
Abstract
Physiological brain temperature is an important determinant of brain function, and it is well established that changes in brain temperature dynamically influence hippocampal neuronal activity. We previously demonstrated that the thermosensor TRPV4 is activated at physiological brain temperature in hippocampal neurons thereby controlling neuronal excitability in vitro. Here, we examined whether TRPV4 regulates neuronal excitability through its activation by brain temperature in vivo. We locally cooled the hippocampus using our novel electrical device and demonstrated constitutive TRPV4 activation in normal mouse brain. We generated a model of partial epilepsy by utilizing kindling stimuli in the ventral hippocampus of wild type (WT) or TRPV4-deficient (TRPV4KO) mice and obtained electroencephalograms (EEG). The frequencies of epileptic EEG in WT mice were significantly larger than those in TRPV4KO mice. These results indicate that TRPV4 activation is involved in disease progression of epilepsy. We expected that disease progression would enhance hyperexcitability and lead to hyperthermia in the epileptogenic foci. To confirm this hypothesis, we developed a new device to measure exact brain temperature only in a restricted local area. From the recording results by the new device, we found that the brain temperatures in epileptogenic zones were dramatically elevated compared with normal regions. Furthermore, we demonstrated that the temperature elevation was critical for disease progression. Based on these results, we speculate that brain cooling treatment at epileptogenic foci would effectively suppress epileptic discharges through inhibition of TRPV4. Notably, the cooling treatment drastically suppressed neuronal discharges dependent on the inactivation of TRPV4.
Similar content being viewed by others

Introduction
Small changes in brain temperature are known to affect brain function [1, 2]. Clinical evidence suggests that brain temperature is a critical determinant of cognitive function. It has also been reported that therapeutic hypothermia leads to cognitive dysfunction in heart disease patients [3], and cooling of the prefrontal cortex in monkeys causes performance impairment during a delayed matching-to-sample task [4]. In rodents, brain cooling significantly impairs spatial learning [2]. These reports indicate that the maintenance of brain temperature is important for healthy brain function. However, the underlying molecular mechanisms for this regulation remain unknown.
TRPV4 is a nonselective cation channel, first described as an osmosensor for detecting hypotonic stimuli [5,6,7,8]. The expression of TRPV4 in kidney, cochlea, sweat glands, sensory nerve terminals, and osmosensory cells in the brain is in agreement with the osmo- and/or mechano-sensing functions of TRPV4 [9,10,11,12]. Mice lacking the TRPV4 gene exhibit defects in high-threshold mechano-sensation and pressure sensation [13]. TRPV4 was reported to be necessary for the response to changes in osmotic pressure and to function as an osmotic sensor in the CNS [14, 15]. TRPV4 can also be activated by heat ( >27 °C–34 °C), the phorbol ester derivative 4α-phorbol 12,13 didecanoate (4αPDD), or lipids such as metabolites of arachidonic acid [16,17,18,19]. In addition, we found that TRPV4 was strongly expressed in hippocampal neurons and constitutively activated by physiological brain temperature to control neuronal excitability in vitro [20]. Furthermore, we reported that TRPV4 is also expressed in microglia and specific subtypes of astrocytes in the brain, where it regulates synaptic activity through gliotransmitter release [21,22,23]. These results indicate that TRPV4 is a key molecule regulating neuronal excitability. Since TRPV4 is activated at physiological brain temperature in vitro [20], we speculated that TRPV4 might be constitutively active under normothermic conditions in vivo, and might also affect neuronal excitability and behavior.
Therapeutic temperature management can be necessary when a hemodynamically stable cardiac arrest survivor is uneventfully cooled and rewarmed, or when a spontaneously breathing patient with traumatic brain injury develops persistent neurogenic fever and shivers uncontrollably [24].
It is well known that hypothermia provides a strong neuroprotective effect on the brain following various types of brain insult in experimental animals. The representative neuroprotective effects are: i) the hypothermia reduces the metabolic rates and energy depletion of brain tissue; ii) attenuates excitatory amino acid release and the synthesis of free radicals; iii) prevents blood brain barrier disruption and brain edema [25]. Brain cooling has been well established as an effective treatment for suppressing epileptic events; [26, 27] however, the molecular mechanisms by which cooling reduces epileptic events are still unknown. If TRPV4 is constitutively activated by brain temperature, which serves to enhance brain activity, then TRPV4 is a good candidate molecule for the suppression of epileptic events by cooling.
In this study, we examined the functional significance of TRPV4 activation at brain temperature for the regulation of hippocampal activity. We also examined whether inactivation of TRPV4 by cooling of epileptogenic foci or injection of a TRPV4-specific inhibitor into epileptic foci effectively suppresses epileptic discharges in mouse models.
Materials and methods
Animals
TRPV4KO mice were generated by crossing heterozygous mice, and the genotypes were determined by PCR. TRPV4KO mice were mated to C57BL/6 J mice to the fourth generation and we gained mutant line. We used the 25th generation of TRPV4 knockout mice, which had been backcrossed at every 5th generation. All animal care and experimental procedures were performed according to the guidelines of the Gunma University Animal Care and Experimentation Committee. All animal experiments were approved by the Gunma University Animal Care and Experimentation Committee.
