patients had absolute monocytosis at the time of emergence of secondary Philadelphia chromosome, and four of them had monocytosis before acquisition of the Philadelphia chromosome. Absolute eosinophilia, basophilia, and left-shifted granulocytes, features of chronic myeloid leukemia, were observed in only one patient (#13).
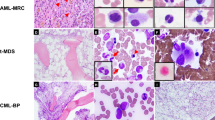
Bone marrow smears both before and after detection of secondary Philadelphia chromosome were available for retrospective review in nine patients. There was no change in the morphology of blasts before versus after emergence of secondary Philadelphia chromosome (Fig. 1), except in patient #1 whose blasts appeared more monoblastic at detection of secondary Philadelphia chromosome [62]. No immunophenotypic shift in the blasts was detected by flow cytometric analysis in 14 patients with available data (#1, 2, 4, 5, 6, 7, 9, 11, 12, 13, 16, 17, 18, and 19). Given the evolving nature of flow cytometry methods over the course of this study, however, blasts were evaluated with only a limited number of markers in early cases.

Morphology of blasts before and after emergence of secondary BCR-ABL1. a, b Blast morphology of patient with acute myeloid leukemia (#8) before (a) and after (b) the emergence of secondary BCR-ABL1. c, d Blast morphology of patient with myelodysplastic syndrome (#16) before (c) and after (d) the emergence of secondary BCR-ABL1. e, f Blast morphology of patient with T lymphoblastic leukemia (#19) before (e) and after (f) the emergence of secondary BCR-ABL1. Original magnification: x1000
Clonal relatedness between diseases before and at emergence of secondary Philadelphia chromosome
Detailed cytogenetic and molecular findings at initial diagnosis, and before and after the emergence of secondary Philadelphia chromosome are provided in Tables 2 and 3.
Karyotypes at initial diagnosis were available in 16 patients (Table 2). Seven patients had a normal karyotype and nine had an abnormal karyotype. In seven of nine patients with an abnormal karyotype at initial diagnosis, the initial chromosomal alterations were present in the secondary Philadelphia chromosome-positive clones, thus the diseases before and at the emergence of the Philadelphia chromosome were clonally related, and the emergence of secondary Philadelphia chromosome may represent clonal evolution.
In five of seven patients with a normal karyotype, tests for molecular mutations with a limited panel were performed in bone marrow samples obtained before and at time of emergence of secondary Philadelphia chromosome. Common molecular mutations were detected before and at emergence of secondary Philadelphia chromosome in four patients (Table 3). FLT3-ITD mutation in patients #2 and 10, NPM1 mutation in patient #4, and NRAS mutation in patient #6. Of note, the post-transplant microsatellite polymorphism pattern showed a mixed chimera in three patients tested (#2, 4, and 5), supporting the recipient origin of relapsed disease.
BCR-ABL1 transcript subtype and ABL1 mutation
FISH analyses were performed in 16 patients, and BCR-ABL1 rearrangement was confirmed positive in all. In 14 patients tested by reverse transcription-quantitative PCR for BCR-ABL1 transcript subtype, the e1a2 fusion transcripts (P190) were identified in 12 patients (86%). In the other two patients (#3 and 6), b2a2 or simultaneous b2a2 and b3a3 transcripts were identified, respectively. Of five patients analyzed for ABL1 kinase domain mutations (#2, 3, 7, 16, and 17), only one (#7) showed a mutation (p.G254E) (Table 3).
Evolution of secondary Philadelphia chromosome-positive clones during therapy
Before the emergence of secondary Philadelphia chromosome detectable by chromosomal banding analysis, BCR-ABL1 rearrangement or fusion transcripts were tested in eight patients by either FISH (n = 3) or reverse transcription-quantitative PCR (n = 4) or both (n = 1). A low level of BCR-ABL1 fusion transcripts was detected in three patients either at initial presentation (#1, 0.11%; #11, 0.01%) or later during therapy (#18, 0.70%) by reverse transcription-quantitative PCR, but not by FISH. The BCR-ABL1 transcript subtype was of e1a2 in all three patients. The median time from the detection of low level of BCR-ABL1 by reverse transcription-quantitative PCR to the emergence of secondary Philadelphia chromosome detected by karyotyping was 9 months (range, 5–11 months).
