
Abstract
Acute myeloid leukemia (AML) with intrachromosomal amplification of chromosome 21 (iAMP21) is rare and has not been well characterized. We report 13 patients, 7 men and 6 women, with a median age of 65 years. Eleven patients presented with AML with myelodysplasia-related changes, and two patients had therapy-related AML. Cytopenias were detected in all patients (11 pancytopenia and two bi-lineage cytopenia). Myelodysplastic changes were observed in all 11 patients with adequate cells to evaluate. Myelofibrosis was present in ten patients. All patients had a complex karyotype, including abnormalities of chromosomes 5, 7, 17, and hsr(21)(q22), and ten patients showed TP53 deletion and/or mutation. Eleven patients received AML-based chemotherapy, one of whom also received hematopoietic stem cell transplant. By the end of the last follow-up, eight patients died with median survival of 3.2 months, four patients were alive with persistent AML, and one was in complete remission. The median overall survival was 6 months for all patients. We conclude that AML with iAMP21 is often associated with cytopenias, myelodysplasia, a complex karyotype, TP53 mutation/deletion, and a poor prognosis despite current therapies.
Similar content being viewed by others
Introduction
The Runt domain of transcription factor RUNX1, also known as AML1 and CBFA2, located on chromosomal 21q22. RUNX1 is frequently dysregulated in leukemias as results of chromosomal translocations, amplification, and point mutations [1]. The most frequent translocations involving RUNX1 are t(8;21)(q22;q22)/RUNX1T1-RUNX1 in acute myeloid leukemia (AML) and t(12;21)(p12.3;q22)/ETV6-RUNX1 in B-lymphoblastic leukemia (B-ALL) [2,3,4]. Intrachromosomal amplification of chromosome 21 (iAMP21) is an uncommon finding in hematologic neoplasms that is most often detected by fluorescence in situ hybridization (FISH) analysis using a RUNX1 probe. iAMP21 is recognized as ≥5 copies of RUNX1 within one cell or ≥3 extra copies of RUNX1 on a single abnormal chromosome 21 [5,6,7]. iAMP21 has been well characterized in B-ALL and recognized as a provisional entity by the current World Health Organization (WHO) classification [5]. B-ALL with iAMP21 accounts for ~2% of pediatric B-ALL, with a median age of 9 years, and is often associated with low white blood cell (WBC) count [5,6,7,8]. iAMP21 has been recognized as a poor risk factor in B-ALL and patients with B-ALL iAMP21 should be treated with more intensive therapy to overcome this adverse risk [7].
iAMP21 has been rarely reported in AML, with <15 cases being reported in the literature, mainly in single case reports [1, 2, 9,10,11,12,13]. Patients with AML iAMP21 have been mainly adults, who always had a complex karyotype. Clinicopathologic findings, molecular mutation profiles, and outcomes were largely unavailable in the reported cases.
In this study, we report 13 patients with AML iAML21 from our institution. We describe the clinicopathologic, immunophenotypical, and conventional cytogenetics findings in this study group and we performed next-generation sequencing (NGS) analysis in 11 patients.
Materials and methods
Case selection
We searched the cytogenetics archives of our institution for cases of AML with RUNX1 ≥ 5 copies during the years of January 2010 through December 2019. Thirteen cases of AML iAMP21 were identified. Clinical and laboratory data and patient outcomes were obtained by review of medical records. This study was conducted in accordance with the Declaration of Helsinki.
Morphologic examination
Wright–Giemsa-stained peripheral blood (PB) and bone marrow (BM) aspirate smears, and hematoxylin and eosin-stained sections of BM core biopsy specimens were assessed in all cases. Myelofibrosis was evaluated by reticulin and trichrome stains performed on the BM core biopsy when available. The grade of myelofibrosis was based on the European Consensus on grading of BM fibrosis [14].
