
Abstract
Angiogenesis is involved in ischemic heart disease as well as the prognosis of heart failure (HF), and endothelial cells are the main participants in angiogenesis. In this study, we found that miR-221-3p is highly expressed in vascular tissue, especially in endothelial cells, and increased miR-221-3p was observed in heart tissue of HF patients and transverse aortic constriction (TAC)-induced HF mice. To explore the role of miR-221-3p in endothelial cells, microRNA (miRNA) mimics and inhibitors were employed in vitro. Overexpression of miR-221-3p inhibited endothelial cell proliferation, migration, and cord formation in vitro, while inhibition of miR-221-3p showed the opposite effect. Anti-argonaute 2 (Ago2) coimmunoprecipitation, dual-luciferase reporter assay, and western blotting were performed to verify the target of miR-221-3p. Hypoxia-inducible factor-1α (HIF-1α) was identified as a miR-221-3p target, and the adverse effects of miR-221-3p on endothelial cells were alleviated by HIF-1α re-expression. In vivo, a mouse model of hindlimb ischemia (HLI) was developed to demonstrate the effect of miR-221-3p on angiogenesis. AntagomiR-221-3p increased HIF-1α expression and promoted angiogenesis in mouse ischemic hindlimbs. Using the TAC model, we clarified that antagomiR-221-3p improved cardiac function in HF mice by promoting cardiac angiogenesis. Furthermore, serum miR-221-3p was detected to be negatively correlated with heart function in chronic heart failure (CHF) patients. Our results conclude that miR-221-3p inhibits angiogenesis of endothelial cells by targeting HIF-1α and that inhibition of miR-221-3p improves cardiac function of TAC-induced HF mice. Furthermore, miR-221-3p might be a potential prognostic marker of HF.
Similar content being viewed by others


Introduction
Heart failure (HF) is defined as the inability of the heart to supply enough blood and oxygen to the surrounding tissue to meet its metabolic needs [1]. Increased pressure loading on the heart can lead to pathological cardiac hypertrophy and eventually HF. It has been reported that cardiomyocyte apoptosis [2] and myocardial fibrosis [3] are the leading causes of HF. In addition to the direct damage to cardiomyocytes due to stress, angiogenesis of endothelial cells also plays an important role in development of the hypertrophic heart [4]. The degree of cardiac hypertrophy is correlated with vascular density, and blockage of angiogenesis can cause the hypertrophic heart to further progress to HF. For instance, p-53 can inhibit angiogenesis by reducing the expression of hypoxia-inducible factor-1α (HIF-1α), accelerating the development of HF [5]. In contrast, promoting angiogenesis can protect against heart dysfunction and HF. Canopy FGF signaling regulator 2 (Cnpy2) and Rho family GTPase 3 (Rnd3) were found to have protective effects which can attenuate the transition from compensatory hypertrophy to dilated HF through the HIF-1α/vascular endothelial growth factor (VEGF) pathway [6, 7].
HIF is a heterodimeric protein of the PER–ARNT–SIM (PAS) subfamily [8]. It is composed of two subunits—one of either hypoxia-inducible α-subunits, HIF-1α or HIF-2α, and the constitutive HIF-1β subunit [9]. HIF-1α plays a critical role in angiogenesis, and many angiogenesis-related genes such as nitric oxide synthases, VEGF, and genes regulating matrix metabolism (urokinase-type plasminogen activator receptor) are directly or indirectly induced by HIF-1α [10]. HIF-1α is stable for a very short time under normal oxygen conditions and is rapidly degraded by ubiquitination. The prolyl-4-hydroxylase domain (PHD)-containing enzymes hydroxylate HIF-1α at two proline sites in its oxygen-dependent degradation domain, which results in HIF-1α being targeted by the von Hippel–Lindau tumor suppressor protein and further degraded by the ubiquitin–proteasome pathway. However, PHD is inhibited under hypoxic conditions [11, 12]. Although ubiquitin-mediated degradation is the classic regulation mode of HIF-1α expression, other regulatory mechanisms are also involved; these include HIF-1α downregulation by microRNAs (miRNAs), such as miR-199a and miR-20a, which target the Hif-1α 3′UTR region [13,14,15].
MiRNAs are endogenous 22-nt RNAs that play critical regulatory roles in animals and plants by targeting messenger RNAs (mRNAs) for cleavage or translational repression [16]. Many miRNAs are involved in the regulation of angiogenesis [17]. The antiangiogenic properties of miR-221-3p were first identified by targeting c-kit in vitro [18]. Since then, there has been some literature on the role of miR-221-3p in angiogenesis [19,20,21,22]. However, whether the role of miR-221-3p in angiogenesis is significantly related to the development of HF has not been investigated. In this study, we detected the expression profile of miR-221-3p and explored the role of miR-221-3p in angiogenesis in HF mice. The results suggested that inhibition of miR-221-3p alleviated HF by improving endothelial cell angiogenesis.