After the emergence of secondary Philadelphia chromosome detected by karyotyping, the Philadelphia chromosome-positive subclones were the major one in 11 patients, simultaneously with the emergence of secondary Philadelphia chromosome in eight patients (#1, 6, 7, 9, 13, 16, 17, and 19) and shortly after the emergence of secondary Philadelphia chromosome in three patients (#4, 12, and 18). The Philadelphia chromosome-positive subclones were a minor clone in the remaining eight patients. Twelve patients had follow-up information on the evolution of Philadelphia chromosome-positive subclones after initial detection. The Philadelphia chromosome-positive subclones were persistent by FISH analysis in six patients (#2, 6, 8, 11, 12, and 16) despite chemotherapy with or without tyrosine kinase inhibitors. In the remaining six patients (#3, 4, 7, 10, 17, and 18), the Philadelphia chromosome-positive subclones were undetectable by FISH analysis, including three patients (#4, 10, and 17) with undetectable BCR-ABL1 transcripts by reverse transcription-quantitative PCR. Of those six patients with undetectable Philadelphia chromosome-positive subclones, five were treated with tyrosine kinase inhibitors and one received conventional chemotherapy. Interestingly, all 12 patients had persistent or refractory acute myeloid leukemia or acute lymphoblastic leukemia regardless of the presence or absence of the Philadelphia chromosome-positive subclones, including one patient (#18) with refractory therapy-related acute myeloid leukemia resulting from treatment of B lymphoblastic leukemia with secondary Philadelphia chromosome.
Treatment and outcome
Before the emergence of secondary Philadelphia chromosome, all 19 patients received chemotherapy (Table 1). In addition, five patients received allogeneic hematopoietic stem cell transplantation. Only nine patients achieved morphologic remission or deeper response, and all nine patients developed relapsed disease.
After the emergence of secondary Philadelphia chromosome, 15 patients had treatment and follow-up information available; the remaining four patients were transferred to other institutions for treatment and were lost to follow-up (Table 1). Seven patients were treated with chemotherapy only, five treated with chemotherapy plus tyrosine kinase inhibitors, two treated with tyrosine kinase inhibitors only, and one not treated due to other comorbid conditions. Thirteen patients died with refractory disease after a median follow-up of 3 months (range, 1–9 months) after the emergence of secondary Philadelphia chromosome. Two patients were living; one (#11) had an initial diagnosis of acute myeloid leukemia, and had refractory disease at last follow-up 8 months after the emergence of secondary Philadelphia chromosome. The other patient (#17) had an initial diagnosis of B lymphoblastic leukemia, and received chemotherapy combined with tyrosine kinase inhibitors, as well as subsequent allogeneic hematopoietic stem cell transplantation after the emergence of second Philadelphia chromosome. The patient was free of residual disease at last follow-up 6 months after the emergence of secondary Philadelphia chromosome. Another patient (#18) with an initial diagnosis of B lymphoblastic leukemia achieved remission with tyrosine kinase inhibitors after the emergence of secondary Philadelphia chromosome. However, this patient developed therapy-related acute myeloid leukemia and died shortly thereafter.
Discussion
We reported 19 patients with Philadelphia chromosome-negative de novo acute myeloid leukemia, myelodysplastic syndrome, B lymphoblastic leukemia, and T lymphoblastic leukemia who secondarily acquired the Philadelphia chromosome during therapy. The emergence of secondary Philadelphia chromosome-positive clones by karyotyping occurred concomitantly with relapsed or refractory disease in de novo acute leukemia or secondary acute myeloid leukemia arising from myelodysplastic syndrome. In most patients, the BCR-ABL1 transcript subtype was of the e1a2 (encoding P190 BCR-ABLl protein), and the Philadelphia chromosome-positive disease and the original leukemia were clonally related. The Philadelphia chromosome-positive clones were the dominant clones in more than half of the patients when secondary Philadelphia chromosome emerged. Despite the absence of the Philadelphia chromosome-positive clones by karyotyping in 6 patients, 13 of 15 patients with follow-up information died with refractory disease after a median follow-up of 3 months, including one patient with therapy-related acute myeloid leukemia.