Electrophysiology
Male WT or TRPV4KO mice (4–5 weeks old) were deeply anaesthetized with halothane then decapitated. The brain was quickly removed and transferred to ice-cold medium saturated with 95% O2 and 5% CO2. The medium contained (in mM): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.0 NaH2PO4, 26.2 NaHCO3, and 11 glucose. Transverse hippocampal slices (400 μm thickness) were prepared in the medium with a tissue slicer (VT-1200S, Leica, Germany) and placed in a humidified interface-type holding chamber for at least 1 h before recordings. A single slice was then transferred to the recording chamber and submerged with a nylon net stretched out on a U-shaped piece of flattened platinum wire beneath a continuously perfusing medium (flow rate: ∼2 ml min−1) that had been saturated with 95% O2 and 5% CO2. The composition of the medium used for the recordings was the same as that used for slice preparations, unless otherwise stated, with the exception that all perfusing solutions contained 100 μM picrotoxin to block GABAA receptor-mediated inhibitory synaptic responses. Whole-cell recording data were sampled at 10 kHz and filtered at 5 kHz for analysis (Axon 200B amplifier with pCLAMP10 software, Axon Instruments, Foster City, CA). All patch-clamp experiments were performed at 25 oC, 30 oC or 35 oC. Chamber temperature was monitored with a thermocouple placed within 100 μm of the patch-clamped cells. To measure evoked fEPSPs, we put glass electrodes (filled with 3 M NaCl) onto the medial perforant path, stimulated at 0.1 Hz for 200 μsec, and recorded fEPSPs from the DG. For the analysis of fEPSPs, we measured their peak amplitudes.
Electrode implantation
Surgeries were similarly performed as previously described [28]. The animal was anaesthetized with isofluorane and placed in a stereotaxic frame. After a skin incision to expose the skull surface, three small holes (∼0.5 mm in diameter) were drilled through the skull. Electrodes were inserted into the brain using micromanipulators and then glued onto the skull. Twisted bipolar electrodes (tips of ∼100 μm apart) were placed into bilateral hippocampal DG areas (bregma: −2.2 mm, lateral 1.2 mm and depth 2.0 mm). A reference electrode was positioned at a frontal area (bregma + 1.0 mm, lateral 1.0 mm and depth 0.5 mm). All electrodes were made of polyamide-insulated stainless steel wires (outer diameter 200 μm; Plastics One, Ranoake, VA, USA). Implanted animals were allowed to recover for ≥ 1 week prior to further experimentation. The locations of implanted electrodes were verified by behavioral state-dependent hippocampal EEG activities and/or by later histological assessments.
Hippocampal DG kindling
Unilateral DG kindling was conducted using a standard protocol [28]. Constant current pulses with monophasic square waveforms, pulse duration of 0.5 ms and current intensities of 10–100 μA were generated by a Grass stimulator and delivered through an isolation unit (model S88, Grass Medical Instruments, Warwick RI, USA). Initially, an ascending series was performed to determine afterdischarges (AD) threshold for individual animals. In the ascending series, a train of current pulses (60 Hz for 2 s) with incremental intensities (10 μA per step) were applied every 30 min. The lowest stimulation intensity by which an AD event of ≥ 5 s was elicited was considered the AD threshold. The animal was then stimulated at 125% of the AD threshold daily for several weeks. The animal was considered kindled when stage 5 motor seizures were elicited over three consecutive days [28]. An ascending series was similarly conducted in each kindled animal.
EEG recording, local brain temperature measurement, and artificial brain cooling
EEG electrodes connected to a 5 mm2 computer circuit board were implanted into the hippocampal DG or on the skull (for ECoG). In addition, electrodes for EMG were implanted on the neck muscle. The mice were then housed separately for a recovery period of at least 7 days. After 7 days, the mice were connected to the EEG machine (EEG4214, Nihon Koden, Japan), and the analog output was connected to PowerLab8/30 (AD Instruments, CO, U.S.A.). The digital EEG data were analyzed by Labchart8.0 software. Epileptic discharges were isolated and calculated as EEG patterns and amplitudes by Labchart8.0 software (AD Instruments, CO, U.S.A.). To measure local brain temperature, we made thin thermistors (diameter < 500 μm), and these were connected to a 5 mm2 computer circuit board, which was also connected to EEG and EMG electrodes. To avoid significant measurement errors, we established a new temperature measurement system based on a compensation circuit by utilizing voltage input (Fig. 3a). Firstly, we performed the calibration of the temperature. We recorded the two water voltages (at 35 °C or 40 °C), and these voltage values were input to Labchart8.0 software, and measured the local brain temperature by PowerLab8/30. For artificial brain cooling, we modified brain cooling devices [29] based on a Peltier device, and these were implanted into the DG. The core body temperatures were measured using transmitter devices (Mini Mitter Company) surgically implanted in the peritoneal cavity under anesthesia (50 mg/kg sodium pentobarbital). After surgery, each animal was housed alone in a recording cage and allowed to recover from surgery for more than 1 week. Data were recorded by placing each cage containing an animal implanted with a radiotransmitter on a receiver plate (ER-4000, Mini Mitter Company).
Statistical analysis
Statistical analyses were conducted using StatView (SAS Institute, Cary, NC). Statistical analysis was performed using Student’s t tests or repeated measures (RM) ANOVA followed by Dunnett’s post hoc test. P values less than 0.05 were considered significant. Values in graphs are expressed as mean ± s.e.m.