Materials and methods
Reagents
Human umbilical vein endothelial cells (HUVECs) and human embryonic kidney 293 (HEK293) cells were obtained from the American Type Culture Collection (Washington, USA). Dulbecco’s Modified Eagle Medium (DMEM), Roswell Park Memorial Institute (RPMI)1640, and Opti-MEM™ I Reduced Serum Medium were purchased from GIBCO (Life Technologies Corporation, Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Transgen Biotech (Beijing, China). Mouse monoclonal antibody against β-actin (AC004) was purchased from ABclonal Technology (Wuhan, China). Rabbit monoclonal antibody against HIF-1α (14179s) was purchased from Cell Signaling Technology (Danvers, MA, USA). Polyvinylidene difluoride membranes were obtained from Millipore (Merck KGaA, Darmstadt, Germany). Prestained protein ladders were purchased from Thermo Fisher Scientific (Glen Burnie, MD). Mimics/inhibitors, negative controls, and primers for miRNAs were obtained from RiboBio (Guangzhou, China). The sequences of oligos are as follows; miRNA mimic control sense strand: 5′-UUUGUACUACACAAAAGUACUG-3′, antisense strand 3′-AAACAUGAUGUGUUUUCAUGAC-5′, miRNA inhibitor control: 5′-mCmAmGmUmAmCmUmUmUmUmGmUmGmUmAmGmUmAmCmAmAmA-3′, and m stands for methylation modification. Endotoxin-free DNA plasmid purification kits were purchased from TIANGEN (Beijing, China), and DNA ladders were purchased from Thermo Fisher Scientific.
Human serum samples
Ten heart samples were collected from cardiac transplant patients with chronic heart failure (CHF), and ten heart samples were obtained from accident victims. Thirty serum samples were from patients with CHF. The study of humans was approved by the Clinical Research Committees of Tongji Medical College and adhered to the guidelines of the Declaration of Helsinki. Written informed consent was obtained from all study participants.
Animals
Male C57BL/6 mice (8–10 weeks old) were purchased from the Experimental Animal Center of Hubei (Wuhan, China). All animals were raised in the animal care facility of Tongji Medical College, in specific-pathogen-free (SPF) level conditions. The study was approved by the Institutional Animal Research Committee of Tongji Medical College. All animal experimental protocols complied with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health.
RNA isolation and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using TRIzol reagent (Transgen Biotech). Total RNA (2 µg) was reverse transcribed into cDNA using a reverse transcription kit (Thermo Fisher scientific) and specific RT primers (RiboBio). Quantitative PCR was performed using KAPA SYBR® FAST qPCR Kits (Kapa Biosystems, Wilmington, USA) and specific primers (RiboBio) on a 7900HT Fast Real-Time PCR system (Applied Biosystems). The U6 small nuclear RNA was used as an internal standard of miRNAs. Each reaction was performed in triplicate, and analysis was performed with the 2−∆∆Ct method.
RNA Fluorescence in situ hybridization (FISH)
Mouse heart tissue was fixed with 4% paraformaldehyde, dehydrated, embedded in paraffin, and sectioned into 4-µm-thick slices. The sections were deparaffinized, rehydrated, and subjected to antigen retrieval. Then, the sections were hybridized with the Locked Nucleic Acid detection probe (Qiagen) in hybridization buffer (Qiagen) at 52 °C for 1 h. The sections were then washed, as described earlier [23]. Fluorescence signal detection was performed using anti-digoxin antibody (11333062910) (Sigma, St. Louis, MO USA) and the TSA amplification system (Thermo Fisher Scientific). Then, immunofluorescence staining of CD31 (ab29364) (Abcam, Cambridge, MA, USA) and nuclear DAPI staining were performed [24].
Cell culture and treatments
HUVECs were cultured in RPMI 1640 supplemented with 10% FBS, and HEK293 cells were maintained in DMEM supplemented with 10% FBS at 37 °C and a humidified atmosphere of 5% CO2. MiRNA mimic (100 nM), miRNA inhibitors (100 nM), and their controls were transfected into HUVECs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Cells were collected 48 h after transfection. HEK293 cells were cotransfected with 200 ng of pMIR-Report Luciferase plasmid constructs, 10 ng of pRL-TK plasmid (Promega, Madison, WI), and miR-221-3p mimic (100 nM) or mimic control (100 nM) using Lipofectamine 2000.
Cell viability assay
HUVECs were seeded in 96-well plates and treated with miR-221-3p mimic, inhibitor and their controls. After 48 h, the culture medium was replaced with 90 µL of serum-free medium and 10 µL of cell count kit-8 (CCK-8) reagent (Beyotime, Shanghai, China) per well. The cells were cultured at 37 °C for 2 h. The absorbance was detected at 450 nm following the manufacturer’s recommendations.
EdU assay of cell proliferation
The proliferation of HUVECs was measured using an EdU kit (RiboBio) following the manufacturer’s protocol.
Migration assay
After treatment with miR-221-3p mimic, inhibitor and their controls, 2 × 105 HUVECs were transferred to the upper chamber of a transwell insert with an 8 μm pore membrane (Corning Life Sciences, Corning, NY). Cells were incubated for 6–8 h and then fixed and stained with crystal violet. The upper surface of the membrane was wiped, and the cells on the lower surface were counted in five random fields under a microscope.
Cord formation in matrigel
Angiogenesis was assayed using the µ-slide angiogenesis kit (ibidi, Martinsried, Germany). The µ-slide was precoated with 10 µL of growth factor-reduced Matrigel matrix (Corning Life Sciences) per well, and 1 × 105 HUVECs were seeded per well. After 4–6 h of incubation, phosphate-buffered solution (PBS) with Calcein-AM was added, and the cells were incubated for 15 min. The cells were then imaged using an inverted microscope.