Secondary Philadelphia chromosome acquired during therapy of myeloid or lymphoid neoplasms is rare; 36 such cases have been reported in the form of single-case reports in the English literature (Table 4 and Supplemental Table 2) [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. We reviewed these published cases and reclassified them, if necessary or possible, according to the 2016 update of World Health Organization classification [63]. The 36 cases included the following: acute myeloid leukemia (n = 14), myelodysplastic syndrome (n = 10), B lymphoblastic leukemia (n = 6), T lymphoblastic leukemia (n = 5), and chronic myelomonocytic leukemia (n = 1). There were 20 men and 16 women with a median age of 43 years (range, 3–71 years), approximately 10 years younger than that in our study. Similar to our cases, secondary Philadelphia chromosome was detected at relapse or at a terminal stage of refractory disease in patients with acute leukemia. In patients with myelodysplastic syndrome, the Philadelphia chromosome emerged at time of or after acute transformation in 8 of 10 patients. The median interval from initial diagnosis to secondary acquisition of the Philadelphia chromosome was 15 months (range, 1–120 months). Twenty-one patients had an abnormal karyotype at initial diagnosis or anytime during therapy before secondary Philadelphia chromosome emerged; the abnormalities present before the emergence of secondary Philadelphia chromosome were also observed in the Philadelphia chromosome-positive clones in 19 patients, including one patient with NRAS mutation observed before and at emergence of the Philadelphia chromosome. Additionally, one patient with a normal karyotype before secondary acquisition of the Philadelphia chromosome had NPM1 mutations before and at emergence of the Philadelphia chromosome. The BCR-ABL1 fusion transcript encoded P190 in 16/24, P210 in 5/24, and both P210 and P190 both in three patients. Of note, in the latter three patients, the Philadelphia chromosome was not detected by karyotyping but P210-encoding transcripts were detected by reverse transcription-quantitative PCR, and P190/e1a2 transcripts emerged later 3, 4, and 14 months before the detection of the Philadelphia chromosome by karyotyping, suggesting that the secondary Philadelphia chromosome encoded P190. BCR-ABL1 at the onset of the disease was detected in only 2 of 17 patients tested with reverse transcription-quantitative PCR, FISH, or both. Twenty-three patients had treatment information available, 21 patients received chemotherapy, 8 received tyrosine kinase inhibitors, and 6 received allogeneic hematopoietic stem cell transplantation. Of 33 patients with follow-up information available regarding the disease status, 30 had relapsed or refractory disease and 26 were dead at last follow-up, with a median follow-up of 5 months (range, 1–22 months). For the 16 patients with follow-up information available regarding the evolution of the Philadelphia chromosome-positive clones, the Philadelphia chromosome-positive clones were persistent and/or became the major clone in most patients despite intensive chemotherapy, and only 3 patients achieved cytogenetic remission [32, 52, 53].
The emergence of secondary Philadelphia chromosome can be explained by multiple mechanisms. It may indicate clonal evolution and disease progression. This hypothesis is supported by the findings that the diseases before and after the emergence of secondary Philadelphia chromosome were clonally related by karyotyping or molecular analyses in the vast majority of patients (86%) in our series and in patients reported previously (Table 4 and Supplemental Table 2). This apparently underestimates the clonal relatedness given the limited panel used to detect molecular mutations. Alternatively, in a subset of patients the emergence of secondary Philadelphia chromosome could be due to the expansion of a minute Philadelphia chromosome-positive subclone not detectable initially by cytogenetic methods. The detection of the same transcript subtype of e1a2 before and at time of emergence of the Philadelphia chromosome-positive subclones in three patients (#1, 11, and 18) in this study would substantiate this theory. In all three patients, interestingly, the Philadelphia chromosome-positive subclones clonally related to the previous disease before the emergence of secondary Philadelphia chromosome.
The persistence or refractoriness of disease despite the disappearance of secondary Philadelphia chromosome-positive clones in some patients suggests that BCR-ABL1 is not essential for the maintenance of the leukemic process. However, the persistence of Philadelphia chromosome-positive clones despite chemotherapy and/or tyrosine kinase inhibitors in some patients and predilection of the e1a2 (P190) subtype rather than the typical b2a2/b3a2 (P210) in most patients support a role of BCR-ABL1 in disease progression. Both P210 and P190 BCR-ABL1 fusion transcripts result in new chimeric proteins with increased tyrosine kinase activity. However, P190 was shown to have at least fivefold higher tyrosine kinase activity than P210 in a murine model [64]. Transgenic mice bearing the e1a2 (P190) fusion transcript develop acute leukemia [65], whereas transgenic mice bearing P210 develop chronic myeloid leukemia [66]. Patients with myelodysplastic syndrome with P190 at initial presentation show rapid progression to acute myeloid leukemia [67]. P190 is also associated with transformation of chronic myeloid leukemia to a more aggressive terminal phase disease [68]. In addition, patients with de novo acute myeloid leukemia with P190 have a higher rate of relapse than patients with P210 [19]. Overall, these findings correlate with our finding that P190 is highly associated with disease progression in myeloid and lymphoid neoplasms.
