
Abstract
We investigated data from 180 consecutive patients with myelodysplastic/myeloproliferative neoplasms with SF3B1 mutation and thrombocytosis (MDS/MPN-SF3B1-T) who were diagnosed according to the 2022 World Health Organization (WHO) classification of myeloid neoplasms to identify covariates associated with survival. At a median follow-up of 48 months (95% confidence interval [CI] 35–61 months), the median survival was 69 months (95% CI 59–79 months). Patients with bone marrow ring sideroblasts (RS) < 15% had shorter median overall survival (OS) than did those with bone marrow RS ≥ 15% (41 months [95% CI 32–50 months] versus 76 months [95% CI 59–93 months]; P < 0.001). According to the univariable analyses of OS, age ≥ 65 years (P < 0.001), hemoglobin concentration (Hb) < 80 g/L (P = 0.090), platelet count (PLT) ≥ 800 × 10E + 9/L (P = 0.087), bone marrow RS < 15% (P < 0.001), the Revised International Prognostic Scoring System (IPSS-R) cytogenetic category intermediate/poor/very poor (P = 0.005), SETBP1 mutation (P = 0.061) and SRSF2 mutation (P < 0.001) were associated with poor survival. Based on variables selected from univariable analyses, two separate survival prediction models, a clinical survival model, and a clinical-molecular survival model, were developed using multivariable analyses with the minimum value of the Akaike information criterion (AIC) to specifically predict outcomes in patients with MDS/MPN-SF3B1-T according to the 2022 WHO classification.
Similar content being viewed by others
Introduction
Refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T) was proposed as a provisional entity in 2001 for the World Health Organization (WHO) classification of hematopoietic tumors and classified as “myelodysplastic/myeloproliferative neoplasm (MDS/MPN), unclassifiable” [1, 2]. RARS-T remained in the 2008 revision of the WHO classification [3,4,5]. In the 2016 revision, RARS-T was renamed MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) and became a well-characterized, distinct MDS/MPN overlap entity [6, 7].
MDS/MPN-RS-T is characterized by ≥15% bone marrow ring sideroblasts (RS) and sustained thrombocytosis [6]. The median age at diagnosis is ~75 years [8,9,10,11,12,13]. Approximately 80% of patients have normal cytogenetics, and approximately one-half have SF3B1 and JAK2 mutations [14]. In those with SF3B1 mutations, the percentage of patients with bone marrow RS specified for the diagnosis of MDS/MPN-RS-T was unchanged [6]. This is unlike myelodysplastic syndrome with ring sideroblasts (MDS-RS), where only 5% bone marrow RS is required in the presence of SF3B1 mutation [6].
The median survival of people with MDS/MPN-RS-T is ~75 months [8, 10, 11]. Previous studies reported that age >80 years, abnormal cytogenetics, hemoglobin concentration (Hb) ≤ 100 g/L, and ASXL1 and/or SETBP1 mutations were correlated with worse survival [9,10,11]. There is no widely accepted survival prediction model.
Because of the high frequency of SF3B1 mutations in MDS/MPN-RS-T and the correlation between SF3B1 mutation and bone marrow RS, in the 2022 WHO classification, people with SF3B1 mutation and <15% bone marrow RS were reclassified as myelodysplastic/myeloproliferative neoplasms with SF3B1 mutation and thrombocytosis (MDS/MPN-SF3B1-T) [15,16,17,18,19].
Previous studies of MDS/MPN-RS-T were based on the 2008 or 2016 revision of the WHO classification and were restricted to people with ≥15% bone marrow RS [8,9,10,11,12, 20]. We aimed to describe the clinical and molecular landscapes of MDS/MPN-SF3B1-T patients, compare the prognoses of people with < or ≥15% bone marrow RS, and develop prognostic models.