Coimmunoprecipitation of RNA with Ago2 antibody
HUVECs were lysed and then immunoprecipitated with an Anti-argonaute 2 (Ago2) antibody (H00027161-M01) (Abnova, Taiwan, China) or IgG antibody (sc-2025) (Santa Cruz Biotechnology, Santa Cruz, CA) using protein G Sepharose beads (Santa Cruz Biotechnology) as previously described [25]. After washing, a fraction of the beads was used for western blotting to detect Ago2. The remainder of the beads was extracted with TRIzol and quantified by qPCR to detect Hif-1α, Igf1, and Sema3c. The primers of Hif-1α were forward primer: 5′-AAGTTCACCTGAGCCTAATAGTC-3′, and reverse primer: 5′-AGTCTAAATCTGTGTCCTGAGTAG-3′. The primers of Igf1 were forward primer: 5′-ATGCCCAAGACCCAGAAG-3′, and reverse primer: 5′-GGTGGCATGTCACTCTTCACT-3′. The primers of Sema3C were forward primer: 5′-TCAGGAAACTTAAAAGGATGGCA-3′ and reverse primer: 5′- ACGCAAATTGTCCGGAATGC-3′.
Dual-luciferase reporter assay
Luciferase activity was analyzed by the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s protocol. Transfection efficiency was normalized by Renilla luciferase activity.
Western blotting analysis
Cells were homogenized using a lysis buffer (Boster, Wuhan, China) on ice. The total protein was estimated using the BCA protein detection reagent (Boster), and 20 µg of total protein was loaded. Membranes were blocked for 2 h with 5% bovine serum albumin (BSA). After incubation with primary and secondary antibodies (Jackson Immune Research Laboratory), the bands were visualized using enhanced chemiluminescence reagents, and the band intensity was quantified by mean gray value using ImageJ.
Construction of plasmids
To overexpress HIF-1α in HUVECs, the full-length sequence of its protein-coding sequence (CDS) was amplified by PCR and then ligated into the pcDNA3.1 vector. The primers were as follows: forward primer 5′-GGGACCGATTCACCATGGAG-3′, reverse primer 5′-TAATGAGCCACCAGTGTCCAA-3′ (Tianyihuiyuan, Wuhan, China).
Animal models
A mouse hindlimb ischemia (HLI) model was generated as previously described [26]. We ligated the femoral artery of the left hindlimb of the mouse, and the right hindlimb was used as the control group. The HLI mice were randomly divided into two groups (n = 5 per group), including the HLI + antagomiR-negative control (NC) group and the HLI + antagomiR-221-3p group. AntagomiR-NC (RiboBio) or antagomiR-221-3p (RiboBio) (10 mg/kg) was dissolved in 100 µL of PBS and intramuscularly injected at four different sites into the adductor muscle of the ischemic limbs immediately after the surgery, followed by injections twice a week for three weeks. Laser doppler perfusion imaging (PeriCam PSI NR, Perimed, Sweden) was conducted as previously reported [26]. All the mice were sacrificed 21 days after the surgery, and the ischemic side and normal side adductor muscles of the hindlimbs were collected for immunohistochemical staining to evaluate angiogenesis.
Transverse aortic constriction (TAC) surgery was performed under isoflurane anesthesia according to previous studies [27]. For the antagomiR experiment, the mice were divided into three groups, including the sham group, TAC + antagomiR-NC group, and TAC + antagomiR-221-3p group (n = 5 per group). Immediately after TAC surgery, antagomiR-NC (20 mg/kg) and antagomiR-221-3p (20 mg/kg) dissolved in 100 µL of PBS were injected into the tail vein of mice twice a week for 4 weeks, and the mice were sacrificed 4 weeks after the TAC surgery.
Echocardiography
The mice were anesthetized by isoflurane inhalation, and echocardiogram was obtained using a Vevo2100 Imaging System (Toronto, Canada) with a 30-MHz high-frequency scan head [28].
Histology
For histological analyses, the adductor muscles and hearts were fixed in 4% paraformaldehyde for 24 h, dehydrated, embedded in paraffin, and sectioned. Hematoxylin and eosin staining was performed with standard methods [29]. Picrosirius red staining was conducted per the product description (Abcam). For immunohistochemical staining, the sections were first dewaxed. After antigen repair, the sections were incubated with a CD31 or HIF-1α (ab16066) (Abcam) antibody followed by appropriate secondary antibodies and then counterstained with hematoxylin.
Statistical analyses
Data are presented as the mean ± SEM. GraphPad Prism was used for data analyses. Comparisons between two groups were performed using two-tailed Student’s t test. For comparisons of more than two groups, one-way ANOVA was used. Pearson correlation analysis was used for linear correlation analysis. Values of p < 0.05 were considered statistically significant.
Results
MiR-221-3p is mainly expressed in vascular endothelial cells and shows high expression in heart tissue of HF humans and mice
We detected the expression of miR-221-3p in different tissue samples in mice and found high expression of miR-221-3p in vascular tissue (Fig. 1a). In addition, to further clarify the expression profile of miR-221-3p, we isolated the mouse cardiomyocytes, smooth muscle cells, fibroblasts, and endothelial cells as described in previous studies [30,31,32] and detected the expression of miR-221-3p in each of these cell types. The highest expression of miR-221-3p was observed in endothelial cells (Fig. 1b). To observe the expression of miR-221-3p more directly, we used FISH to further detect the expression of miR-221-3p in heart tissue; we found that miR-221-3p expression is mainly located in blood vessels but not in myocardial tissue (Fig. 1c).