Results
The hippocampal dentate gyrus (DG) in TRPV4KO exhibits abnormal neuronal excitability dependent on TRPV4 activation by brain temperature
We reported that TRPV4 is expressed in hippocampal pyramidal neurons in addition to internerons and DG neurons [20]. Furthermore, we found the abnormal behaviors in TRPV4KO mice [30]. These abnormal behaviors are due to lack of sensing brain temperature by neuronal TRPV4. We searched which type of neurons is responsible for the behavior by c-Fos mapping between WT and TRPV4KO mice. We found that DG granule neurons are the critical neurons to regulate normal behaviors by TRPV4 activation at brain temperature [30]. Our previous report [20] led us to consider whether abnormal excitability in TRPV4KO mice might result from abnormal synchronization of firings in DG neurons. To confirm this, we measured population spikes in granule neurons from slice preparations at 35 °C, since the maintenance of physiological temperature (37 °C) was intractable in hippocampal slices (neurons showed hypoxic damage caused by high oxygen consumption at 37 °C). Comparison of the firing states at 30 °C (non-TRPV4 activation) or 35 °C (TRPV4 activation), should reveal that the 35 °C condition causes firing only in WT mice. We recorded population spikes at 30 °C or 35 °C in both WT and TRPV4KO DGs (Fig. 1). Our electrophysiological equipment has chamber-temperature control system (Warner Instrument TC-344B). We heat the slice temperature through bath solution by the rate (approximately 5 °C increase/90~120 sec). We put an electrode on the medial perforant path (MPP) for voltage input, and recorded the spikes from the soma of DG granule neurons. We adjusted the voltage-stimulus to generate only a single spike at 30 °C in both WT and TRPV4KO slices (Fig. 1). In contrast to those results, elevation of the recording temperature to 35 °C increased spike number, and reduced the latency to evoke the 1st spike in WT but not TRPV4KO slices (Fig. 1a–c). These results indicate that TRPV4 activation under physiological temperature significantly enhances neuronal excitability through the depolarization of resting membrane potential (RMP) [20].

The dentate gyrus of TRPV4KO mice exhibits abnormal neuronal firing at physiological temperature. a Recording of field EPSPs (fEPSPs) at medial perforant path (MPP)-DG granule synapses near the cell bodies at 30 °C or 35 °C. Representative traces are shown in the top panels for both WT and TRPV4 hippocampal slices. The observed population spikes are indicated by arrowheads. The timing of the stimuli is shown by arrows. b The number of population spikes were quantified (n = 16 slices) at 30 °C or 35 °C. c The latency of the 1st population spike after the stimulus was quantified (n = 16 slices) at 30 °C or 35 °C. The asterisks represent statistical significance (p < 0.05) compared with the result at 30 °C in the same mouse strain group
Local brain temperature shows dynamic changes and affects brain activity in vivo independent of behavioral movements
To date, effective tools to accurately monitor local brain temperature have not been available. Therefore, we developed a new monitoring system utilizing a combination of fine thermistors and detection equipment (Fig. 2a). Using our novel device, we could detect local brain temperature (within 1 mm3) with very fine accuracy (±0.02 °C). We performed the calibration of the thermistor, before local brain temperature recordings. We recorded the two water voltages (at 35 °C or 40 °C), and these voltage values were input to Labchart8.0 software, and the voltage values were converted to the temperature values by PowerLab8/30 (based on two points calibration). In order to confirm the accuracy of the measured temperature, we put our thermistor on a heat block which was set at 30.0 °C, 35.0 °C or 40.0 °C, and confirmed our temperature value showed those temperature values. For the measurement of local brain temperature, we surgically placed the thermistors and electroencephalogram (EEG) electrodes into the DG then recorded the local brain temperature. Local DG temperature dynamically changed independent from behavioral movements, which could be detected by electromyography (EMG) recordings (Fig. 2b, arrows). Several large changes in local DG temperature correlated with EEG activity (Fig. 2b, arrowheads). We quantified the changes in core body or brain (DG) temperature during non-REM sleep-wake transitions (Fig. 2c). In regards to core body temperature, wakefulness increased temperature by approximately 2 °C (Fig. 2c, sleep; 36.3 ± 0.4 °C vs wakefulness; 38.3 ± 0.6 °C in WT mice). In contrast, wakefulness only increased brain temperature by about 0.8 °C (Fig. 2c, sleep; 36.9 ± 0.2 °C vs wakefulness; 37.7 ± 0.2 °C in WT mice), indicating that local brain temperature was precisely controlled compared to changes in core body temperature. This also suggested that TRPV4 converts temperature elevation into brain activity. We did not observe any differences in local brain temperature changes between WT and TRPV4KO mice (sleep; 36.7 ± 0.3 °C vs wakefulness; 37.4 ± 0.2 C in TRPV4KO mice), indicating that TRPV4 deficiency did not directly affect core body or brain temperature.

