
Abstract
Early-onset developmental and epileptic encephalopathy (DEE) is a group of devastating disorders that appear during the neonatal and infantile periods. Despite great progress in the discovery of genes leading to early-onset DEE, many cases with unexplained etiology remain. Furthermore, to date, the association of copy number variations (CNVs) with early-onset DEE has seldom been addressed. Here, we investigated the contribution of CNVs to epilepsy in a cohort of Japanese children with a variety of early-onset DEEs. Single nucleotide polymorphism (SNP) array analysis was performed for 83 cases that were previously negative for pathogenic single nucleotide variants (SNVs) in 109 genes known or suspected to cause epileptic seizures. Rare CNVs were detected in a total of 12 cases (14.4%), of which three cases (3.6%) involved clearly pathogenic CNVs and nine cases (10.8%) were CNVs of uncertain significance. The three pathogenic CNVs included two de novo heterozygous deletions involving known epileptic encephalopathy genes, such as GABRG2 and PCDH19, and one maternally inherited duplication encompassing MECP2. Our findings indicate rare CNVs are also relevant for the diagnosis of early-onset DEEs, highlighting the importance of not relying only on the investigation of SNVs/small indels at the risk of missing large deletions and duplications.
Similar content being viewed by others


Introduction
According to the International League Against Epilepsy (ILAE), epileptic encephalopathy (EE) is a group of epilepsy syndromes in which the epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone, with potential worsening of these impairments over time [1]. Early-onset developmental and epileptic encephalopathy (DEE) is an EE in which the onset occurs during the neonatal or infantile periods. Common early-onset DEE syndromes include Ohtahara syndrome (Online Mendelian Inheritance in Man [2] number, OMIM# 308350), West syndrome (OMIM# 308350), Dravet syndrome (OMIM# 607208) and Malignant Migrating Partial Seizures in Infancy (MMPSI, OMIM# 614959). In Far-East Asia, a total of 47% of infants with DEEs were classified into West syndrome [3]. Febrile seizures (FS) occur more frequently in Japanese young children, ranging between 3.4 and 9.3% [4], in contrast to the frequency of 2–5% found in Caucasian patients [5].
The refractory epileptic activity itself interferes with brain development and causes severe neurological consequences, especially in the immature brain [6]. Accordingly, earlier ages of disease onset, especially those in the first year of life, are likely to have much poorer outcomes [7]. Therefore, early intervention and control of the seizures are beneficial, particularly for younger children [8]. The genetic component in the etiology of epilepsy has long been recognized, with estimates of it accounting for 32% of all epilepsies, 23% of focal epilepsy and 36% of nonfocal epilepsy [9]. Approximately 20–30% of epilepsy cases are caused by extraneous factors such as head trauma or stroke [10]. However, without genetic analysis, the clinical diagnosis of epilepsy syndromes is challenging due to unspecific and overlapping phenotypes [11]. Recently, a large number of causative epileptic genes have been detected, and approximately 30–50% of infants with EEs have been reported to have causative mutations [12, 13]. Nonetheless, many cases remain unsolved.
Copy number variations (CNVs) are DNA segments that differ in copy number at least 1 kb in size compared to a reference genome [14]. Several studies have documented correlations between CNVs and various neurodevelopmental disorders, such as intellectual disability (ID), schizophrenia and autism, where CNVs were determined to play an important role in their genetic etiology [15]. In epilepsy genetics, the focus has been put on the investigation of monogenic epilepsies caused by single nucleotide variants (SNVs). Consequently, the involvement of CNVs in the pathogenesis of epilepsy, particularly to early-onset DEEs, has seldom been addressed. Targeted resequencing, either by custom or commercial gene panels, has been the predominant approach for the diagnosis of several disorders, including epilepsy. However, a major limitation is that only SNVs and small indels can be identified, while large duplications and deletions are ignored in most cases [16].
In this study, we investigated the contributions of CNVs in a cohort of Japanese children with early-onset DEEs of unknown etiology who were previously investigated for point mutations in known epilepsy genes.
Materials and methods
Subjects
Our cohort consisted of 83 unrelated subjects (26 males and 57 females) of Japanese origin, except for two subjects of British and one of Bengali origin. The subjects were previously examined for the presence of pathogenic SNVs using a sequencing screen with a custom targeted 109-gene panel [17]. This panel included around 70% of well-established causative early-onset DEE genes such as SCN1A and KCNQ2, as well as other genes suspected to cause epileptic seizures. A flowchart showing the strategy of this study is displayed in Fig. 1. The subjects were clinically diagnosed based on clinical features, characteristic electroencephalogram (EEG) patterns or magnetic resonance imaging (MRI). The majority of the patients were clinically classified as Dravet syndrome or symptomatic epilepsy (Table 1).