Materials and methods
Subjects
MDS/MPN-SF3B1-T was diagnosed according to the 2022 WHO classification of myeloid neoplasms [19]. A total of 180 consecutive patients with MDS/MPN-SF3B1-T in our hospital between June 2006 and August 2023 were enrolled in the study. We interrogated clinical (sex, age at diagnosis, date of leukemia transformation, and date of death or last follow-up) and laboratory data (blood cell count, bone marrow aspirate smear, bone marrow biopsy, bone marrow RS, karyotypes, and sequencing analysis) from the subjects at the time of diagnosis. Bone marrow aspirate smears at diagnosis were available for all subjects, and cytogenetic data were available and evaluable for 169 subjects. To explore the differences between our cohort and previous cohorts of MDS/MPN-RS-T patients, we compared clinical and laboratory parameters and molecular signatures between the Mayo–Moffitt cohort (patients with MDS/MPN-RS-T, n = 158) and our cohort (patients with MDS/MPN-SF3B1-T, n = 180) [11]. In our study, we developed two survival prediction models, namely, the clinical survival model and the clinical-molecular model. The clinical model was constructed using data from all patients in our cohort, designated training cohort 1 (n = 180). Moreover, a clinical-molecular model was generated based on patients with next-generation sequencing (NGS) information in our cohort, which was defined as training cohort 2 (n = 122). There was no significant difference in clinical or laboratory parameters between training cohort 1 and training cohort 2 (Supplementary Table S1). Data from 43 patients with MDS/MPN-RS-T in the International Prognostic Scoring System (IPSS)-Molecular cohort were obtained from the cBioPortal platform [21, 22]. This cohort, referred to as the validation cohort (n = 43), was used as an external dataset for validation. The study was approved by the Ethics Committees of the Institute of Hematology, Chinese Academy of Medical Science, and Peking Union Medical College according to the guidelines of the Declaration of Helsinki.
Bone marrow evaluation
Wright‒Giemsa staining was performed on the bone marrow and blood slides of patients at the time of diagnosis. Five hundred nucleated cells from each bone marrow slide and 200 nucleated cells from each blood slide were enumerated and classified for histological assessment. Prussian blue staining was performed on bone marrow slides to identify and enumerate the RS. Hematoxylin–eosin and Gomori methenamine silver staining were routinely performed on bone marrow biopsy sections. The degree of bone marrow fibrosis was classified using European consensus guidelines [23].
Cytogenetics
Chromosome analyses were performed on metaphase cells from unstimulated bone marrow aspirates after 24 h of culture using R-banding techniques. Cytogenetic abnormalities were analyzed and reported using the International System for Human Cytogenetic Nomenclature (2013) and classified according to the Revised International Prognostic Scoring System (IPSS-R) [24].
Targeted gene sequencing
DNA from the bone marrow or peripheral blood of patients at diagnosis was used for NGS as described [25]. The sequences of 122 patients with MDS/MPN-SF3B1-T were sequenced with a 14-gene panel using NGS at diagnosis (Supplementary Table S2). In addition, in patients without NGS data, the SF3B1 mutation (at the 666th and 700th amino acid sites) status in 28 patients with MDS/MPN-SF3B1-T was detected using Sanger sequencing, and the JAK2 V617F status in 42 patients with MDS/MPN-SF3B1-T was tested using allele-specific PCR with a sensitivity of 1%.
Relative mutation dominance
In patients harboring both SF3B1 and myeloproliferative neoplasm (MPN)-driver mutations (JAK2/CALR/MPL mutations) in MDS/MPN-SF3B1-T, a cutoff of ≥5% difference between variant allele frequencies (VAFs) of the two mutations was used to evaluate the relative dominant mutation [26]. Consequently, these patients were classified into three groups: SF3B1 mutation dominance, MPN driver mutation dominance, and no dominance.
Treatment and follow-up
The treatment regimens of 160 patients were documented during follow-up. A total of 111 (69.4%) patients received agents to improve anemia, including recombinant human erythropoietin, lenalidomide, androgens, cyclosporin or luspatercept; 17 (10.6%) patients received only cytoreductive agents, such as hydroxyurea or interferon; 25 (15.6%) patients received the above combination therapies; and 7 (4.4%) patients underwent watch and wait protocols. With a median follow-up of 48 months (95% confidence interval [CI] 35–61 months), the last follow-up was on March 16th, 2024, and 16 subjects (8.9%) were lost to follow-up. Thrombosis information and leukemia transformation status information were available for 100 and 126 subjects, respectively, according to medical records or telephone follow-up.