Adding tyrosine kinase inhibitors to the therapeutic regimens has significantly altered the outcome of patients with chronic myeloid leukemia [69, 70] and Philadelphia chromosome-positive acute lymphoblastic leukemia [71, 72], and the efficacy has also been shown in Philadelphia chromosome-positive de novo acute myeloid leukemia [19]. It is worth mentioning that tyrosine kinase inhibitors are effective in rare cases of myelodysplastic syndrome with the Philadelphia chromosome [73] and rare cases with concurrent BCR-ABL1 and CBFB rearrangements [31, 74, 75]. However, the effect of tyrosine kinase inhibitors along with or in place of chemotherapy in patients with late-acquired Philadelphia chromosome in relapsed or refractory disease is not well understood. We reported seven such patients treated with tyrosine kinase inhibitors. In this study, eradication of the Philadelphia chromosome-positive clones detected by FISH analysis was observed in five of six patients with follow-up information although four of them had refractory Philadelphia chromosome-negative disease. Eight such patients reported in the literature received tyrosine kinase inhibitors; seven showed partial or complete response to tyrosine kinase inhibitors although four patients had relapsed or refractory disease at last follow-up. These findings show that incorporating tyrosine kinase inhibitors in the treatment is an effective approach to treat the Philadelphia chromosome-positive clones in most patients. Overall however, prognosis of these patients is still poor due to other factors.
In conclusion, the Philadelphia chromosome, usually seen as a primary change in chronic myeloid leukemia, can also occur secondarily as a late-developing chromosomal abnormality during therapy of myeloid and lymphoid neoplasms. The transcript subtype associated with the late-occurring Philadelphia chromosome is overwhelmingly of the e1a2 (P190), and the disease before and after the emergence of the Philadelphia chromosome are clonally related. Incorporating tyrosine kinase inhibitors into treatment may eradicate the Philadelphia chromosome-positive clones and could potentially change the disease prognosis in a small subset of patients, but most patients do not respond to the treatment and the overall survival is poor.
References
Vardiman JW, Melo JV, Baccarani M, et al. Chronic myelogenous leukaemia, BCR-ABL1 positive, In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. IARC: Lyon; 2008. p. 32–37.
Borowitz MJ, Chan, JKC. B-lymphoblasticleukemia/lymphoma with recurrent genetic abnormalities. In: Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman, JW, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. IARC: Lyon; 2008. p. 5.
Konoplev S, Yin CC, Kornblau SM, et al. Molecular characterization of de novo Philadelphia chromosome-positive acute myeloid leukemia. Leuk Lymphoma. 2013;54:138–44.
Nacheva EP, Grace CD, Brazma D, et al. Does BCR/ABL1 positive acute myeloid leukaemia exist? Br J Haematol. 2013;161:541–50.
Soupir CP, Vergilio JA, Dal Cin P, et al. Philadelphia chromosome-positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol. 2007;127:642–50.
Carbonell F, Swansbury J, Min T, et al. Cytogenetic findings in acute biphenotypic leukaemia. Leukemia. 1996;10:1283–7.
Killick S, Matutes E, Powles RL, et al. Outcome of biphenotypic acute leukemia. Haematologica. 1999;84:699–706.
Raanani P, Trakhtenbrot L, Rechavi G, et al. Philadelphia-chromosome-positive T-lymphoblastic leukemia: acute leukemia or chronic myelogenous leukemia blastic crisis. Acta Haematol. 2005;113:181–9.
You MJ, Medeiros LJ, Hsi ED. T-lymphoblastic leukemia/lymphoma. Am J Clin Pathol. 2015;144:411–22.
Melo JV, Myint H, Galton DA, et al. P190BCR-ABL chronic myeloid leukaemia: the missing link with chronic myelomonocytic leukaemia? Leukemia. 1994;8:208–11.
Gong Z, Medeiros LJ, Cortes JE, et al. Clinical and prognostic significance of e1a2 BCR-ABL1 transcript subtype in chronic myeloid leukemia. Blood Cancer J. 2017;7:e583.