Flowchart of the strategy of this study and summary of SNP array results. CNV copy number variation, DEE developmental and epileptic encephalopathy, VUS variant of uncertain significance
Genomic DNA was extracted from peripheral blood following standard procedures. Written informed consent was obtained for all patients from their parents or legal guardians to participate in the project. This study was approved by the ethics committees at Tokyo Medical and Dental University (Approval number 2015-2) and Fukuoka University (Approval number 06-51).
Single nucleotide polymorphism (SNP) array analysis
An SNP array was performed on genomic DNA samples from 83 cases using an Illumina HumanOmniExpress 24-v1-1 BeadChip kit (Illumina, San Diego, CA, USA), which contains 713,599 markers and has a mean overall spacing of 4.08 kb. All experiments were carried out following the manufacturer’s instructions. Data extraction and CNV calling was performed with KaryoStudio v1.4 software (Illumina) using the cnvPartition v3.0.7 plug-in algorithm. Genomic coordinates refer to the GRCh37/hg19 human genome assembly. A threshold of >50 kb and seven markers were set for the analysis of duplications, whereas a threshold of >10 kb and five markers were set for deletions. For the analysis of copy-neutral loss of heterozygosity (CNLOH), the cut-off length was set at 3 Mb.
Data interpretation
CNVs were evaluated in accordance to the standards and guidelines proposed by the American College of Medical Genetics (ACMG) [18] for the interpretation of postnatal constitutional CNVs, being ultimately classified as pathogenic, variant of uncertain significance (VUS) or benign. The following criteria were considered substantially indicative of a pathogenic CNV: (1) the presence of genes known to cause epileptic seizures; (2) a phenotype identical to previously reported cases; (3) de novo occurrence; and (4) rare, i.e., not reported or present at a low frequency in public CNV databases, such as the Database of Genomic Variants (DGV) [19], or in our internal MCG CNV database (http://www.cghtmd.jp/CNVDatabase). For the remaining CNVs that did not include a known epilepsy gene, we considered several characteristics regarding their gene content (e.g., gene function, expression patterns, and knockout mouse model phenotype). For genes included in heterozygous losses, pLI scores from the constraint metrics provided by the Exome Aggregation Consortium (ExAC) [20] database were also examined. High pLI scores (≥0.9) indicate that a gene might be extremely intolerant to a loss-of-function mutation, thus suggesting haploinsufficiency.
Confirmation of the results by quantitative real-time PCR (qPCR)
Candidate CNVs were validated by qPCR, which was performed with samples from the patients and their parents. Briefly, qPCR was carried out with the KAPA SYBR FAST qPCR Master Mix (KAPA Biosystems, Wilmington, MA, USA) on a 7500 Real-time PCR System (Applied Biosystems, Carlsbad, CA, USA). As controls for copy number calibration, samples obtained from healthy donors and the qPCR values for GAPDH were used for normalization. All samples were run in triplicate, and the data were analyzed using the comparative cycle threshold method, which assumes that the calibrator DNA has two copies of the control gene [21]. The qPCR primer sequences are available upon request.
Androgen receptor methylation assay
The methylation status of the human androgen receptor (AR) gene at Xq12 was assessed to infer X chromosome inactivation using blood-derived DNA samples from the female patient with a deletion that disrupts PCDH19 and the mother carrier of a MECP2 duplication. The primers and protocol for restriction digestion with the methylation-sensitive HpaII enzyme (New England Biolabs, Ipswich, MA, USA) are described in Kiedrowski et al. [22]. The PCR method was adapted from that developed by Schuelke [23] to fluorescently label PCR fragments. The PCR products were separated by capillary electrophoresis on an ABI 3730xl DNA Analyzer (Applied Biosystems) with the GeneScan 500 LIZ size standard (Applied Biosystems), and fragment analysis was performed with GeneMapper software (Applied Biosystems).
Results
CNV analysis using an SNP array for 83 patients with epileptic disorders detected rare CNVs that might be potentially linked to the phenotype of 12 cases (14.4%): pathogenic CNVs in three cases (3.6%) and VUS in nine cases (10.8%) (Fig. 1, Table 2). The 12 CNVs were further confirmed by qPCR. The analysis of CNLOH did not indicate an excess of homozygous regions; therefore, no case of uniparental disomy (UPD) was detected in our cohort.
Pathogenic CNVs
The CNVs detected in patients 1−3 included genes that are well known to cause epilepsy (Figs. 2–4, Table 2), and the clinical findings of these patients were consistent with the phenotypes generally correlated with aberrations involving the same genes. Therefore, we concluded the rearrangements to be clearly associated with the epileptic phenotypes of these three cases.