Statistical analysis
Continuous variables were presented as medians and interquartile ranges (IQRs), and nominal variables were presented as counts and relative frequencies. Differences in continuous variables between the two groups were analyzed using the Mann–Whitney U-test. Nominal variables from different groups were compared using the Pearson chi-square test or Fisher’s exact test. Spearman’s correlation analysis was used to evaluate correlations between mutations in any two genes in the targeted NGS panel. P values were adjusted by Benjamini–Hochberg method in multiple comparisons. A two-tailed P value < 0.05 was considered significant. Overall survival (OS) was calculated from the date of diagnosis to the date of death or last follow-up. The median follow-up was calculated using the reverse Kaplan‒Meier method. Univariable analyses of survival were performed using the log-rank test, and variables with P values < 0.1 were selected for Cox multivariable analyses. In multivariable analyses, backward stepwise elimination was used for variable selection. Independent variables incorporated in the final survival models were based on the multivariable Cox regression model with the minimum Akaike information criterion (AIC) value in the training cohort [27,28,29]. Variables in the survival model were assigned weights based on the regression coefficients derived from the Cox regression model. Kaplan–Meier curves and Harrell’s concordance index (C-index) were used to assess the discrimination of survival models. Statistical analyses were performed with SPSS software version 26.0 (IBM Corp., Armonk, NY, USA), GraphPad Prism version 9.0 (GraphPad Inc., San Diego, CA, USA), and R software version 4.2.3 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Clinical features and molecular landscape of MDS/MPN-SF3B1-T according to the 2022 WHO classification
There were 180 patients with MDS/MPN-SF3B1-T in our cohort. The median age of patients with MDS/MPN-SF3B1-T at diagnosis was 65 years (IQR, 57–70 years). The median Hb, median platelet count (PLT), and median bone marrow RS at diagnosis were 74 g/L (IQR, 63–88 g/L), 552 × 10E + 9/L (IQR, 487–718 × 10E + 9/L), and 38% (IQR, 24–57%), respectively. The clinical features and laboratory details at diagnosis of all patients with MDS/MPN-SF3B1-T were comprehensively documented in Table 1.
In our cohort, cytogenetic data were available and evaluable for 169 patients. A total of 135 (79.9%) patients harbored normal karyotypes and complex karyotypes were detected in 5 (3.0%) patients. The most common mutated gene in MDS/MPN-SF3B1-T was SF3B1. SF3B1 mutations were detected in 134 (89.3%) patients, using either NGS or direct Sanger sequencing. The specific mutations of SF3B1 included K700E (61.2%), K666N/R/E/M/Q/T (18.7%), H625C/G/H/L (9.0%) and H662Q/Y (8.2%) (Fig. 1A). Among the 122 patients who underwent NGS, the commonly mutated genes in MDS/MPN-SF3B1-T included SF3B1 (93.4%), JAK2 (32.8%), TET2 (29.5%), DNMT3A (21.3%), ASXL1 (17.2%), TP53 (8.2%), MPL (6.6%), SETBP1 (6.6%), and SRSF2 (4.9%) (Fig. 1B and Supplementary Table S3). Correlation analysis of different gene mutations in MDS/MPN-SF3B1-T revealed that the correlation coefficients of mutations in any two genes were lower than 0.3 (all adjusted P > 0.05), indicating weak correlations (Supplementary Fig. S1). NGS revealed that 46 (37.7%) patients harbored both SF3B1 and MPN driver mutations, 2 of whom had simultaneous mutations in SF3B1, JAK2, and MPL. Among patients with both SF3B1 and MPN driver mutations, 22 (47.8%), 13 (28.3%) and 11 (23.9%) patients exhibited SF3B1 mutation dominance, no dominance, and MPN driver mutation dominance, respectively (Supplementary Fig. S2).

A Distribution of specific mutation sites in the SF3B1 gene in 134 patients with SF3B1 mutations detected by NGS or Sanger sequencing. There were 4 patients with 2 mutation sites and 1 patient with 3 mutation sites in SF3B1; therefore, a total of 140 mutations in SF3B1 were detected in 134 SF3B1-mutated patients. B Molecular landscape of 122 patients with MDS/MPN-SF3B1-T detected by NGS. MDS/MPN-SF3B1-T myelodysplastic/myeloproliferative neoplasm with SF3B1 mutation and thrombocytosis, NGS next-generation sequencing.
Compared with patients diagnosed with MDS/MPN-RS-T within the Mayo–Moffitt cohort (n = 158), those diagnosed with 2022 WHO-defined MDS/MPN-SF3B1-T in our cohort (training cohort 1, n = 180) had a younger median age (65 versus 71 years), a lower median hemoglobin level (74 versus 95 g/L) and a greater percentage of TET2 mutations (29.5% versus 7.0%, P = 0.003) at diagnosis (Supplementary Table S4) [11].