Flow cytometric immunophenotyping
BM aspirate specimens were subjected to standard eight-color flow cytometry immunophenotypic analysis as described previously [15], using antibodies against the following antigens: CD2, CD3, CD4, CD5, CD7, CD9, CD10, CD11b, CD13, CD14, CD19, CD20, CD22, CD25, CD34, CD38, CD41, CD56, CD64, CD79b, CD117, CD123, Human Leukocyte Antigen-DR isotype (HLA-DR), myeloperoxidase (MPO), and terminal deoxynucleotide transferase (TdT; BD Biosciences, San Jose, CA). Cytochemical stains for MPO were performed on diagnostic BM aspirate smears.
Conventional cytogenetics and FISH analyses
Conventional G-banded chromosomal analysis was performed on unstimulated 24-h and 48-h BM aspirate cultures using standard techniques described previously [16]. Twenty metaphases from each sample were analyzed and results were reported according to the 2016 International System for Human Cytogenetics Nomenclature (ISCN 2016). A complex karyotype was defined as ≥3 chromosomal abnormalities.
FISH analysis was performed on BM aspirate smears or cultured cells with RUNX1T1/RUNX1 dual-color dual-fusion FISH probes (Abbott Molecular) according to the manufacturer’s instructions. The presence of ≥5 and/or clusters of RUNX1 signals was considered as evidence of RUNX1 amplification. Of a note, our laboratory started to perform FISH for RUNX1T1/RUNX1 on all new AML patients presented at our institution in 2018.
Molecular mutation studies
Molecular analyses were performed as a part of the routine clinical work-up. Targeted NGS studies using panels of genes commonly altered in hematopoietic neoplasms were performed in nine patients, using a 28-gene, 53-gene, or 81-gene panel (Supplemental 1) as described previously [17].
Statistical analysis
The Kaplan–Meier method was used to estimate overall survival (OS) from the date of iAMP21 detection. The clinical follow-up end points included the date of death from any cause, or censored at time of hematopoietic stem cell transplant (SCT) or last follow-up for alive patients.
Results
Patients
We identified 25 patients with iAMP21 during the study period, 12 with B-ALL and 13 with AML, the later 13 patients constitute this study cohort, accounting for ~0.1% of total AML cases at our institution.
A summary of the clinical features of these patients is shown in Table 1. All patients were adults, six women and seven men, with a median age of 65 years (range, 34–83 years). Nine patients had a history of malignancy and had been treated at somewhere outside of our institutions (Table 1), the intervals from diagnosis of the first (prior) malignancy to the detection of iAMP21 were from 5 months to 294 months. For two patients who had therapy-related AML (t-AML), patient #12 received adriamycin, cytoxan, gemzar, paclitaxel, herceptin/trastuzumab, abraxane, and carboplatin for her breast cancer, and patient #13 was treated with revlimid, velcade, and dexamethasone for plasma cell myeloma. Of note, five patients had karyotypic and/or chromosomal information of the prior malignancies. Patient #6 had a karyotype of 46,XX,−5,del(7)(q22q36),+8,−21,+mar[7]/46,XX[8]; patient #13 had a normal diploid karyotype; patient #7 was reported to have a complex karyotype; patient #10 had monosomy 7 and add(11q); and patient #11 had del(5q). However, no detail karyotype was available for the latter three patients (cases #7, 10, and 11). None of the patients had FISH for RUNX1 performed at outside hospitals. iAMP21 was detected in the first BM specimen at our institution for all patients.
PB and BM findings
Complete blood count (CBC) data are available for all patients (Table 1). All patients had cytopenias, 11 with pancytopenia and 2 with bi-lineage cytopenia. The median WBC count was 1.3 × 109/L (range, 0.1–15.4 × 109/L), median hemoglobin level 8.2 g/dL (range, 7.2–9.6 g/dL), median platelet count 21 × 109/L (range, 4–163 × 109/L), and median blast count in the PB 8.5% (range, 0–67%).
The findings in BM core biopsy specimens and aspirate smears are summarized in Supplemental 2. The BM cellularity ranged from 5% to 95%. The median blast count was 40% (range, 20–80%). Blasts were medium sized to large with fine chromatin and distinct nucleoli. Cytoplasmic vacuoles were observed in seven cases. Auer rods were rare or absent. Dysplasia of one or multiple lineages was observed in 11 patients, and dysplasia could not be evaluated in the other 2 patients (#2 and #4) due to lack of adequate hematopoietic elements. Using the French–American–British (FAB) classification system [18], the neoplasms were classified as M0 (n = 1), M1 (n = 5), M2 (n = 6), and M6 (n = 1). Using the WHO classification, 11 neoplasms were classified as AML with myelodysplasia-related changes (AML-MRC), including two patients with relapsed AML; two were classified as t-AML.