Local brain temperature is dynamically changed in relation to brain activity. a A schematic drawing of a newly designed local temperature detection system. The picture of right black bar (red arrowhead) represents our original thermistor is very tiny compared with a commercial thermistor (left silver bar; for mouse body temperature detection, Physitemp Co. Ltd., TCAT-2DF). The two lead cables of our thermistors were connected to a 5 mm2 computer circuit board, which was also connected to our local temperature detection system. b Representative traces of local brain temperature, EEG, and EMG in WT hippocampal DGs under freely moving conditions. c: The average core body or brain (DG) temperature (n = 6 WT mice) was quantified during sleep or wakefulness. The asterisks represent statistical significance (p < 0.05) compared with the sleep period
Constitutive activation of TRPV4 by brain temperature enhances hippocampal excitability in vivo
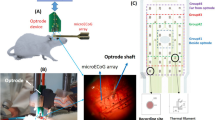
We expected that TRPV4KO mice would exhibit abnormal brain activity since they displayed abnormal behavior [30] and their neurons displayed altered excitability in acute slices (Fig. 1). The above findings demonstrated that TRPV4 is an important molecular determinant for electrical excitability (Fig. 1); however, it was still unknown whether TRPV4 could be constitutively activated by brain temperature. To demonstrate this in vivo, we developed a novel device for localized brain cooling (Fig. 3a). The device is equipped with a Peltier device, a probe to implant into the brain, and four funnels (Fig. 3a). Once the switch is turned on, voltage input is supplied to the Peltier device through a cable. The probe temperature can then be dropped to ~12 °C within a few seconds (Fig. 3b). Since the voltage input significantly heats the Peltier device, it must be cooled down to avoid propagation of heat to the cooling probe. Hence, four big funnels were incorporated into our device to effectively release this heat (Fig. 3a). These funnels prevent heating of the cooling probe, and contribute to the cooling of the probe (Fig. 3b). After performing surgery on WT or TRPV4KO mice to insert the brain-cooling devices into the DGs, the mice were connected to cables (Fig. 3c) with input to an A/D converter connected to a computer. Under normal conditions, the basal EEG power spectra of the theta (θ) waves in WT DGs were significantly larger than those in TRPV4KO DGs (Fig. 4a–c, high magnifications are represented as green traces, indicating that WT hippocampal activity is enhanced by TRPV4 activation in vivo. Cooling of the Peltier device (to 12 °C) effectively reduced DG temperature to around 30 °C (Fig. 4a). The artificial cooling significantly reduced DG activity in WT mice (Fig. 4a). These results suggested that brain temperature enhanced hippocampal activity through TRPV4-dependent mechanisms. Notably, the θ power reduction rates (cooling effects) in WT mice were much larger than those observed in TRPV4KO mice (Fig. 4d), indicating that the activation of TRPV4 by brain temperature is an important determinant of brain activity. Thus, we demonstrated that constitutive activation of TRPV4 occurs in the brain through its activation by brain temperature, with the result being an enhancement of neuronal excitability (Fig. 4).

A new device for local brain cooling. a A photo showing our novel brain cooling device. The thin, black bar is surgically implanted into the mouse brain. The square, black piece is equipped with a Peltier device. To prevent heating of the Peltier device, the cooling device is equipped with four funnels. The six-pin unit is connected to a cable (as shown in panel C) for measurement of brain temperature and EEG. b Properties of our brain cooling device. After turning on the switch, the black bar (shown in A) can be immediately cooled to around 12 °C. c Appearance of the mice post-operation

TRPV4 contributes to hippocampal activity through its activation by brain temperature. a Representative EEG traces of WT or TRPV4KO hippocampal DGs under freely moving conditions. Local DG temperature was also measured, and was artificially lowered using the implanted cooling devices (indicated by the blue line). Green insets represent high magnifications of the basal DG EEG. Red insets represent high magnifications of the cooled DG EEG. b Representative EEG power spectrum of Figure A. The blue trace represents WT basal activity. The red trace represents TRPV4KO basal activity. The light green trace represents WT activity under cooling. The purple trace represents TRPV4KO activity under cooling. c: Quantification of EEG θ wave power spectra. d: Reduction of theta power under cooling conditions was calculated using an operation (basal – cooling), and the ratio was also quantified compared with WT results. The asterisks represent statistical significance (p < 0.05) compared with the WT group
Interaction between epilepsy and brain temperature
Our brain cooling device clearly demonstrated that inactivation of TRPV4 by cooling treatment reduced neuronal activity (Fig. 4), indicating that brain activity was significantly affected by brain temperature. We therefore sought to identify if elevation of brain temperature was associated with known diseases. We focused on epilepsy given that it is evoked by abnormal hyper-neuronal activities such as epileptic discharges. We generated epileptic mice utilizing a hippocampal kindling model as previously described [28]. After generating the mice (confirmed by epileptic discharge EEG patterns, Fig. 5a), we measured localized hippocampal temperature and quantified the temperature average (Fig. 5b). Interestingly, the hippocampal temperature of the epileptic mice (Fig. 5b, epilepsy; 37.9 ± 0.1 °C) was significantly elevated by approximately 1 °C over that of control mice (Fig. 5b, control; 36.9 ± 0.1 °C). These results suggested that temperature elevation at epileptic foci might accelerate epileptic discharges due to neuronal hyperactivity. To confirm this, we prepared acute hippocampal slices from the epileptic mice, then measured epileptic discharges using whole cell patch clamp configurations at two different temperatures: 37 °C (normal brain temperature) or 38 °C (epileptic brain temperature). Compared with the 37 °C condition (Fig. 5c), 38 °C (Fig. 5d) significantly increased epileptic firing (Fig. 5e). Thus, for the first time, we demonstrate that brain temperature is significantly altered by epilepsy, and this change accelerates epileptic discharges.