Comparison of MDS/MPN-SF3B1-T patients with <15% bone marrow RS and ≥15% bone marrow RS
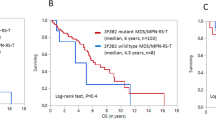
Compared with MDS/MPN-RS-T, one of the key modifications of the 2022 WHO classification for MDS/MPN-SF3B1-T was that patient with <15% bone marrow RS and SF3B1 mutations were newly involved in the disease. We divided 180 patients with MDS/MPN-SF3B1-T into two groups: patients with <15% bone marrow RS (n = 24) and those with ≥15% bone marrow RS (n = 156). At least one SF3B1 mutation was detected in 24 patients with <15% bone marrow RS using either NGS or Sanger sequencing. Patients with <15% bone marrow RS had a greater proportion of the male (83.3% versus 54.5%, P = 0.008) and had a higher median lactic dehydrogenase level (300.5 U/L [IQR, 218.4–343.5 U/L] versus 204.2 U/L [IQR, 161.0–252.2 U/L], P = 0.002). No significant differences in age at diagnosis, blood cell count, karyotype stratification, or molecular landscape were noted between the two groups (Table 1). One patient with <15% bone marrow RS and 15 patients with ≥15% bone marrow RS were lost to follow-up. The median OS of patients with <15% BM RS (n = 23; 41 months, 95% CI 32–50 months) was obviously inferior to that of patients with ≥15% BM RS (n = 141; 76 months, 95% CI 59–93 months; P < 0.001) (Fig. 2). Furthermore, we also divided patients with <15% bone marrow RS and SF3B1 mutations into another two groups according to 5% bone marrow RS and found no difference in survival for patients with 5–15% BM RS (n = 12; median OS 41 months, 95% CI 31–50 months) compared with those with <5% BM RS (n = 11; median OS 38 months, 95% CI 6–70 months; P = 0.905; Supplementary Fig. S3).

BM bone marrow, RS ring sideroblasts, MDS/MPN-SF3B1-T myelodysplastic/myeloproliferative neoplasm with SF3B1 mutation and thrombocytosis.
Prognostic factors for overall survival in MDS/MPN-SF3B1-T patients
The median OS of all patients with MDS/MPN-SF3B1-T was 69 months (95% CI 59–79 months). Leukemia transformations and thrombotic events after diagnosis were documented in 11 patients (8.7%) and 4 (4.0%) patients, respectively.
Univariable analyses of OS showed that age ≥ 65 years (P < 0.001), Hb < 80 g/L (P = 0.090), PLT ≥ 800 × 10E + 9/L (P = 0.087), bone marrow RS < 15% (P < 0.001), IPSS-R cytogenetic category intermediate/poor/very poor (P = 0.005), SETBP1 mutation (P = 0.061) and SRSF2 mutation (P < 0.001) were associated with poor survival (Supplementary Table S5).
In training cohort 1 (n = 180), age ≥ 65 years, Hb < 80 g/L, PLT ≥ 800 × 10E + 9/L, bone marrow RS < 15%, and IPSS-R cytogenetic category intermediate/poor/very poor were included in the multivariable analysis (Supplementary Table S5). The multivariable Cox regression model with the minimum AIC value included all five variables mentioned above. Considering these five variables, we developed a clinical survival model in which age ≥ 65 years, Hb < 80 g/L, PLT ≥ 800 × 10E + 9/L, bone marrow RS < 15%, and IPSS-R cytogenetic category intermediate/poor/very poor were worth 1 point, 1 point, 1 point, 2 points, and 1 point, respectively (Table 2). Then, we classified patients into three survival categories: low (0–1 point, n = 66, median OS 96 months, 95% CI unavailable), intermediate (2–3 points, n = 73, median OS 64 months, 95% CI 56–72 months) and high risk (≥4 points, n = 15, median OS 36 months, 95% CI 18–55 months), among which survival was significantly different (low risk versus intermediate risk, P < 0.001; low risk versus high risk, P < 0.001; intermediate risk versus high risk, P = 0.001; Fig. 3A).

A Kaplan–Meier probability estimates of OS in training cohort 1 (n = 180) were presented across the clinical survival model risk categories. The cytogenetic information or survival status of 26 patients in training cohort 1 was not available. Thus, 154 patients were included in the clinical survival model. Patients were divided into three categories: low (median OS 96 months, 95% CI unavailable), intermediate (median OS 64 months, 95% CI 56–72 months) and high risk (median OS 36 months, 95% CI 18–55 months) (low versus intermediate risk, P < 0.001; low versus high risk, P < 0.001; intermediate versus high risk, P = 0.001). B Kaplan–Meier probability estimates of OS in training cohort 2 (n = 122) were presented across the clinical-molecular survival model risk categories. The cytogenetic information and survival status of 12 patients were not available for training cohort 2. Thus, 110 patients were included in the clinical-molecular survival model. Patients were divided into three categories: low (median OS not reached), intermediate (median OS 65 months, 95% CI 55–76 months) and high risk (median OS 30 months, 95% CI 23–36 months) (low versus intermediate risk, P = 0.011; low versus high risk, P < 0.001; intermediate versus high risk, P < 0.001). OS overall survival.