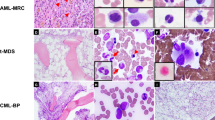
Cytochemical staining for MPO was performed on BM aspirate smears of nine patients: unequivocally positive in two, rare positive blasts in two, and negative in five. Reticulin and trichrome stains were performed on BM biopsy specimens in all cases and ten patients showed variable degree myelofibrosis, MF-1 (n = 6), MF-2 (n = 3), MF-3 (n = 1), and MF-0 (n = 3), (Fig. 1, Supplemental 2).

a–c (Case #1), a: bone marrow (BM) aspirate smear showed blasts (1000×). b: BM biopsy specimen showed sheets of immature cells (400×). c: Reticulin stain showed increased reticulin fibers (400×). d–f (Case #5), d: BM aspirate smear showed blasts (1000×). e BM biopsy specimen showed sheets of immature cells (400×). f: Reticulin stain showed mild increase in reticulin fibers (MF-1) in a loose network (400×).
Immunophenotypic findings
The immunophenotypes of the blasts are summarized in Table 2. The blasts were variably positive for following markers: CD13 (11/13), CD33 (11/13), CD117 (11/13), MPO (7/12), CD7 (6/13), CD4 (4/11), and TdT (4/12). The blasts were negative for CD2, sCD3, cCD3, CD5, CD14, and CD19 in all cases assessed
Cytogenetic and molecular findings
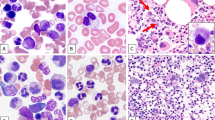
Karyotype and copies of RUNX1 detected by FISH analysis are summarized in Table 3. All patients had a complex karyotype; 12 patients showed a high degree of genomic complexity as indicated by ≥5 cytogenetic alterations; and 10 had a monosomal karyotype. The most common additional cytogenetic alterations were: −5/del[5q] (n = 9), −7/del[7q] (n = 6), and −17/del[17p] (n = 6). High copy numbers of RUNX1 were present as: hsr[21](q22) (n = 4), extra copies of chromosome 21 and/or i[21q], insertion of 21q22, double minutes (dmin), and ring or marker chromosomes. FISH analysis revealed ≥5 RUNX1 copies in interphase cells in 12 out of 13 cases, except case #9, in which 3–4 copies of RUNX1 were observed in interphases; but it showed hsr[21](q22) on chromosomal analysis and clusters of RUNX1 signals on the metaphase FISH (Fig. 2).

Left side: conventional cytogenetic analysis; right side: FISH analysis with RUNX1T1 (red)/RUNX1 (green) dual-color dual-fusion probe. a Case #9, karyotype: 46,XY,del(5)(q13q33),+10,del(11)(q12),der(11;20)(q10;p10),del(17)(p11.2),−18,hsr(21)(q22). FISH showed two copies of RUNX1T1, four copies (interphase) or cluster (metaphase) of RUNX1 signals. b Case #10, karyotype: 54,XX,+1,+2,−4,+6,+8,+10,+14,add(17)(p11.2),+add(21)(p11.1),+i(21)(q10),+mar. FISH showed three copies of RUNX1T1 (+8) and >5 or cluster of RUNX1 signals.
Molecular mutation data are summarized in Table 3. Mutation of TP53 was detected in 9 out of 11 patients tested, 6 of them showed only TP53 mutation without any other gene mutations. Of a note, two patients (cases #6 and #7) showed TP53 mutation at the initial diagnosis of MDS and chronic myelomonocytic leukemia (CMML-1), respectively. Other mutations in AML iAMP21 were uncommon and included DNMT3A (n = 2), RUNX1 (n = 1), PRPF40B (n = 1), and NF1 (n = 1). None of the fusion transcripts associated with AML, such as t(8;21), t(9;22), and inv [16], were detected in any of these cases. None of the 13 patients tested showed FLT3, NRAS, or KRAS mutation.