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–84.
Pane F, Frigeri F, Sindona M, et al. Neutrophilic-chronic myeloid leukemia: a distinct disease with a specific molecular marker (BCR/ABL with C3/A2 junction). Blood. 1996;88:2410–4.
Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–9.
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am J Hematol. 2016;91:252–65.
Wassmann B, Pfeifer H, Goekbuget N, et al. Alternating versus concurrent schedules of imatinib and chemotherapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph + ALL). Blood. 2006;108:1469–77.
Fielding AK, Rowe JM, Buck G, et al. UKALLXII/ECOG2993: addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood. 2014;123:843–50.
Fielding AK. Treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia in adults: a broader range of options, improved outcomes, and more therapeutic dilemmas. Am Soc Clin Oncol Educ Book. 2015:e352–e359.
Reboursiere E, Chantepie S, Gac AC, et al. Rare but authentic Philadelphia-positive acute myeloblastic leukemia: two case reports and a literature review of characteristics, treatment and outcome. Hematol Oncol Stem Cell Ther. 2015;8:28–33.
Kohn G, Manny N, Eldor A, et al. De novo appearance of the ph-1 chromosome in a previously monosomic bone marrow (45,XX,-6): conversion of a myeloproliferative disorder to acute myelogenous leukemia. Blood. 1975;45:653–7.
Abe S, Sandberg AA. Chromosomes and causation of human cancer and leukemia. XXXII. Unusual features of Ph1-positive acute myeloblastic leukemia (AML), including a review of the literature. Cancer. 1979;43:2352–64.
Smadja N, Krulik M, De Gramont A, et al. Acquisition of a Philadelphia chromosome concomitant with transformation of a refractory anemia into an acute leukemia. Cancer. 1985;55:1477–81.
Jacobsen RJ, Himoe E, Sacher RA, et al. Late appearance of Philadelphia chromosome. Br J Haematol. 1986;63:392–4.
Vandenberghe E, Martiat P, Baens M, et al. Megakaryoblastic leukemia with an N-ras mutation and late acquisition of a Philadelphia chromosome. Leukemia. 1991;5:683–6.
Shimamoto T, Ohyashiki K, Ohyashiki JH, et al. Late appearance of a Philadelphia translocation with minor-BCR/ABL transcript in a t(7;11)(p15; p15) acute myeloid leukemia. Leukemia. 1995;9:640–2.
Fujimura T, Ohyashiki K, Ohyashiki JH, et al. Two additional cases of acute myeloid leukemia with t(7;11)(p15; p15) having low neutrophil alkaline phosphatase scores. Cancer Genet Cytogenet. 1993;68:143–6.
Matsue K, Miyamoto T, Ito M, et al. Late appearance of the Philadelphia chromosome with monosomy 7 in a patient with de novo AML with trilineage myelodysplasia. Am J Hematol. 1995;49:341–6.
Chen Z, Morgan R, Notohamiprodjo M, et al. The Philadelphia chromosome as a secondary change in leukemia: three case reports and an overview of the literature. Cancer Genet Cytogenet. 1998;101:148–51.
Najfeld V, Geller M, Troy K, et al. Acquisition of the Ph chromosome and BCR-ABL fusion product in AML-M2 and t(8;21) leukemia: cytogenetic and FISH evidence for a late event. Leukemia. 1998;12:517–9.
Shah N, Leaker MT, Teshima I, et al. Late-appearing Philadelphia chromosome in childhood acute myeloid leukemia. Pediatr Blood Cancer. 2008;50:1052–3.
Bacher U, Haferlach T, Alpermann T, et al. Subclones with the t(9;22)/BCR-ABL1 rearrangement occur in AML and seem to cooperate with distinct genetic alterations. Br J Haematol. 2011;152:713–20.
Aoki J, Kakihana K, Kobayashi T, et al. Tyrosine kinase inhibitor therapy for acute myeloid leukemia with late-appearing Philadelphia chromosome. Leuk Res. 2012;36:e41–e42.
Neuendorff NR, Schwarz M, Hemmati P, et al. BCR-ABL1(+) acute myeloid leukemia: clonal selection of a BCR-ABL1(-) subclone as a cause of refractory disease with nilotinib treatment. Acta Haematol. 2015;133:237–41.