In training cohort 2 (n = 122), age ≥ 65 years, Hb < 80 g/L, PLT ≥ 800 × 10E + 9/L, BM RS < 15%, IPSS-R cytogenetic category intermediate/poor/very poor, SRSF2 mutation and SETBP1 mutation were included in the subsequent multivariable analyses based on the results of the preceding univariable analyses (Supplementary Table S5). The Cox proportional hazards regression model including all seven variables showed the lowest AIC value. We developed another clinical–molecular survival model including the above seven variables (Table 2) and classified patients in training cohort 2 into three survival categories: low risk (0–1 point, n = 45, median OS not reached), intermediate risk (2–3 points, n = 47, median OS 65 months, 95% CI 55–76 months) and high risk (≥4 points, n = 18, median OS 30 months, 95% CI 23–36 months), among which survival was significantly different (low risk versus intermediate risk, P = 0.011; low risk versus high risk, P < 0.001; intermediate risk versus high risk, P < 0.001) (Fig. 3B).
To compare the discriminative power of the clinical and clinical-molecular survival models, we also applied the clinical survival model to training cohort 2 and found that the clinical survival model could stratify patients in training cohort 2 into three risk categories with significantly different survival rates (Supplementary Fig. S4). In training cohort 2, the discriminative power of the clinical-molecular survival model, as measured using the C-index, was superior to that of the clinical survival model (C-index, 0.722 [±0.051] versus 0.695 [±0.053]).
We applied the clinical survival model and clinical-molecular survival model to the validation cohort. OS data were available for 41 patients in the validation cohort. Although subjects in the validation cohort classified as high risk were rare, survival among different risk categories was significantly different according to both the clinical survival model (P = 0.027, Supplementary Fig. S5A) and the clinical–molecular survival model (P = 0.013, Supplementary Fig. S5B). The clinical-molecular survival model also performed better than the clinical survival model in the validation cohort (C-index, 0.688 [±0.059] versus 0.677 [±0.059]).
Discussion
MDS/MPN-RS-T is characterized by bone marrow RS ≥ 15% and sustained thrombocytosis with the proliferation of atypical megakaryocytes; these features partly overlap with those of MDS-RS and essential thrombocytosis [8, 14, 30, 31]. We compared clinical signatures and molecular landscapes between our cohort and the Mayo–Moffitt cohort and detected differences in several clinical parameters and TET2 mutation frequencies (Supplementary Table S4) [11]. The mutational landscapes of 75 patients in another cohort of 92 RARS-T patients were described, and the frequency of TET2 mutations was 25.3%, which was comparable to that in our cohort [20]. The difference in clinical and molecular genetic signatures between the Mayo–Moffitt cohort and our cohort may be partly due to differences in the ethnic groups. The SF3B1 mutation is the most common mutation in the disease entity, and nearly 90% of patients with sequencing information in our study harbored the mutation. The specific mutation sites of SF3B1 in our study were mainly distributed at the 700th, 666th, 625th, and 662nd amino acid sites, which was similar to the findings of other studies on RARS-T or MDS/MPN-RS-T [9,10,11]. Previous studies on RARS-T or MDS/MPN-RS-T reported that ~30–75% of patients have both SF3B1 and JAK2 mutations [9,10,11, 20, 32]. Co-mutations of SF3B1 and JAK2 or/and MPL mutations were present in 37.7% of patients who underwent NGS in our cohort, which is a relatively low percentage compared with that in some previous studies. This finding may be partly due to a lack of NGS information for some patients. CALR mutations are infrequent in MDS/MPN-RS-T patients, and CALR mutations were not detected in any of the patients in our cohort [10, 20, 33]. Several studies on MDS/MPN-RS-T have shown that the SF3B1 mutation is more ancestral than MPN driver mutations according to comparisons of the VAFs of these mutations or Sanger sequencing of individual colonies [26, 31, 34]. In our study of patients with co-mutation of SF3B1 and MPN driver mutations, we found that the VAF of SF3B1 mutations was greater than (a cutoff of at least 5%) or equal to the VAF of JAK2 or MPL mutations in almost 80% of patients, indicating that SF3B1 mutations might occur before JAK2 or MPL mutations in most patients with MDS/MPN-SF3B1-T [26].
The 2022 WHO classification of myeloid neoplasms revised the diagnostic criteria for MDS/MPN-RS-T based on SF3B1 mutation and was renamed MDS/MPN-SF3B1-T [19]. The greatest change in the criteria was that patients with <15% bone marrow RS and SF3B1 mutations were included in the disease entity. To the best of our knowledge, few studies have investigated patients with <15% bone marrow RS in MDS/MPN-SF3B1-T. Comparisons between patients with <15% bone marrow RS and those with ≥15% bone marrow RS illustrated that survival in the former subgroup was significantly inferior to that in the latter subgroup. Although the number of patients with <15% bone marrow RS was relatively low, heterogeneity between the two groups was obvious.