Outcomes
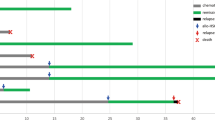
After detection of iAMP21, ten patients received multi-regimen chemotherapy. One patient achieved remission and received subsequent allogeneic SCT (at 4 months); the other nine patients were refractory to chemotherapy. At time of last follow-up, eight patients died from disease, four were alive with persistent disease, and one patient (case #5) was alive in complete remission after SCT. The median OS for this group of patients was 6 months (Fig. 3). Of a note, cases #4, #7, #11, and #13 had a short follow-up.

Overall survival of 13 patients.
We also compared the OS of these 13 patients with iAMP21 to 20 patients who had AML with complex karyotype (14 had monosomal karyotype) but no iAMP21. Though patients with iAMP21 had a shorter OS, it was not significant (median OS, 6 months vs. 8.4 months, p = 0.2624).
Discussion
Amplification is a genomic alteration that typically results in overexpression of particular oncogenes located within the amplicon. Gene amplifications are uncommon events in AML and are often associated with refractoriness to chemotherapy and an aggressive clinical course [16, 19, 20]. KMT2A (MLL) and MYC are the most two frequently amplified genes in AML [16, 19]. Amplification of genes can locate extrachromosomally as dmin, intrachromosomally as homogeneously staining regions (hsr), or as a ring, marker/derivative chromosome, or isochromosome [12, 13, 16, 19, 21, 22]. Except hsr[21](q22) that can be easily recognized as iAMP21, other forms of amplification of RUNX1 could be misinterpreted as unclarified ring, mark, or derivative chromosomes and “missed” by chromosomal analysis. Therefore, the true frequency of iAMP21 in AML may be underestimated. The most reliable assay to detect iAMP21 is FISH analysis with RUNX1 probe. In our institution, we identified 13 cases in the past 10 years, 9 of them were from the recent 3 years. The “increased” rate in the recent 3 years is likely due to the initiation of FISH analysis for RUNX1T1/RUNX1 in all newly diagnosed AML, which started 3 years ago.
RUNX1 is expressed in all hematopoietic lineages and acts to regulate the expression of various genes specific to hematopoiesis. RUNX1 is one of the genes most frequently dysregulated in leukemia through different mechanisms, including translocations, amplifications, and mutation [1]. B-ALL with iAMP21 has been added as a new provisional entity in the 2017 WHO classification [23]. AML iAMP21 often exhibits several features that differ from B-ALL iMAP21. Patients with AML iAMP21 are almost all adults, with only one child reported who had constitutional r(21)[11]. AML iAMP21 is almost always associated with complex karyotype [2, 9,10,11,12,13], most of which being highly complex. AML iAMP21 also shows high frequency of TP53 deletion and/or mutation: 3/3 patients reported in the literature with mutation information available [9, 10] and 10/13 patients in our cohort had TP53 mutation and/or deletion. Patients with AML iAMP21 often present pancytopenia: 3/3 reported patients who had CBC information available [2, 9, 11] and 11/13 in this study group. Lastly, patients with AML iAMP21 are highly refractory to standard AML-based induction therapy and only one patient in this cohort (case #5) achieved remission after induction.
Outcome information was available in only two patients reported in the literature. One patient responded to treatment initially, but relapsed after 9 months and died 3 months later after SCT [2]. The other patient was reported to be in complete remission after SCT [11]. In our cohort, eight patients died with a median survival of 3.2 months; five patients were alive but four of them (#4, #7, #11, and #13) were recently diagnosed and had relatively short follow-up periods (<5 months), only one patient (#5) achieved remission after induction and has been in remission for ~6 year after SCT. Comparing to other patients in this cohort, this patient (#5) had a relatively less complex, non-monosomal karyotype, with a very low percentage (4%) of interphases showing RUNX1 amplification, and no gene mutation (including TP53) by 53-gene NGS panel. All these features may contribute to the good responses to therapies and a better outcome.