Verhoef G, Meeus P, Stul M, et al. Cytogenetic and molecular studies of the Philadelphia translocation in myelodysplastic syndromes. Report of two cases and review of the literature. Cancer Genet Cytogenet. 1992;59:161–6.
Kohno T, Amenomori T, Atogami S, et al. Progression from myelodysplastic syndrome to acute lymphoblastic leukaemia with Philadelphia chromosome and p190 BCR-ABL transcript. Br J Haematol. 1996;93:389–91.
Roumier C, Daudignon A, Soenen V, et al. p190 bcr-abl rearrangement: a secondary cytogenetic event in some chronic myeloid disorders? Haematologica. 1999;84:1075–80.
Kelemen K, Galani K, Conley CR, et al. Secondary Philadelphia chromosome and erythrophagocytosis in a relapsed acute myeloid leukemia after hematopoietic cell transplantation. Cancer Genet. 2014;207:268–71.
Yamashita S, Umemura T, Sadamura S, et al. Acute leukemias expressing p210-and p 190-type bcr/abl mRNAs: report of two cases and review of the literature. Acta Haematol. 1996;96:99–104.
Nakamura K, Inaba T, Nishimura J, et al. Molecular analysis of BCR/ABL products in a case of myelodysplastic syndrome with late appearing Philadelphia chromosome. Br J Haematol. 1991;78:130–2.
Katsuno M, Yamashita S, Sadamura S, et al. Late-appearing Philadelphia chromosome in a patient with acute nonlymphocytic leukaemia derived from myelodysplastic syndrome: detection of P210- and P190-type bcr/abl fusion gene transcripts at the leukaemic stage. Br J Haematol. 1994;87:51–56.
Keung YK, Beaty M, Powell BL, et al. Philadelphia chromosome positive myelodysplastic syndrome and acute myeloid leukemia-retrospective study and review of literature. Leuk Res. 2004;28:579–86.
Prebet T, Michallet AS, Charrin C, et al. Secondary Philadelphia chromosome after non-myeloablative peripheral blood stem cell transplantation for a myelodysplastic syndrome in transformation. Bone Marrow Transplant. 2004;33:247–9.
Hamaguchi H, Suzukawa K, Nagata K, et al. Establishment of a novel human myeloid leukaemia cell line (HNT-34) with t(3;3)(q21; q26), t(9;22)(q34;q11) and the expression of EVI1 gene, P210 and P190 BCR/ABL chimaeric transcripts from a patient with AML after MDS with 3q21q26 syndrome. Br J Haematol. 1997;98:399–407.
Miller BA, Reid MM, Nell M, et al. T-cell acute lymphoblastic leukaemia with late developing Philadelphia chromosome. Br J Haematol. 1984;56:139–46.
Coad JE, Arthur DC, Gajl-Peczalska KJ, et al. Late-developing Philadelphia chromosomes in a case of T-cell acute lymphoblastic leukemia. Leukemia. 1994;8:889–94.
Tsuchiya H, Migita M, Yamamori S, et al. A late-appearing Philadelphia chromosome in acute lymphoblastic leukemia confirmed by expression of BCR-ABL mRNA. Leukemia. 1995;9:1689–93.
Tchirkov A, Bons JM, Chassagne J, et al. Molecular detection of a late-appearing BCR-ABL gene in a child with T-cell acute lymphoblastic leukemia. Ann Hematol. 1998;77:55–59.
Campbell LJ, Martinow A, Michael PM, et al. Correlation of cytogenetics, BCR-ABL PCR studies and fluorescence in situ hybridisation (FISH) in adult acute lymphoblastic leukaemia. Aust N Z J Med. 1999;29:707–12.
Sessarego M, Defferrari R, Dejana A, et al. Late-appearing Philadelphia chromosome in acute lymphoblastic leukemia. Cancer Genet Cytogenet. 1990;48:35–38.
Inokuchi K, Shinohara T, Futaki M, et al. Establishment of a cell line with variant BCR/ABL breakpoint expressing P180BCR/ABL from late-appearing Philadelphia-positive acute biphenotypic leukemia. Genes Chromosomes Cancer. 1998;23:227–38.
Chen L, Stamatoullas A, Bastard C, et al. Secondary Philadelphia chromosome in a patient with acute lymphoblastic leukemia. Cancer Genet Cytogenet. 2004;152:132–5.