a QPCR of miR-221-3p in different organs of the mouse, n = 5. b MiR-221-3p expression in cardiomyocytes, SMCs, fibroblasts, and endothelial cells of mouse measured by qPCR, n = 3. c Representative FISH images of miR-221-3p expressed in CD31-positive endothelial cells of the mouse heart. Scale bar: 20 µm. d MiR-221-3p expression in human normal heart tissue and failing heart tissue detected by qPCR. Control, n = 10, DCM, n = 10. e Expression change of miR-221-3p in heart tissue of TAC-induced mice detected by qPCR, n = 5 per group. Data shown as the mean ± SEM. QPCR quantitative polymerase chain reaction, SMCs smooth muscle cells, FISH fluorescence in situ hybridization, DCM dilated cardiomyopathy, TAC transverse aortic constriction, W weeks, HF heart failure.
To clarify the role of high expression of miR-221-3p in cardiovascular tissue, we collected ten heart samples from patients who underwent heart transplantation due to end-stage HF and ten normal hearts of traffic accident victims. We found that the expression of miR-221-3p was elevated in the heart tissue of HF patients compared with that in the control group (Fig. 1d). We further examined the expression of miR-221-3p in heart tissue from TAC-induced HF mice. We found that miR-221-3p expression was reduced in hypertrophic hearts 2 weeks after TAC, but it increased at 4 weeks after TAC (n = 5 per group) (Fig. 1e). Meanwhile, echocardiographic analyses revealed that TAC-induced HF occurred at 4 weeks but not at 2 weeks after TAC surgery in mice (Supplementary Fig. 1A–C). Further, we found that miR-221-3p expression was also increased in the aorta 4 weeks after TAC (n = 5 per group) (Supplementary Fig. 1D). These results suggested that miR-221-3p played an essential role in the progression of HF.
MiR-221-3p regulates endothelial cell angiogenesis in vitro
To explore the effect of miR-221-3p on endothelial cells, gain/loss-of-function assays were conducted by transfection of miR-221-3p mimic and inhibitor in HUVECs in vitro. CCK-8 assays confirmed that miR-221-3p overexpression reduced cell viability, while miR-221-3p inhibition had the opposite effect (Fig. 2a). The EdU incorporation assay and quantification indicated that overexpression of miR-221-3p inhibited HUVEC proliferation, while blockage of miR-221-3p expression dramatically enhanced cell proliferation (Fig. 2b, c). Furthermore, a transwell chamber experiment was employed to determine the migration of endothelial cells. As expected, HUVECs with miR-221-3p mimic transfection showed impaired cell migration (Fig. 2d, e), while miR-221-3p inhibition significantly increased cell migration (Fig. 2d, e). Since miR-221-3p upregulation impaired HUVEC proliferation and migration, it is implicated that miR-221-3p might be involved in angiogenesis. To confirm this hypothesis, we further valuated HUVEC angiogenesis with or without miR-221-3p treatment. As shown in Fig. 2f, g, miR-221-3p mimic impaired HUVEC cord formation, while inhibition of miR-221-3p significantly promoted HUVEC angiogenesis in vitro.

a Quantification of CCK-8 assay OD value in HUVECs transfected with miR-221-3p mimic, miR-221-3p inhibitor or controls, n = 4. b Representative images of EdU incorporation assay in HUVECs transfected with miR-221-3p mimic, miR-221-3p inhibitor or controls and c quantification of proliferation ratio of HUVECs. Scale bar: 100 µm, n = 3. d Representative images of transwell chamber experiments in HUVECs transfected with miR-221-3p mimic, miR-221-3p inhibitor or controls and e quantification of migrated cell number. Scale bar: 100 µm, n = 5. f Representative images of cord formation in Matrigel of HUVECs transfected with miR-221-3p mimic, miR-221-3p inhibitor or controls and g quantification of tube length. Scale bar: 1000 µm, n = 5. Data shown as the mean ± SEM. CCK-8 cell count kit-8, con control.