Brain temperature elevation at epileptogenic foci accelerates epileptic events. a We generated epileptic mice utilizing a hippocampal kindling method. After generating the mice, we confirmed the generations of model by epileptic discharge EEG patterns. b Quantification of hippocampal temperature in sham-operated (control) or kindling epileptic model mice (n = 6). The asterisks represent statistical significance (p < 0.01) compared with the control group. c, d Neuronal firing was recorded from acute hippocampal slices (n = 8) at the surgical sites at two different temperatures (C; 37 °C or D; 38 °C). e Quantification of firing frequency at two different temperatures (n = 8). The asterisks represent statistical significance (p < 0.01) compared with the 37 °C condition
TRPV4 is a molecular target for a cure of epilepsy
The observations described above led us to hypothesize that the brain cooling treatment (Figs. 4 and 5), which we used to inhibit TRPV4 activity in vivo, at epileptogenic foci could efficiently suppress epileptic discharges. To examine this possibility, WT or TRPV4KO epileptic mice, whose epilepsy was evoked by local injection of kainic acid (0.1 μl of a 3 mg/ml kainite solution), had brain-cooling devices (Fig. 3a) surgically implanted into their DGs, then the kainite-induced epileptogenic foci were cooled (Fig. 6). We first compared the epileptic discharges between WT and TRPV4KO mice at normal brain temperature. The number of epileptic events and amplitude of epileptic EEG was significantly reduced in TRPV4KO mice compared with WT mice (Fig. 6a, and physiological conditions in Fig. 6b, c). These results indicate that constitutive TRPV4 activation by brain temperature, which results in the enhancement of neuronal excitability, significantly affects epileptic events. To further confirm this, we inactivated TRPV4 utilizing our cooling device. We cooled the temperature of the epileptogenic foci to around 30 °C (Fig. 6a). The artificial cooling significantly reduced epileptic discharges in both WT and TRPV4KO mice. Notably, the reduction rates (cooling effects) in WT mice were much larger than those observed in TRPV4KO mice (Fig. 6), indicating that the constitutive activation of TRPV4 by brain temperature significantly exacerbated the epilepsy. These results indicate that TRPV4 activation by brain temperature is significantly correlated to epileptic progression, and TRPV4-independent mechanisms are also involved in this progression. These observations led us consider if TRPV4 inactivation by injection of a TRPV4-specific inhibitor instead of brain cooling might also suppress epileptic events. We therefore injected 0.1 μl of a 1 mM solution of HC067047 (a TRPV4-specific inhibitor) into epileptic foci in the mouse hippocampal kindling model described above. The injection significantly reduced epileptic events and the amplitude of epileptic discharges in WT mice, but not in TRPV4KO mice (Fig. 7). Hence, we demonstrated that inhibition of TRPV4 by brain cooling or a TRPV4 inhibitor at epileptogenic foci is an effective therapeutic strategy.

Brain cooling suppresses epileptic discharges through inhibition of TRPV4. a Representative EEG traces of WT or TRPV4KO epileptic mice, whose epilepsy was evoked by local injection of kainic acid (0.1 μl of a 3 mg/ml kainite solution) into the hippocampus. The brain-cooling devices were also placed into the DGs, then the kainite-induced epileptogenic foci were cooled. Local DG temperature was also measured. b, c Quantification of the number (B) or amplitude (C) of epileptic discharges under basal or cooling conditions in WT or TRPV4KO epileptic mice (n = 6). The asterisk represents statistical significance (p < 0.01) compared with the WT basal condition. The hash mark represents statistical significance (p < 0.01) compared with the WT basal condition

A TRPV4-specific inhibitor suppresses epileptic discharges. a Representative EEG traces of WT or TRPV4KO epileptic mice, whose epilepsy was evoked by kindling treatment into the hippocampus. Local injection of HC067047 (a TRPV4-specific inhibitor) into the epileptogenic foci was performed during EEG recordings. b, c Quantification of the number (B) or amplitude (C) of epileptic discharges under basal or HC067047-injected conditions in WT or TRPV4KO epileptic mice (n = 6). The asterisk represents statistical significance (p < 0.05) compared with the WT basal condition. The hash mark represents statistical significance (p < 0.05) compared with the WT basal condition
Discussion
Although we previously reported physiological temperature activation of TRPV4 and enhanced neuronal excitability through TRPV4 activation in dissociated hippocampal neurons [20], we did not have corresponding in vivo evidence. In this study, we demonstrated that TRPV4 is constitutively active in a temperature-dependent manner and enhances neuronal excitability in vivo (Fig. 4), consistent with our previous in vitro results [20]. Furthermore, we demonstrated that loss of the thermo-sensing function of TRPV4 leads to abnormal hippocampal activity (Figs. 1, 4, 6). These results indicate that brain temperature is an important factor contributing to brain function, and that TRPV4 is a critical determinant of neuronal excitability through brain temperature in mammals. TRPV4KOs displayed abnormal depression-like and social behaviors [30]. Interestingly, the behavioral abnormalities were very similar to schizophrenia patients [30, 31], suggesting that disruption of TRPV4 function might lead to psychiatric symptoms. Indeed, it was reported that whole-genome sequencing of 85 quartet families of autism revealed that frame-shift mutations of TRPV4 were occurred in the autism patients [32]. Furthermore, genome-wide association studies in control and major depressive disorder revealed that TRPV4 mutation is a risk factor for the depression [33]. Much clinical evidence suggests that brain temperature is a critical determinant of cognitive function as we described above. The mechanisms underlying the importance of brain temperature on brain function have not been previously determined. Here, we identified that TRPV4 is involved in this mechanism using precise combinational methods including slice patch clamp recordings, EEG recordings and artificial control of brain temperature (Figs. 1–7). We demonstrated that a TRPV4-independent pathway could still contribute to brain activity in TRPV4KOs (Fig. 4) as we previously reported [20]. However, our results (Figs. 1 and 4) suggest that maintenance of TRPV4 function is required for normal brain function. Thus, our research might contribute to the examination of the molecular mechanisms underlying suppression of brain activity focusing on brain temperature or TRPV4 activity.
