
Abstract
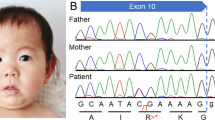
We analyzed our two new cases of infantile-onset epilepsy with developmental delay with de novo variant in TUBB2A and review the related literatures. Our two probands were both girls with infantile-onset epilepsy and global developmental delay. Case 1 had a novel de novo heterozygous missense variant: c.728C>T [p.Pro243Leu] (NM_001069.2). Her brain magnetic resonance imaging (MRI) showed nonspecific white matter myelination delay and slightly enlarged anterior horn of lateral ventricle. Her epilepsy had been controlled by TPM monotherapy. Case 2 had a reported de novo variant c.743C>T [p.Ala248Val] (NM_001069.2). Her brain MRI showed bilateral microgyria and corpus callosum dysplasia. A total of seven TUBB2A mutations cases had been published previously in five papers, therefore, until now, there were nine patients with TUBB2A mutations. All patients had developmental delay, among them seven cases also with infantile-onset epilepsy, one case with abnormal EEG but without clinical seizures. There are six cases that have different degree of cortical dysplasia, one case with cerebellar vermis atrophy and brainstem sacsinopathy, the rest two cases have no obvious brain structural abnormalities. There was one case with variant c.1249G>A (p.D417N) that had atypical clinical presentation, including prominent progressive spastic ataxia, sensory motor axonal neuropathy, and bilateral optic macular dystrophy, but relatively mild intellectual disability, his MRI showed cerebellar atrophy, thinning of the corpus callosum and pons sacsinopathy, but no cortical malformation. The p.A248V mutation was the most common mutation occurred in three patients (3/9). The clinical phenotypes of these three patients were similar, all of them had global developmental delay with no language and corpus callosum dysplasia, two cases with epilepsy and the other one only have EEG epileptic discharges without clinical seizure, two cases with cortical dysplasia and the other one without obvious brain malformation. In brief, global developmental delay was the most common phenotype of TUBB2A mutation-related disease, most cases also had infantile-onset epilepsy and cortical dysplasia and corpus callosum dysplasia. The region between seventh and eighth alpha-helix of TUBB2A may be a “hot spot” mutation domain.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


References
Sferra A, Fattori F, Rizza T, Flex E, Bellacchio E, Bruselles A, et al. Defective kinesin binding of TUBB2A causes progressive spastic ataxia syndrome resembling sacsinopathy. Hum Mol Genet. 2018;27:1892–904.
Romaniello R, Arrigoni F, Fry AE, Bassi MT, Rees MI, Borgatti R, et al. Tubulin genes and malformations of cortical development. Eur J Med Genet. 2018;61:744–54.
Ejaz R, Lionel AC, Blaser S, Walker S, Scherer SW, Babul-Hirji R, et al. De novo pathogenic variant in TUBB2A presenting with arthrogryposis multiplex congenita, brain abnormalities, and severe developmental delay. Am J Med Genet A. 2017;173:2725–30.
Rodan LH, Achkar CME, Berry GT, Poduri A, Prabhu SP, Yang E, et al. De novo TUBB2A variant presenting with anterior temporal pachygyria. J Child Neurol. 2017;32:127–31.
Cushion TD, Paciorkowski AR, Pilz DT, Mullins JGL, Seltzer LE, Marion RW, et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am J Hum Genet. 2014;94:634–41.
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-River F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. J Am Med Assoc. 2014;312:1880–87.
Rodan LH, Achkar CME, Berry GT, Poduri A, Prabhu SP, Yang E, et al. De novo TUBB2A variant presenting with anterior temporal pachygyria. J Child Neurol. 2017;32:127–31.
Ejaz R, Lionel AC, Blaser S, Walker S, Scherer SW, Babul-Hirji R, et al. De novo pathogenic variant in TUBB2A presenting with arthrogryposis multiplex congenita, brain abnormalities, and severe developmental delay. Am J Med Genet A. 2017;173:2725–30.
Leandro-García LJ, Leskelä S, Landa I, Montero-Conde C, López-Jiménez E, Letón R, et al. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton. 2010;67:214–23.
Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WBA. developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–69.
Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873–87.
Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell. 2014;157:1117–29.
Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–42.
Acknowledgements
We sincerely thank the patients’ families for their kind support for this study.
Author information
Authors and Affiliations
Contributions
Study design, analysis and revision of the manuscript, YJ. Follow-up of patient’s information, SC and JL. Draft preparation, SC and JL. Collection of clinical and WES data, SC, JL, YW, and YJ.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cai, S., Li, J., Wu, Y. et al. De novo mutations of TUBB2A cause infantile-onset epilepsy and developmental delay. J Hum Genet 65, 601–608 (2020). https://doi.org/10.1038/s10038-020-0739-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-020-0739-5