In studies related to MDS/MPN, unclassifiable (MDS/MPN-U), MDS/MPN-U patients with ≥15% bone marrow RS who did not meet the MDS/MPN-RS-T criteria were defined as MDS/MPN-U patients with bone marrow RS ≥ 15% (MDS/MPN-U-RS) [11, 35]. The aforementioned studies indicated that MDS/MPN-U-RS had similar outcomes to MDS/MPN-RS-T, whereas the survival of MDS/MPN-U patients with <15% bone marrow RS was inferior to that of MDS/MPN-RS-T patients, suggesting that bone marrow RS ≥ 15% might predict favorable outcomes in MDS/MPN-U patients [11, 35]. In our cohort of MDS/MPN-SF3B1-T patients, bone marrow RS ≥ 15% was also considered a favorable prognostic factor for survival, whereas SF3B1 mutation was not confirmed to have prognostic significance. Previous studies have demonstrated that the molecular landscape is very different among SF3B1-mutated MDS, MDS/MPN and acute myeloid leukemia patients and that co-mutations contribute to diverse clinical and morphological features [31, 36]. Although it has been proven that SF3B1 mutation can define a specific category in MDS, SF3B1 mutation-based criteria in MDS/MPN-SF3B1-T still need further study [19, 37].
Previously, there were some studies on patients with RARS-T or MDS/MPN-RS-T that aimed to explore prognostic factors of survival [9,10,11]. Age > 80 years, wild-type JAK2 and wild-type SF3B1 were identified as the main adverse factors for survival in a cohort of 111 patients with RARS-T, whereas the prognostic significance of SF3B1 and JAK2 mutations has not been validated in other studies, including ours [9]. Another study on patients with RARS-T developed a hazard ratio weighted prognostic model based on abnormal karyotype, ASXL1 and/or SETBP1 mutations, and Hb < 100 g/L [10]. The above two studies enrolled patients based on the 2008 revision of the WHO classification of myeloid neoplasms [4]. A more recent study on 158 patients with MDS/MPN-RS-T was based on the 2016 WHO criteria, in which abnormal karyotype (excluding -Y) and Hb ≤ 100 g/L were found to be independent predictors of survival [11]. We developed a clinical survival model and clinical-molecular survival model using independent factors for the prediction of survival, which were determined not by a significant P value (P < 0.05) but by the minimum value of the AIC in multivariable Cox regression models [27, 38,39,40]. Compared with previous studies, such as the Mayo–Moffitt cohort published in 2022, we included patients with <15% bone marrow RS in the prognostic analysis of MDS/MPN-SF3B1-T for the first time [9,10,11]. We found that bone marrow RS < 15% was a predictor of adverse survival in patients with this disease. Thus, whether patients with <15% bone marrow RS and SF3B1 mutations should be included in MDS/MPN-SF3B1-T needs further validation. SRSF2 mutation (~50%) is one of the most frequent mutations in chronic myelomonocytic leukemia, but it has not been reported to be associated with prognosis [41]. However, previous studies have shown that SRSF2 mutation predicts an adverse prognosis in patients with MDS [42]. A study on SRSF2-mutated neoplasms revealed that co-mutation of SRSF2 and other splicing factors had a clear predominance of blastic phenotype [43]. In our cohort, SF3B1 mutations were detected in all SRSF2-mutated patients (n = 6) (4 with the SF3B1 K666E/T/M/R mutation and 2 with the SF3B1 K700E mutation). Among the six patients with SRSF2 mutations, one patient progressed to leukemia and subsequently died, whereas another three patients died from unknown causes. Based on the findings of previous studies and our data, we believe that SRSF2 mutation may be associated with leukemia transformation and inferior survival in MDS/MPN-SF3B1-T patients. In addition, age, Hb, karyotype, and SETBP1 mutation have been proposed as predictors of survival in other studies, whereas differences in the cutoff values of some variables may be attributed to differences in ethnic group or time of diagnosis [9,10,11]. A prior study reported that the frequency of thrombotic events in MDS/MPN-RS-T patients was comparable to that in patients with essential thrombocytosis but more frequent than that in patients with MDS-RS [8, 14]. In a Mayo Clinic cohort comprising 82 RARS-T patients, multivariable analysis of thrombosis-free survival revealed that the presence of SF3B1 mutations independently predicted inferior thrombosis-free survival; however, the underlying mechanism involved remains unclear [44]. In our study, we did not identify significant factors for predicting thrombotic predisposition, potentially due to the limited documentation of thrombotic events and the short duration of follow-up.