MiR-221-3p reduces HIF-1α expression by targeting its 3′UTR
Next, we used the RNA-hybrid server (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/submission.html) to predict the possible targets of miR-221-3p and found three potential target genes including insulin-like growth factor-1 (Igf1), semaphorin 3 C (Sema3C), and Hif-1α. To determine the target of miR-221-3p, we conducted anti-Ago2 coimmunoprecipitation in HUVECs after transfection of miR-221-3p mimic or mimic control. MiRNAs can inhibit the translation of mRNAs by forming the RNA-induced silencing complex (RISC), and the Ago protein family is thought to be an important component of RISC [16]. MiRNAs function by directing Ago proteins to a complementary site in targeted mRNAs, and among the Ago proteins, Ago2 has catalytic activity and participates in the mRNA cleavage process [33, 34]. After overexpressing miR-221-3p, the anti-Ago2 coimmunoprecipitation experiment can be used to find target genes that bind to miR-221-3p. Coimmunoprecipitation with Ago2 and IgG antibody demonstrated that Ago2 was specifically isolated using the Ago2 antibody (Fig. 3a). We found that the association between Ago2 and Hif-1α, but not Igf1 or Sema3C, was increased after miR-221-3p mimic transfection in HUVECs (Fig. 3b). Subsequently, multiple sequence alignment of miR-221-3p and Hif-1α indicated a binding site within the 3′UTR of the Hif-1α gene in both human and mouse (Fig. 3c). To confirm this, we cloned the 3′UTR of Hif-1α which included a wild-type seed region sequence or a mutated seed region sequence to pMIR-report Luciferase plasmid (Fig. 3d). Dual-luciferase reporter assays showed that miR-221-3p mimic transfection significantly suppressed luciferase activity in pMIR-Hif-1α 3′UTR-treated HEK293 cells compared with those transfected with the mimic control, but luciferase activity in pMIR-Hif-1α 3′UTR mutation-treated HEK293 cells remained unaltered after miR-221-3p mimic and mimic control transfection (Fig. 3e). Moreover, western blotting showed that miR-221-3p mimic reduced HIF-1α protein expression in HUVECs, while miR-221-3p inhibitor increased HIF-1α expression in HUVECs (Fig. 3f, g).

a The IP assay showing Ago2 protein expression in coimmunoprecipitated products of HUVECs transfected with mimic con or miR-221-3p mimic, n = 3. b QPCR analyses of mRNAs in association with Ago2 in HUVECs transfected with mimic con or miR-221-3p mimic, n = 3. c Sequence alignment between miR-221-3p and the Hif-1α 3′UTR in human and mouse. d Schematic diagram of the pMIR-report luciferase plasmids of pMIR-Hif-1α 3′UTR and pMIR-Hif-1α 3′UTR mut. e MiR-221-3p binding to Hif-1α 3′UTR in HEK293 cells detected by a dual-luciferase reporter assay, n = 3. f Representative western blotting images of HIF-1α and β-actin and g quantification of HIF-1α expression in HUVECs transfected with miR-221-3p mimic, miR-221-3p inhibitor or controls, n = 3. Data shown as the mean ± SEM. UTR untranslated region, IP immunoprecipitation, qPCR quantitative polymerase chain reaction, ns not significant, mut mutation, con control.
Restored HIF-1α eliminates the miR-221-3p induced endothelial cell impairments
To verify whether HIF-1α could induce endothelial cell dysfunction, siRNA against Hif-1α was transfected into HUVECs. The knockdown efficiency of the siRNA was almost 50% at the protein level (Supplementary Fig. 2A, B). Knockdown of HIF-1α resulted in decreased cell viability (Supplementary Fig. 2C). Moreover, knockdown of HIF-1α induced endothelial dysfunction, as demonstrated by reduced migration (Supplementary Fig. 2D, E) and damaged cord formation in Matrigel (Supplementary Fig. 2F, G).
Further, to determine whether HIF-1α was involved in the miR-221-3p-modulated HUVEC impairments, we constructed a pcDNA3.1-Hif-1α plasmid containing the CDS sequence of the Hif-1α gene without the 3′UTR, which cannot be degraded by miR-221-3p. As expected, pcDNA3.1-Hif-1α transfection significantly increased HIF-1α expression in HUVECs treated with mimic control. However, pcDNA3.1-Hif-1α transfection could only rescue the HIF-1α expression to normal levels in HUVECs treated with miR-221-3p mimic (Fig. 4a, b). The rescued HIF-1α expression in miR-221-3p mimic-treated HUVECs restored cell viability and proliferation (Fig. 4c–e). Similarly, the transwell chamber experiment and cord formation in the Matrigel assay showed that restored HIF-1α expression markedly improved cell migration and cord formation in miR-221-3p mimic-treated HUVECs (Fig. 4f–i).

a Representative western blotting images of HIF-1α and β-actin and b quantification of HIF-1α expression in HUVECs transfected with or without mimic con, miR-221-3p mimic, pcDNA3.1 plasmid, and pcDNA3.1-Hif-1α plasmid, n = 3. c Quantification of CCK-8 assay OD value in HUVECs transfected with or without mimic con, miR-221-3p mimic, pcDNA3.1 plasmid, and pcDNA3.1-Hif-1α plasmid, n = 3. d Representative images of EdU incorporation assay in HUVECs transfected with or without mimic con, miR-221-3p mimic, pcDNA3.1 plasmid, and pcDNA3.1-Hif-1α plasmid and e quantification of proliferation ratio of HUVECs. Scale bar: 100 µm, n = 3. f Representative images of transwell chamber experiments in HUVECs transfected with or without mimic con, miR-221-3p mimic, pcDNA3.1 plasmid, and pcDNA3.1-Hif-1α plasmid and g quantification of migrated cell number. Scale bar: 100 µm, n = 3. h Representative images of cord formation in Matrigel of HUVECs transfected with or without mimic con, miR-221-3p mimic, pcDNA3.1 plasmid, and pcDNA3.1-Hif-1α plasmid and i quantification of tube length. Scale bar: 1000 µm, n = 3. Data shown as the mean ± SEM. CCK-8 cell count kit-8, ns not significant, con control.