According to the heat activation properties of TRPV4 ( > 27–34 °C) in mammals [16, 20], we hypothesize that our observations on the contribution of TRPV4 to neuronal excitability also applies to homeothermic animals in general [34, 35]. Compared with cold-blooded animals, birds and mammals must perform many complicated behaviors such as constructing shelter and nurturing offspring. Brain structure has substantially evolved along several lines from cold-blooded animals to homeothermic animals. In addition to brain structure, our ancestors acquired specific characteristics to maintain body temperature as homeothermic animals. Thus, brains have evolved effective mechanisms to utilize thermal energy for neuronal excitability at any given time. These features might accelerate species-specific behaviors in birds and mammals, and TRPV4 might be crucial in these evolutionary steps as a major contributing factor for electrical excitability.
We demonstrated that TRPV4KO mice exhibit abnormal neuronal excitability (Fig. 1) and hippocampal activity (Fig. 4). What is the molecular mechanism related to these abnormalities? We reported that TRPV4KO hippocampal neurons did not exhibit significant morphological differences compared with WT neurons [20], indicating that neuronal maturation was normal in TRPV4KO hippocampus. In addition to the normal morphology of TRPV4KO neurons, the cells had normal synaptic structures and pre-synaptic activity [20]. Thus, we can exclude the possibility that TRPV4KO neurons display abnormal differentiation, morphology, or synapses to explain the EEG abnormalities. TRPV4 is a well-characterized, non-selective cation channel [5, 6]. The activation of TRPV4 leads to an influx of cations [22] into cells and depolarizes the RMP [20]. The depolarization of RMP by TRPV4 activation accelerates hippocampal firing (Fig. 1) and EEG activity (Fig. 4). These results suggest that brain temperature constitutively activates TRPV4, and enhances brain activity. Here we can raise two possibilities for the constitutive activation of TRPV4. One is that TRPV4 can be directly activated by brain temperature in a dynamic temperature-dependent manner as we observed (Fig. 4). The other is that TRPV4 can be constitutively regulated by both temperature and other endogenous ligands such as arachidonic acids and anandamide [19, 36]. Thermosensitive TRP channels have two different features: activation by temperature and activation by other ligands [37]. We recently reported that TRPV2 in embryos promoted axon outgrowth and was involved in the regulation of intestinal movements as a mechanosensor [38, 39], although it is well known that TRPV2 is also a noxious heat sensor [23, 40]. Furthermore, we reported that TRPV4 in urinary bladder detects increased urinary volume, acting as a mechanosensor [41], while TRPV4 acts as a thermosensor in skin or oesophageal keratinocytes [16, 42, 43] and brain [20]. These results indicate that multiple ligands exist for thermosensitive TRP channels, and some of our results might involve temperature-independent activation of TRPV4. These results indicate that multiple ligands exist for thermosensitive TRP channels. Indeed, we reported that arachidonic acid activates astrocytic TRPV4 in the hippocampus and that excited astrocytes release ATP as a gliotransmitter [22]. However, we consider the contribution of these effects to be very small in our experiments. Notably, heat application caused TRPV4-independent responses in addition to TRPV4-dependent responses (Figs. 1 and 4), while artificial DG cooling also suppressed brain activity in TRPV4KOs (Fig. 4). We previously reported that physiological temperature contributes to electrical excitation by both TRPV4 and TRPV4-independent pathways [20]. Recently, other ion channels were identified as thermosensors in addition to thermo-TRP channels, including TRPV4 [44,45,46]. These channels might be involved in the TRPV4-independent pathways. We reported that the post-synaptic clustering of TRPV4 is strictly regulated depending on the state of neuronal maturation, and the post-synaptic targeting of TRPV4 required specific triggering signals only in matured neurons [47]. During neonatal development, mice cannot maintain constant body temperature without their mother’s care. Usually, the mothers heat their babies under their body until the babies reach a critical age (~10 days old). If the mother leaves in search of food, the body temperature of babies is drastically decreased. Under such situations, the brain thermo-sensor TRPV4 might perturb neuronal excitation, since TRPV4 activation and inactivation sequentially occur in neonatal brain. To prevent the perturbation, immature neurons might not have TRPV4 in post-synaptic sites. Thus, our finding that TRPV4 is a critical determinant of neuronal excitability through brain temperature, can be restricted in adult animals.