The study is not devoid of limitations: (i) it involves a relatively small sample size and a brief follow-up period, (ii) it operates on a retrospective basis, and (iii) the external validation cohort comprises a relatively small number of patients and lacks those with <15% BM RS.
In conclusion, patients with <15% bone marrow RS and SF3B1 mutations exhibited worse survival rates than those with ≥15% bone marrow RS in MDS/MPN-SF3B1-T patients. The inclusion of this patient group requires further validation. Additionally, we developed survival models for MDS/MPN-SF3B1-T patients based on the 2022 WHO classification, which should help physicians estimate survival in persons with MDS/MPN-SF3B1-T in the 2022 WHO classification.
Data availability
The datasets supporting the findings of this study are available from the corresponding authors upon reasonable request.
References
Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. World Health Organization classification of tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2001.
Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–302.
Wardrop D, Steensma DP. Is refractory anaemia with ring sideroblasts and thrombocytosis (RARS-T) a necessary or useful diagnostic category? Br J Haematol. 2009;144:809–17.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts and RARS with thrombocytosis. Am J Hematol. 2015;90:549–59.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts (RARS) and RARS with thrombocytosis: “2019 update on diagnosis, risk-stratification, and management”. Am J Hematol. 2019;94:475–88.
Broseus J, Florensa L, Zipperer E, Schnittger S, Malcovati L, Richebourg S, et al. Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica. 2012;97:1036–41.
Broséus J, Alpermann T, Wulfert M, Florensa Brichs L, Jeromin S, Lippert E, et al. Age, JAK2(V617F) and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia. 2013;27:1826–31.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, King RL, Ketterling RP, et al. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am J Hematol. 2016;91:492–8.
Mangaonkar AA, Lasho TL, Ketterling RP, Reichard KK, Gangat N, Al-Kali A, et al. Myelodysplastic/myeloproliferative neoplasms with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T): Mayo-Moffitt collaborative study of 158 patients. Blood Cancer J. 2022;12:26.
Edahiro Y, Ochiai T, Hashimoto Y, Morishita S, Shirane S, Inano T, et al. Clinical characteristics of Japanese patients with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Int J Hematol. 2023;118:47–53.
Vardell VA, Peters DJ, Tantravahi SK, Patel AB. Myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: a national analysis of overall survival and demographic features. Blood. 2022;140:1350–2.
Patnaik MM, Tefferi A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T) - “2021 update on diagnosis, risk-stratification, and management”. Am J Hematol. 2021;96:379–94.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95.
Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jädersten M, Jansson M, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233–41.
Malcovati L, Stevenson K, Papaemmanuil E, Neuberg D, Bejar R, Boultwood J, et al. SF3B1-mutant MDS as a distinct disease subtype: a proposal from the International Working Group for the prognosis of MDS. Blood. 2020;136:157–70.
Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26:542–5.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19.
Jeromin S, Haferlach T, Weissmann S, Meggendorfer M, Eder C, Nadarajah N, et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica. 2015;100:e125–127.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. 2022;1:EVIDoa2200008.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–32.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Li B, Liu J, Jia Y, Wang J, Xu Z, Qin T, et al. Clinical features and biological implications of different U2AF1 mutation types in myelodysplastic syndromes. Genes Chromosom Cancer. 2018;57:80–88.
Palomo L, Meggendorfer M, Hutter S, Twardziok S, Ademà V, Fuhrmann I, et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood. 2020;136:1851–62.
Cui Y, Zhang J, Li Z, Wei K, Lei Y, Ren J, et al. A CT-based deep learning radiomics nomogram for predicting the response to neoadjuvant chemotherapy in patients with locally advanced gastric cancer: a multicenter cohort study. eClinicalMedicine. 2022;46:101348.
McFarlane MR, Hochstedler KA, Laucis AM, Sun Y, Chowdhury A, Matuszak MM, et al. Predictors of pneumonitis after conventionally fractionated radiotherapy for locally advanced lung cancer. Int J Radiat Oncol Biol Phys. 2021;111:1176–85.
Wu J, Zhang H, Li L, Hu M, Chen L, Xu B, et al. A nomogram for predicting overall survival in patients with low-grade endometrial stromal sarcoma: a population-based analysis. Cancer Commun. 2020;40:301–12.
Montalban-Bravo G, Kanagal-Shamanna R, Darbaniyan F, Siddiqui MT, Sasaki K, Wei Y. et al. Clinical, genomic, and transcriptomic differences between myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) and myelodysplastic syndrome with ring sideroblasts (MDS-RS). Am J Hematol. 2021;96:E246–E249.