AntagomiR-221-3p increases HIF-1α expression and improves angiogenesis in ischemic hindlimbs
To further demonstrate the role of miR-221-3p in angiogenesis in vivo, we developed a hindlimb artery ligation model in mice. We observed hindlimb blood flow at 0, 7, 14, and 21 days after surgery using laser speckle imaging, as shown in Fig. 5a. We observed angiogenesis on the seventh day after surgery, and the blood flow of the ischemic hindlimbs gradually recovered over time. On day 21, the blood flow of the ischemic hindlimbs in the antagomiR-221-3p group had recovered to a much greater extent than in the antagomiR-NC group. The ischemic hindlimb blood flow ratio in the antagomiR-221-3p group was significantly higher than that in the antagomiR-NC group throughout the study period (n = 5 per group) (day 7: 61.3 ± 3.48% vs 27.5 ± 3.221%, p < 0.0001; day 14: 71.6 ± 3.14 vs 33.8 ± 2.087, p = 0.0006; day 21: 91.25 ± 4.854 vs 61.5 ± 1.848%, p = 0.001) (Fig. 5b).

a Representative laser speckle perfusion images and b statistical analysis of left/right (L/R) hindlimb perfusion ratios at different time points after femoral artery ligation in the HLI + antagomiR-NC group and HLI + antagomiR-221-3p group. ****p < 0.0001 vs HLI + antagomiR-NC, ***p = 0.0006 vs HLI + antagomiR-NC, **p = 0.001 vs HLI + antagomiR-NC, n = 5 per group. c H&E staining, CD31 and HIF-1α immunohistochemistry staining obtained from transverse sections of the adductor muscle of the sham group, HLI + antagomiR-NC group, and HLI + antagomiR-221-3p group after 21 days of femoral artery ligation. Scale bar: 50 µm. d Quantification of CD31 (up) and HIF-1α (down) expression in the sham group, HLI + antagomiR-NC group, and HLI + antagomiR-221-3p group, n = 5 per group. Data shown as the mean ± SEM. HLI hindlimb ischemia, NC negative control, H&E hematoxylin and eosin.
Histological analyses of the adductor tissue isolated from the hindlimbs on day 21 showed significantly higher CD31 expression in the antagomiR-221-3p group compared with that in the antagomiR-NC group after HLI surgery, suggesting a significant increase in angiogenesis. Meanwhile, HIF-1α expression was increased in the antagomiR-221-3p group compared with the antagomiR-NC group (Fig. 5c, d).
AntagomiR-221-3p improves cardiac function of TAC-induced HF mice by increasing angiogenesis
To further verify the role of miR-221-3p in HF, we used TAC to establish a mouse HF model. Echocardiography revealed that TAC surgery caused a significant decrease in cardiac function in mice 4 weeks after TAC surgery (Ejection fraction (EF) %: 58.46 ± 2.533 vs 33.35 ± 3.283, p = 0.0003), (Fractional shortening (FS) %: 29.46 ± 2.31 vs 15.78 ± 1.737, p = 0.0015) (Fig. 6a, b), while antagomiR-221-3p significantly improved TAC-induced decrease in cardiac function (EF %: 44.94 ± 3.458 vs 33.35 ± 3.283, p = 0.0413), (FS %: 22.12 ± 2.01 vs 15.78 ± 1.737, p = 0.0439) (Fig. 6a, b). Further histological staining revealed that the cardiomyocyte hypertrophy in the antagomiR-221-3p group was alleviated compared with that in the antagomiR-NC group, and myocardial tissue fibrosis was reduced compared with the antagomiR-NC group 4 weeks after TAC (Fig. 6c, d). To determine whether the improvement in cardiac function was caused by increased angiogenesis in the antagomiR-221-3p group, we performed immunohistochemical staining of CD31 and HIF-1α and found that TAC caused a decrease in angiogenesis of the heart with a significant decrease in CD31 and HIF-1α expression in the antagomiR-NC group compared with the sham group. Injection of antagomiR-221-3p increased CD31 and HIF-1α expression significantly (Fig. 6e, f).

a Four weeks after TAC surgery, the representative echocardiography images of the sham group, TAC + antagomiR-NC group, and TAC + antagomiR-221-3p group. b EF and FS value of the sham group, TAC + antagomiR-NC group, and TAC + antagomiR-221-3p group, n = 5 per group. c Hematoxylin and eosin (H&E) staining and PSR staining obtained from hearts of sham group, TAC + antagomiR-NC group, and TAC + antagomiR-221-3p group. Scale bar (up): 20 µm, scale bar (down): 100 µm. d Quantification of cardiomyocyte area (left) and the degree of cardiac fibrosis (right), n = 5 per group. e CD31 and HIF-1α immunohistochemistry staining obtained from hearts of the sham group, TAC + antagomiR-NC group, and TAC + antagomiR-221-3p group. Scale bar: 20 µm. f Quantification of CD31 (left) and HIF-1α expression level (right), n = 5 per group. Data shown as the mean ± SEM. TAC transverse aortic constriction, NC negative control, EF ejection fraction, FS fractional shortening, H&E hematoxylin and eosin, PSR picrosirius red.