Very recently, we established to measure cellular temperature distribution in the single neuron, and reported that acute ischemia lead the hyperthermia (approximately 2 °C) in the hypoxic regions [35]. The ischemia evoked excessive glutamate release, and those caused the hyperthermia [35]. We revealed that the hyperthermia abnormally activates the TRPV4 and exacerbates brain edema. Hence, the TRPV4 antagonist can abolish the brain edema [35]. Based on this finding, it is highly possible that the epileptogenic foci abnormally elevate neuronal activities through TRPV4 activation by local hyperthermia. The elevation of neuronal activities further increases the temperature of epileptogenic foci as a negative feedback loop. In this study, we found that local DG temperature dynamically changed independent from the movements (Fig. 2b, arrows). The temperature changes resemble neuronal bursts (Fig. 2b, arrows). On the other hand, several spike-like changes in local DG temperature correlated with EEG activity (Fig. 2b, arrowheads). These results indicate that neuronal activity partially evokes the hyperthermia in vivo, and other mechanisms, such as increase of blood flow or cell metabolism can also evoke it. We recently reported that body temperature significantly elevated TRPV4 sensitivity to membrane stretch as a result of a synergistic effect of temperature and mechanical stimuli [34]. We revealed that the Müller glial swelling induced TRPV4 activation triggered Ca2+ influx, and evoked MCP-1 release in retinal detachment. The excessive MCP-1 recruits many macrophages near retina, and those cells kill photoreceptors [34]. This is the critical mechanism to exacerbate photoreceptor death in addition to hypoxia in retinal detachment. The dynamic temperature changes in the detached regions might affect the TRPV4 over-activation based on our finding (Figs. 2 and 5).
The suppressive effect of cooling on epileptic discharges in TRPV4KOs was significantly smaller than in WT mice (Fig. 6). For many years, brain cooling was thought to be an effective way of suppressing epileptic events; [26, 27] however, the underlying molecular mechanisms were unknown. Our results indicate that brain cooling inactivates TRPV4 and exerts suppression of epileptic discharge. Indeed, the injection of a TRPV4-specific inhibitor also effectively suppressed discharge (Fig. 7). Thus, for the first time, we reveal that the molecular mechanisms underlying brain cooling treatment at epileptogenic foci effectively suppress epileptic discharge.
References
Schiff SJ, Somjen GG. The effects of temperature on synaptic transmission in hippocampal tissue slices. Brain Res. 1985;345:279–84.
Moser EI, Andersen P. Conserved spatial learning in cooled rats in spite of slowing of dentate field potentials. J Neurosci. 1994;14:4458–66.
Torgersen J, Strand K, Bjelland TW, Klepstad P, Kvale R, Soreide E, et al. Cognitive dysfunction and health-related quality of life after a cardiac arrest and therapeutic hypothermia. Acta Anaesthesiol Scand. 2010;54:721–8.
Fuster JM, Bauer RH. Visual short-term memory deficit from hypothermia of frontal cortex. Brain Res. 1974;81:393–400.
Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertibrate osmoreceptor. Cell. 2000;103:525–35.
Stortmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nature Cell Biol. 2000;2:695–702.
Wissenbach U, Bodding M, Freichel M, Flockerzi V. Trp12, a novel Trp related protein from kidney. FEBS Letter. 2000;485:127–34.
Nilius B, Prenen J, Wissenbach U, Bodding M, Droogmans G. Differential activation of the volume-sensitive cation channel TRP12 (OTRPC4) and volume-regulated anion currents in HEK-293 cells. Pflugers Arch. 2001;443:227–33.
Mutai H, Heller S. Vertebrate and invertibrate TRPV-like mechanoreceptor. Cell Calcium. 2003;33:471–8.
Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichiling DB, et al. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497–511.
Nillius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiolol Cell Physiol. 2004;286:C195–C205.
Delany NS, Hurle M, Facer P, Alnadaf T, Plumpton C, Kinghorn I, et al. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol Genomics. 2001;4:165–74.
Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. JBiol Chem. 2003;278:22664–8.
Mizuno A, Matsumoto N, Imai M, Suzuki M. Impaired osmotic sensation in mice lacking TRPV4. Am J Physiolol Cell Physiol. 2003;285:C96–C101.
Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA. 2003;100:13698–703.
Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–14.
Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002a;277:13569–77.
Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nillius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002b;277:47044–51.
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nillius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–8.
Shibasaki K, Suzuki M, Mizuno A, Tominaga M. Effects of body temperature on neural activity in the hippocampus: regulation of resting membrane potentials by transient receptor potential vanilloid 4. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:1566–75.
Konno M, Shirakawa H, Iida S, Sakimoto S, Matsutani I, Miyake T, et al. Stimulation of transient receptor potential vanilloid 4 channel suppresses abnormal activation of microglia induced by lipopolysaccharide. Glia. 2012;60:761–70.
Shibasaki K, Ikenaka K, Tamalu F, Tominaga M, Ishizaki Y. A novel subtype of astrocytes expressing TRPV4 (transient receptor potential vanilloid 4) regulates neuronal excitability via release of gliotransmitters. The Journal of biological chemistry. 2014;289:14470–80.
Shibasaki K, Ishizaki Y, Mandadi S. Astrocytes express functional TRPV2 ion channels. Biochem Biophys Res Commun. 2013;441:327–32.
Seder DB, Van der Kloot TE. Methods of cooling: practical aspects of therapeutic temperature management. Crit Care Med. 2009;37:S211–22.
Oku T, Fujii M, Tanaka N, Imoto H, Uchiyama J, Oka F, et al. The influence of focal brain cooling on neurophysiopathology: validation for clinical application. J Neurosurg. 2009;110:1209–17.
Baldwin M, Frost LL. Effect of hypothermia on epileptiform activity in the primate temporal lobe. Science. 1956;124:931–2.
Karlov VA. Focal cooling suppresses continued activity of epileptic focus in patients with partial status epilepticus. Epilepsia. 2003;44:1605.
Stover KR, Lim S, Zhou TL, Stafford PM, Chow J, Li H, et al. Susceptibility to hippocampal kindling seizures is increased in aging C57 black mice. IBRO Rep. 2017;3:33–44.