The regions predominantly amplified in iAMP21 span chromosome 21q22.1q22.3 and ranged from 11.8 to 40.0 Mb [9, 22]. Three genes, APP, ERG, and ETS2 are often amplified in patients with iAMP21 [9, 22, 24]. APP has been linked to Alzheimer’s disease and dementia in adults with Down syndrome. ETS2 and ERG are proto-oncogenic transcription factors that are involved in cell cycle development and regulation. RUNX1 may be a bystander amplified with other significant genes [25]. Although the pathogenesis of AML iAMP21 is not clear, the TP53 gene has important function in maintaining genomic stability and integrity. Mutational inactivation of TP53 has been shown in experimental models to result in gene amplification as well as in aneuploidy [26, 27]. Given that most patients with AML iAMP21 had TP53 deletion/mutation and a very complex karyotype, genome instability and chromothripsis could be the underlying mechanisms of pathogenesis [26, 28]. Two patients (#6 and #7) showed TP53 mutation at the initial diagnosis of MDS and CMML-1, supporting TP53 mutation to be a primary event and likely play an important during the pathogenesis. Interestingly, two patients with AML iAMP21 had constitutional abnormality of r(21) [2, 11], suggesting the “instability” of structurally abnormal chromosome 21 r(21) and leads to iAMP21. Pathogenetic mechanisms in AML with iAMP21 may overlap with AML associated with MLL amplification, which shares some features, such as complex karyotype, TP53 mutation/deletion, and aggressive outcomes [16]. A more comprehensive molecular study may help to understand the pathogenesis of this rare disease, especially for the cases with no or only TP53 mutation.
In summary, AML iAMP21 is often associated with pancytopenia, morphological dysplasia, a complex karyotype, and TP53 deletion/mutation. Patients with AML iAMP21 are often refractory to conventional AML therapy and have very poor outcomes. Early stem cell transplantation as well as novel therapies may improve patients’ outcome.
References
Roumier C, Fenaux P, Lafage M, Imbert M, Eclache V, Preudhomme C. New mechanisms of AML1 gene alteration in hematological malignancies. Leukemia. 2003;17:9–16.
Streubel B, Valent P, Lechner K, Fonatsch C. Amplification of the AML1(CBFA2) gene on ring chromosomes in a patient with acute myeloid leukemia and a constitutional ring chromosome 21. Cancer Genet Cytogenet. 2001;124:42–6.
Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995;9:1985–9.
McLean TW, Ringold S, Neuberg D, Stegmaier K, Tantravahi R, Ritz J, et al. TEL/AML-1 dimerizes and is associated with a favorable outcome in childhood acute lymphoblastic leukemia. Blood. 1996;88:4252–8.
Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th ed; 2017.
Harrison CJ, Moorman AV, Schwab C, Carroll AJ, Raetz EA, Devidas M, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28:1015–21.
Heerema NA, Carroll AJ, Devidas M, Loh ML, Borowitz MJ, Gastier-Foster JM, et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children’s oncology group studies: a report from the children’s oncology group. J Clin Oncol. 2013;31:3397–402.
Robinson HM, Broadfield ZJ, Cheung KL, Harewood L, Harris RL, Jalali GR, et al. Amplification of AML1 in acute lymphoblastic leukemia is associated with a poor outcome. Leukemia. 2003;17:2249–50.
Nguyen D, Li Y, Safah H, Brown TC. RUNX1 deletion/amplification in therapy-related acute myeloid leukemia: a case report and review of the literature. Cancer Genet. 2019;238:37–43.
Andersen MK, Christiansen DH, Pedersen-Bjergaard J. Amplification or duplication of chromosome band 21q22 with multiple copies of the AML1 gene and mutation of the TP53 gene in therapy-related MDS and AML. Leukemia. 2005;19:197–200.
Burillo-Sanz S, Vargas MT, Morales-Camacho RM, Caballero-Velazquez T, Sanchez J, Garcia-Lozano JR, et al. RUNX1 amplification in AML with myelodysplasia-related changes and ring 21 chromosomes. Hematol Oncol. 2017;35:894–9.