Serum miR-221-3p expression is negatively correlated with heart function in HF patients
Since miR-221-3p was highly expressed in heart tissue of HF patients and TAC-induced HF mice, we further examined whether miR-221-3p is expressed in the serum of patients and whether its expression is associated with the progression of HF. The study population consisted of 30 CHF patients, and their baseline characteristics are shown in Supplementary Table 1. Serum miR-221-3p in the patients was detected by qPCR. As shown in Fig. 7a, there was a significant negative correlation between the serum expression of miR-221-3p and heart function of HF patients.

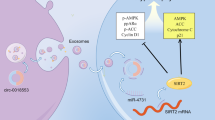
a Linear correlation between serum miR-221-3p expression level and LVEF% in HF patients. n = 30. b The stress load leads to cardiac hypertrophy, which can develop into HF. During this process, the increased expression of miR-221-3p inhibits the expression of HIF-1α, leading to decreased angiogenesis, which in turn promotes HF. LVEF left ventricular ejection fraction, HF heart failure.
Discussion
In our study, high expression of miR-221-3p was observed in endothelial cells, suggesting a prominent role of miR-221-3p in endothelial cell functions. We discovered that miR-221-3p inhibited endothelial cell proliferation, migration, and cord formation, which are the major steps in angiogenesis. Angiogenesis plays a vital role in physiological and pathological blood vessel growth after birth. The former includes physical hypertrophy of the heart, and the latter includes cancer [35] and ischemic heart disease [36]. The association between miRNAs and angiogenesis has been reported recently. MiR-210 improved angiogenesis by inducing the release of angiogenic factors including leptin, interleukin-1-a, and tumor necrosis factor-alpha [37], while miR-17-92, miR-24, and miR-214 showed antiangiogenic effects [17]. Previous studies have reported two conflicting roles of miR-221-3p in endothelial cells—the inhibition of endothelial cell functions [38, 39] and the promotion of blood vessels and lymphatics regeneration [21, 22]. However, the blood vessel regeneration function of miR-221-3p has been mainly associated with exosomal miR-221-3p in cancers, and in this study, we mainly determined the role of miR-221-3p in normal mammalian organs.
We identified that HIF-1α is one of the primary target genes of miR-221-3p in HUVECs. HIF-1α can increase angiogenesis by regulating the VEGF pathway, resulting in more oxygen and nutrients provided to cardiomyocytes [40, 41]. During the progression of HF, the expression of HIF-1α changes at different stages. In cardiac hypertrophy, increased diffusion distance results in reduced oxygen supply in the myocardium, and HIF-1α is upregulated by hypoxia; HIF-1α cannot be maintained at a high level for an extended period and it will be reduced by p-53, which may lead to the progress of HF [5, 42]. Moreover, recent studies have shown that HIF-1α can also be regulated by miRNAs as mentioned above. In the present study, we found that miR-221-3p could reduce the expression of HIF-1α by targeting its 3′UTR.
Through the HLI experiment, we conclude that miR-221-3p can also inhibit angiogenesis in vivo. Further, to verify whether miR-221-3p plays a role in HF, we used TAC surgery to establish an HF mouse model. Our research found that inhibition of miR-221-3p can promote cardiac angiogenesis, alleviate myocardial hypertrophy, and further improve the cardiac function of TAC-induced HF mice. However, antagomiR-221-3p cannot specifically inhibit miR-221-3p in vascular endothelial cells, but instead it broadly inhibits the function of miR-221-3p. MiR-221-3p may be involved in the regulation of the expression of multiple genes other than angiogenesis-related genes, so the effect of antagomiR-221-3p on improving cardiac function may not be entirely dependent on the increase in angiogenesis caused by HIF-1α. Previous studies have found that miR-221-3p can cause cardiomyocyte hypertrophy by inhibiting p27 [43, 44]. Therefore, in addition to increasing endothelial cell angiogenesis, antagomiR-221-3p may also improve the cardiac function of HF mice by alleviating cardiomyocyte hypertrophy directly.
Interestingly, we also observed that the serum expression of miR-221-3p was negatively correlated with the heart function of HF patients. However, previous studies have shown that miR-221-3p can inhibit fibrosis and that the expression of miR-221-3p is negatively correlated with the level of fibrosis, which is not consistent with our results [45]. This can be attributed to differences in the patient groups; our study included patients with a cardiac function of NYHA class II–IV, while the previous study included patients with a cardiac function of NYHA class I–II. In general, our results implicated that miR-221-3p may be an important biomarker in HF. The elevation of miR-221-3p might indicate that the patient is in an advanced stage of HF and may provide some evidence for the treatment of severe HF.
In summary, miR-221-3p is mainly expressed in vascular endothelial cells and blocks endothelial cell angiogenesis by inhibition of HIF-1α. MiR-221-3p is increased during the process of cardiac hypertrophy progression into HF, inhibiting HIF-1α expression, leading to decreased angiogenesis which in turn aggravates the progress of HF (Fig. 7b). Inhibition of miR-221-3p can significantly improve the cardiac function of HF mice, and miR-221-3p may be a new effective molecular target for the progression and treatment of HF.
References
Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. 2015;6:187–214.
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Investig. 2003;111:1497–504.
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–40.
Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565–71.
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–8.
Guo J, Mihic A, Wu J, Zhang Y, Singh K, Dhingra S, et al. Canopy 2 attenuates the transition from compensatory hypertrophy to dilated heart failure in hypertrophic cardiomyopathy. Eur Heart J. 2015;36:2530–40.