Tanaka N, Fujii M, Imoto H, Uchiyama J, Nakano K, Nomura S, et al. Effective suppression of hippocampal seizures in rats by direct hippocampal cooling with a Peltier chip. J Neurosurg. 2008;108:791–7.
Shibasaki K, Sugio S, Takao K, Yamanaka A, Miyakawa T, Tominaga M, et al. TRPV4 activation at the physiological temperature is a critical determinant of neuronal excitability and behavior. Pflugers Archiv: European journal of physiology. 2015;467:2495–507.
Melle I, Barrett EA. Insight and suicidal behavior in first-episode schizophrenia. Expert Rev Neurother. 2012;12:353–9.
Yuen RKC, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21:97–103.
Wong ML, Arcos-Burgos M, Liu S, Velez JI, Yu C, Baune BT, et al. The PHF21B gene is associated with major depression and modulates the stress response. Mol Psychiatry. 2017;22:1015–25.
Matsumoto H, Sugio S, Seghers F, Krizaj D, Akiyama H, Ishizaki Y, et al. Retinal Detachment-Induced Muller Glial Cell Swelling Activates TRPV4 Ion Channels and Triggers Photoreceptor Death at Body Temperature. The. Journal of neuroscience: the official journal of the Society for Neuroscience. 2018;38:8745–58.
Hoshi Y, Okabe K, Shibasaki K, Funatsu T, Matsuki N, Ikegaya Y, et al. Ischemic Brain Injury Leads to Brain Edema via Hyperthermia-Induced TRPV4 Activation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2018;38:5700–9.
Marinelli S, Marzo VD, Berretta N, Matias I, Maccarrone M, Bernardi G, et al. Presynaptic facilitation of glutamatergic synapses to dopoaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci. 2003;23:3136–44.
Tominaga M, Caterina MJ. Thermosensation and pain. J Neurobiol. 2004;61:3–12.
Shibasaki K, Murayama N, Ono K, Ishizaki Y, Tominaga M. TRPV2 enhances axon outgrowth through its activation by membrane stretch in developing sensory and motor neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:4601–12.
Mihara H, Boudaka A, Shibasaki K, Yamanaka A, Sugiyama T, Tominaga M. Involvement of TRPV2 activation in intestinal movement through nitric oxide production in mice. The. Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:16536–44.
Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature. 1999;398:436–41.
Mochizuki T, Sokabe T, Araki I, Fujishita K, Shibasaki K, Uchida K, et al. The TRPV4 cation channel mediates stretch-evoked Ca2+ influx and ATP release in primary urothelial cell cultures. The Journal of biological chemistry. 2009;284:21257–64.
Mandadi S, Sokabe T, Shibasaki K, Katanosaka K, Mizuno A, Moqrich A, et al. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Archiv: European journal of physiology. 2009;458:1093–102.
Mihara H, Boudaka A, Sugiyama T, Moriyama Y, Tominaga M. Transient receptor potential vanilloid 4 (TRPV4)-dependent calcium influx and ATP release in mouse oesophageal keratinocytes. The Journal of physiology. 2011;589:3471–82.
Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15:1015–21.
Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, et al. TREK-1 is a heat-activated background K(+) channel. The EMBO journal. 2000;19:2483–91.
Lamas JA, Fernandez-Fernandez D. Tandem pore TWIK-related potassium channels and neuroprotection. Neural Regen Res. 2019;14:1293–308.
Shibasaki K, Tominaga M, Ishizaki Y. Hippocampal neuronal maturation triggers post-synaptic clustering of brain temperature-sensor TRPV4. Biochem Biophys Res Commun. 2015;458:168–73.
Acknowledgements
We thank Ms. K. Abe (Gunma Univ.) for technical assistance and our lab members for helpful discussions. This study was supported by Grants-in-Aid for Scientific Research from the Takeda Science Foundation, Sumitomo Foundation, the Brain Science Foundation, Narishige Neuroscience Research Foundation, Salt Science Research Foundation No.14C2, the Ichiro Kanehara Foundation, Takano Life Science Research Foundation, MEXT/JSPS KAKENHI JP15H05934 < Thermal Biology > , JP15H03000, JP18H03124, JP18K19418 (all to K.S.) and JP 18H02522 (to Y.I.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shibasaki, K., Yamada, K., Miwa, H. et al. Temperature elevation in epileptogenic foci exacerbates epileptic discharge through TRPV4 activation. Lab Invest 100, 274–284 (2020). https://doi.org/10.1038/s41374-019-0335-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0335-5
This article is cited by
-
Novel therapeutic targets to halt the progression of Parkinson’s disease: an in-depth review on molecular signalling cascades
3 Biotech (2023)
-
Transient Receptor Potential Vanilloid 4: a Double-Edged Sword in the Central Nervous System
Molecular Neurobiology (2023)
-
Blockage of TRPV4 Downregulates the Nuclear Factor-Kappa B Signaling Pathway to Inhibit Inflammatory Responses and Neuronal Death in Mice with Pilocarpine-Induced Status Epilepticus
Cellular and Molecular Neurobiology (2023)
-
TRPV4 contributes to ER stress and inflammation: implications for Parkinson’s disease
Journal of Neuroinflammation (2022)
-
Inhibition of Transient Receptor Potential Vanilloid 4 (TRPV4) Mitigates Seizures
Neurotherapeutics (2022)