Hato A, Murayama T, Nishikawa S, et al. Philadelphia chromosome positive acute lymphoblastic leukemia showing normal karyotype in G-banding chromosomal examination before chemotherapy. Hematology. 2005;10:379–81.
Adriana Z, Al Bahar S, Pandita R. Acute lymphoblastic leukemia with a late- appearing Philadelphia chromosome: case report and review of the literature. Gulf J Oncol. 2013;1:81–86.
Simons A, Shaffer LG, Hastings RJ. Cytogenetic nomenclature: changes in the ISCN 2013 compared to the 2009 edition. Cytogenet Genome Res. 2013;141:1–6.
Merzianu M, Medeiros LJ, Cortes J, et al. inv(16)(p13q22) in chronic myelogenous leukemia in blast phase: a clinicopathologic, cytogenetic, and molecular study of five cases. Am J Clin Pathol. 2005;124:807–14.
Luthra R, Medeiros LJ. TaqMan reverse transcriptase-polymerase chain reaction coupled with capillary electrophoresis for quantification and identification of bcr-abl transcript type. Methods Mol Biol. 2006;335:135–45.
Gabert J, Beillard E, van der Velden VH, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe Against Cancer program. Leukemia. 2003;17:2318–57.
Luthra R, Patel KP, Reddy NG, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465–73.
Ok CY, Patel KP, Garcia-Manero G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54.
Warren M, Luthra R, Yin CC, et al. Clinical impact of change of FLT3 mutation status in acute myeloid leukemia patients. Mod Pathol. 2012;25:1405–12.
Patel KP, Barkoh BA, Chen Z, et al. Diagnostic testing for IDH1 and IDH2 variants in acute myeloid leukemia an algorithmic approach using high-resolution melting curve analysis. J Mol Diagn. 2011;13:678–86.
Bai S, Hu S. BCR-ABL1-negative acute myeloid leukaemia relapsing as BCR-ABL1-positive disease. Br J Haematol. 2017;176:514.
Arber DA, Orazi A, Hasserjian R, et al. The2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Ghaffari S, Daley GQ, Lodish HF. Growth factor independence and BCR/ABL transformation: promise and pitfalls of murine model systems and assays. Leukemia. 1999;13:1200–6.
Voncken JW, Griffiths S, Greaves MF, et al. Restricted oncogenicity of BCR/ABL p190 in transgenic mice. Cancer Res. 1992;52:4534–9.
Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–30.
Lesesve JF, Troussard X, Bastard C, et al. p190bcr/abl rearrangement in myelodysplastic syndromes: two reports and review of the literature. Br J Haematol. 1996;95:372–5.
Dhingra K, Talpaz M, Kantarjian H, et al. Appearance of acute leukemia-associated P190BCR-ABL in chronic myelogenous leukemia may correlate with disease progression. Leukemia. 1991;5:191–5.
Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–52.
Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–9.
Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42.
Ottmann OG, Druker BJ, Sawyers CL, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100:1965–71.
Drummond MW, Lush CJ, Vickers MA, et al. Imatinib mesylate-induced molecular remission of Philadelphia chromosome-positive myelodysplastic syndrome. Leukemia. 2003;17:463–5.
Dai HP, Xue YQ, Wu LL, et al. p210 BCR/ABL1 as a secondary change in a patient with acute myelomonocytic leukemia (M4Eo) with inv(16). Int J Hematol. 2012;96:814–7.
Salem A, Loghavi S, Tang G, et al. Myeloid neoplasms with concurrent BCR-ABL1 and CBFB rearrangements: a series of 10 cases of a clinically aggressive neoplasm. Am J Hematol. 2017;92:520–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kurt, H., Zheng, L., Kantarjian, H.M. et al. Secondary Philadelphia chromosome acquired during therapy of acute leukemia and myelodysplastic syndrome. Mod Pathol 31, 1141–1154 (2018). https://doi.org/10.1038/s41379-018-0014-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0014-x
This article is cited by
-
Secondary chronic myeloid leukemia in a patient with CALR and ASXL1-mutated primary myelofibrosis
International Journal of Hematology (2022)
-
t(11;16)(q23;p13)/KMT2A-CREBBP in hematologic malignancies: presumptive evidence of myelodysplasia or therapy-related neoplasm?
Annals of Hematology (2020)
-
Chronic Myeloid Leukemia: Beyond BCR-ABL1
Current Hematologic Malignancy Reports (2018)
-
Imatinib
Reactions Weekly (2018)