Ok CY, Trowell KT, Parker KG, Moser K, Weinberg OK, Rogers HJ, et al. Chronic myeloid neoplasms harboring concomitant mutations in myeloproliferative neoplasm driver genes (JAK2/MPL/CALR) and SF3B1. Mod Pathol. 2021;34:20–31.
Jeromin S, Haferlach T, Grossmann V, Alpermann T, Kowarsch A, Haferlach C, et al. High frequencies of SF3B1 and JAK2 mutations in refractory anemia with ring sideroblasts associated with marked thrombocytosis strengthen the assignment to the category of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2013;98:e15–17.
Broséus J, Lippert E, Harutyunyan AS, Jeromin S, Zipperer E, Florensa L, et al. Low rate of calreticulin mutations in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia. 2014;28:1374–6.
Inano T, Araki M, Morishita S, Imai M, Yasuda H, Nitta H, et al. JAK2 exon 12 mutation in myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis: not an exclusive mutation to polycythaemia vera. Br J Haematol. 2019;187:e27–e31.
Mangaonkar AA, Swoboda DM, Coltro G, Lasho TL, Novotny PJ, Pophali P, et al. Clinicopathologic characteristics, prognostication and treatment outcomes for myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U): Mayo Clinic-Moffitt Cancer Center study of 135 consecutive patients. Leukemia. 2020;34:656–61.
Aqil B, Sukhanova M, Behdad A, Jennings L, Lu X, Chen Q, et al. Many faces of SF3B1-mutated myeloid neoplasms: concurrent mutational profiles contribute to the diverse clinical and morphologic features. Hum Pathol. 2022;129:81–89.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Steyerberg EW, Van Calster B. Redefining significance and reproducibility for medical research: a plea for higher P-value thresholds for diagnostic and prognostic models. Eur J Clin Investig. 2020;50:e13229.
Wallisch C, Dunkler D, Rauch G, de Bin R, Heinze G. Selection of variables for multivariable models: opportunities and limitations in quantifying model stability by resampling. Stat Med. 2021;40:369–81.
Sutherland C, Hare D, Johnson PJ, Linden DW, Montgomery RA, Droge E. Practical advice on variable selection and reporting using Akaike information criterion. Proc Biol Sci. 2023;290:20231261.
Patnaik MM, Tefferi A. Chronic myelomonocytic leukemia: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:352–72.
Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012;120:3106–11.
Todisco G, Creignou M, Gallì A, Guglielmelli P, Rumi E, Roncador M, et al. Co-mutation pattern, clonal hierarchy, and clone size concur to determine disease phenotype of SRSF2(P95)-mutated neoplasms. Leukemia. 2021;35:2371–81.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, King RL, Ketterling RP, et al. Vascular events and risk factors for thrombosis in refractory anemia with ring sideroblasts and thrombocytosis. Leukemia. 2016;30:2273–5.
Acknowledgements
The authors would like to thank all participants for their commitment and time investment in the study.
Funding
This study is supported in part by National Natural Science Funds (Nos. 82170139, 81530008 and 82070134), CAMS Initiative Fund for Medical Sciences (Nos. 2022-I2M-1-022 and 2022-I2M-C&T-B-093), Clinical research fund from National Clinical Research Centre for Blood Diseases (Nos. 2023NCRCA0117 and 2023NCRCA0103) and Haihe Laboratory of Cell Ecosystem Innovation Fund (22HHXBSS00033).
Author information
Authors and Affiliations
Contributions
ZJX and ZFX designed the study. FHL collected and analyzed the data. TJQ, BL, SQQ, LJP, PHZ, QS, WYC, QYG, MJ, JJL, XFA, and JM recruited patients and collected data. FHL, RPG, ZFX, and ZJX prepared the manuscript. All authors read, approved the final manuscript and agreed to submit for publication.
Corresponding authors
Ethics declarations
Competing interests
RPG is a consultant to NexImmune Inc, Nanexa Pharma, Ascentage Pharm Group and Antengene Biotech LLC. He is Medical Director of FFF Enterprises Inc., Partner in AZAC Inc., is on the Board of Directors of the Russian Foundation for Cancer Research Support and is a member of the Scientific Advisory Board of StemRad Ltd. The other authors have no relevant conflicts of interest to declare about the manuscript.
Informed consent
The patient gave written informed consent compliant with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, F., Qin, T., Li, B. et al. Predicting survival in patients with myelodysplastic/myeloproliferative neoplasms with SF3B1 mutation and thrombocytosis. Leukemia (2024). https://doi.org/10.1038/s41375-024-02262-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41375-024-02262-2