Yue X, Lin X, Yang T, Yang X, Yi X, Jiang X, et al. Rnd3/RhoE modulates hypoxia-inducible factor 1alpha/vascular endothelial growth factor signaling by stabilizing hypoxia-inducible factor 1alpha and regulates responsive cardiac angiogenesis. Hypertension. 2016;67:597–605.
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–4.
Wiesener MS, Jürgensen JS, Rosenberger C, Scholze CK, Hörstrup JH, Warnecke C, et al. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003;17:271–3.
Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621–7.
Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8.
Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H, et al. Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res. 2008;68:5540–5.
Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–86.
Greco S, Gaetano C, Martelli F. HypoxamiR regulation and function in ischemic cardiovascular diseases. Antioxid Redox Signal. 2014;21:1202–19.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Wang H, Cai J. The role of microRNAs in heart failure. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2019–30.
Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108:3068–71.
Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68.
Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–73.
Zhou C-F, Ma J, Huang L, Yi H-Y, Zhang Y-M, Wu X-G, et al. Cervical squamous cell carcinoma-secreted exosomal miR-221-3p promotes lymphangiogenesis and lymphatic metastasis by targeting VASH1. Oncogene. 2018;38:1256–68.
Wu XG, Zhou CF, Zhang YM, Yan RM, Wei WF, Chen XJ, et al. Cancer-derived exosomal miR-221-3p promotes angiogenesis by targeting THBS2 in cervical squamous cell carcinoma. Angiogenesis. 2019;22:397–410.
Gould BR, Damgaard T, Nielsen BS. Chromogenic in situ hybridization methods for microRNA biomarker monitoring of drug safety and efficacy. Methods Mol Biol. 2017;1641:399–412.
de Planell-Saguer M, Rodicio MC, Mourelatos Z. Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nat Protoc. 2010;5:1061.
Wang WX, Wilfred BR, Hu Y, Stromberg AJ, Nelson PT. Anti-argonaute RIP-chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA. 2010;16:394–404.
Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat Protoc. 2009;4:1737–46.
deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:1729.
Gardin Julius M, Siri Francis M, Kitsis Richard N, Edwards John G, Leinwand Leslie A. Echocardiographic assessment of left ventricular mass and systolic function in mice. Circ Res. 1995;76:907–14.
Liu H, Zhu R, Liu C, Ma R, Wang L, Chen B, et al. Evaluation of decalcification techniques for rat femurs using HE and immunohistochemical staining. Biomed Res Int. 2017;2017:1–6.
Pinto AR, Chandran A, Rosenthal NA, Godwin JW. Isolation and analysis of single cells from the mouse heart. J Immunol Methods. 2013;393:74–80.
Gunduz D, Hamm CW, Aslam M. Simultaneous isolation of high quality cardiomyocytes, endothelial cells, and fibroblasts from an adult rat heart. J Vis Exp. 2017;123:55601.
Patel JJ, Srivastava S, Siow RC. Isolation, culture, and characterization of vascular smooth muscle cells. Methods Mol Biol. 2016;1430:91–105.
Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song J-J, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–41.
Schirle NT, Sheu-Gruttadauria J, MacRae IJ. Structural basis for microRNA targeting. Science. 2014;346:608–13.
Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–22.
Shiojima KWI. Cardiac growth and angiogenesis coordinated by intertissue interactions. J Clin Investig. 2007;117:3583–92.
Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124–31.
Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581–8.
Hartmann D, Fiedler J, Sonnenschein K, Just A, Pfanne A, Zimmer K, et al. MicroRNA-based therapy of GATA2-deficient vascular disease. Circulation. 2016;134:1973–90.
Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843.
Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell. 2009;16:167–79.
Gogiraju R, Xu X, Bochenek ML, Steinbrecher JH, Lehnart SE, Wenzel P, et al. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc. 2015;4:e001770.
Su M, Wang J, Wang C, Wang X, Dong W, Qiu W, et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22:986–99.
Wang C, Wang S, Zhao P, Wang X, Wang J, Wang Y, et al. MiR-221 promotes cardiac hypertrophy in vitro through the modulation of p27 expression. J Cell Biochem. 2012;113:2040–6.
Verjans R, Peters T, Beaumont FJ, van Leeuwen R, van Herwaarden T, Verhesen W, et al. MicroRNA-221/222 family counteracts myocardial fibrosis in pressure overload-induced heart failure. Hypertension. 2018;71:280–8.
Acknowledgements
This study was supported by National Science Funding of China (NSFC 81670276, 91739118, and 81870553) to YH and National Science funding of China (NSFC 81770303) to CY. The funding organization had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, Y., Yan, C., Fan, J. et al. MiR-221-3p targets Hif-1α to inhibit angiogenesis in heart failure. Lab Invest 101, 104–115 (2021). https://doi.org/10.1038/s41374-020-0450-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-020-0450-3
This article is cited by
-
Non-coding RNAs and angiogenesis in cardiovascular diseases: a comprehensive review
Molecular and Cellular Biochemistry (2024)
-
Advances in MicroRNA Therapy for Heart Failure: Clinical Trials, Preclinical Studies, and Controversies
Cardiovascular Drugs and Therapy (2023)
-
Factors influencing degradation kinetics of mRNAs and half-lives of microRNAs, circRNAs, lncRNAs in blood in vitro using quantitative PCR
Scientific Reports (2022)