Moosavi SA, Sanchez J, Adeyinka A. Marker chromosomes are a significant mechanism of high-level RUNX1 gene amplification in hematologic malignancies. Cancer Genet Cytogenet. 2009;189:24–8.
Viguie F, Aboura A, Bouscary D, Guesnu M, Baumelou E, Dreyfus F, et al. Isodicentric/pseudoisodicentric chromosome 21 amplification in four cases of acute myelocytic leukemia or myelodysplasia. Cancer Genet Cytogenet. 2002;138:80–4.
Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–32.
Ouyang J, Goswami M, Tang G, Peng J, Ravandi F, Daver N, et al. The clinical significance of negative flow cytometry immunophenotypic results in a morphologically scored positive bone marrow in patients following treatment for acute myeloid leukemia. Am J Hematol. 2015;90:504–10.
Tang G, DiNardo C, Zhang L, Ravandi F, Khoury JD, Huh YO, et al. MLL gene amplification in acute myeloid leukemia and myelodysplastic syndromes is associated with characteristic clinicopathological findings and TP53 gene mutation. Hum Pathol. 2015;46:65–73.
Tang G, Sydney Sir Philip JK, Weinberg O, Tam W, Sadigh S, Lake JI, et al. Hematopoietic neoplasms with 9p24/JAK2 rearrangement: a multicenter study. Mod Pathol. 2019;32:490–8.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–8.
Huh YO, Tang G, Talwalkar SS, Khoury JD, Ohanian M, Bueso-Ramos CE, et al. Double minute chromosomes in acute myeloid leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia are associated with micronuclei, MYC or MLL amplification, and complex karyotype. Cancer Genet. 2016;209:313–20.
Maitta RW, Cannizzaro LA, Ramesh KH. Association of MLL amplification with poor outcome in acute myeloid leukemia. Cancer Genet Cytogenetics. 2009;192:40–3.
Sarova I, Brezinova J, Zemanova Z, Izakova S, Lizcova L, Malinova E, et al. Cytogenetic manifestation of chromosome 11 duplication/amplification in acute myeloid leukemia. Cancer Genet Cytogenetics. 2010;199:121–7.
Baldus CD, Liyanarachchi S, Mrozek K, Auer H, Tanner SM, Guimond M, et al. Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: amplification discloses overexpression of APP, ETS2, and ERG genes. Proc Natl Acad Sci USA. 2004;101:3915–20.
Borowitz M, Chan J, Downing J, Beau ML, Arber D. B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities, In: Swerdlow S, Campo E, Harris N, editors. WHO classification of tumore of hematopoietic and lymphoid tissue. IARC: Lyon; 2017.
Weber S, Haferlach C, Jeromin S, Nadarajah N, Dicker F, Noel L, et al. Gain of chromosome 21 or amplification of chromosome arm 21q is one mechanism for increased ERG expression in acute myeloid leukemia. Genes Chromosomes Cancer. 2016;55:148–57.
Mrozek K, Heinonen K, Theil KS, Bloomfield CD. Spectral karyotyping in patients with acute myeloid leukemia and a complex karyotype shows hidden aberrations, including recurrent overrepresentation of 21q, 11q, and 22q. Genes Chromosomes Cancer. 2002;34:137–53.
Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–48.
Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–35.
Li Y, Schwab C, Ryan S, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508:98–102.
Acknowledgements
We would like to extend our appreciation to our colleagues for the helpful discussions and support throughout this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Xie, W., Xu, J., Hu, S. et al. iAMP21 in acute myeloid leukemia is associated with complex karyotype, TP53 mutation and dismal outcome. Mod Pathol 33, 1389–1397 (2020). https://doi.org/10.1038/s41379-020-0494-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0494-3
This article is cited by
-
Prospective evaluation of genome sequencing to compare conventional cytogenetics in acute myeloid leukemia
Blood Cancer Journal (2023)
-
21q22 amplification detection in three patients with acute myeloid leukemia: cytogenomic profiling and literature review
Molecular Cytogenetics (2022)
-
Optical genome mapping refines cytogenetic diagnostics, prognostic stratification and provides new molecular insights in adult MDS/AML patients
Blood Cancer Journal (2022)