SNP array result of Patient 1. a SNP array profile of the 10-Mb deletion detected at 5q33.2-q34. The deleted region is depicted in black. b Mapping of the deletion using the UCSC Genome Browser. Genes within the deletion previously associated with epileptic encephalopathy (CYFIP2, GABRB2, GABRA1 and GABRG2) are highlighted in red. c Cases with copy number variations involving the cluster of GABAA receptor genes at 5q34 in the literature, represented by red bars (color figure online)

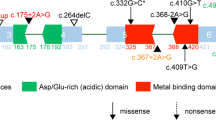
SNP array result of Patient 2. a SNP array profile of the 6.7-Mb deletion detected at Xq21.32-q22.1. The deleted region is depicted in black. b Mapping of the deletion using the UCSC Genome Browser. The deletion disrupts the PCDH19 gene, which is highlighted in red, and includes exons 3–5 of variant 1 (RefSeq NM_001105243). c Cases with copy number variations involving PCDH19 reported in the literature. Red bars represent deletions and the blue bar represents a duplication (color figure online)

SNP array result of Patient 3. a SNP array profile of the 1.1-Mb duplication found at Xq28. The duplicated region is shown in black. b Mapping of the duplication using the UCSC Genome Browser. The gene highlighted in red (MECP2) is thought to be causally linked to the phenotype of Patient 3 (color figure online)
In patient 1, a 10-Mb deletion was detected at 5q33.2-q34, encompassing a total of 39 protein-coding genes annotated in the Reference Sequence [24] (RefSeq) database, of which the following are known to cause epileptic seizures: GABRB2, GABRA1, GABRG2 and CYFIP2 (Fig. 2). The proband is a boy who showed early seizure onset at 1 year and 1 month of age, with febrile seizures (FS) as the initial seizure type. He also presented with generalized tonic-clonic seizures with abnormal EEG findings, slight ventriculomegaly at brain MRI examination and moderate ID. The deletion was found to be de novo.
Patient 2 was found to have a de novo 6.7-Mb deletion at Xq21.32-q22.1 partially involving the PCDH19 gene (Fig. 3), with the breakpoint located within intron 2 of variant 1 (RefSeq NM_001105243). The proband is a girl who had early seizure onset at 1 year and 1 month of age, with an initial seizure type of generalized tonic seizures. The seizures were highly sensitive to fever and occurred in clusters. She also presented with borderline ID. All of the abovementioned features are typically found in patients with PCDH19-related epilepsy (OMIM# 300088). Since PCDH19 is located in the X chromosome, we performed an AR methylation assay to investigate the X chromosome inactivation pattern in the proband, which was highly skewed (87%:13%).
Patient 3 was found to have a 1.1-Mb duplication at Xq28 including the MECP2 gene (Fig. 4). Parental analysis indicated maternal inheritance of the duplication. The proband is a boy whose seizure onset was at 2 years and 4 months of age, with a generalized initial seizure type. Other seizure types included febrile, absence, generalized tonic-clonic and atonic seizures. The patient also presented with severe ID, abnormal EEG findings, hypotonia, impaired speech, loss of ambulation, and recurrent infections, features that are usually encountered in other males with MECP2 duplication syndrome (OMIM# 300260). He also had hypoplasia of the corpus callosum and pons, a typical finding for those who have copy number gains that overlap two low copy repeats (LCRs), LCRK2 and LCRL1 [25]. Since his duplication was maternally inherited, we sought to determine the X inactivation pattern in the reportedly unaffected mother; her X chromosome inactivation was extremely skewed (90%:10%), a pattern often seen in female carriers of X-linked pathogenic mutations.
Variants of uncertain significance
Of the 12 rare CNVs detected in this study, nine were classified as VUS since pathogenicity could not be unequivocally assigned to these variants. This category includes CNVs that overlap with a few previously reported cases that presented with epilepsy, for example, the CNVs involving CNTN6 (patient 5) or TUBB3 (patient 12) in which parental analysis determined they were inherited from an unaffected parent.
Notably, we detected four rearrangements in the 15q11.2 locus of which two were duplications (patients 8 and 9) and two were deletions (patients 10 and 11) (Supplementary Fig. S1). Recurrent microdeletions or microduplications in 15q11.2 are known to increase susceptibility to neuropsychiatric or neurodevelopmental disorders, including seizures [26]. Moreover, previous studies have detected 15q11.2 microdeletions in patients with idiopathic generalized epilepsies and other types of epilepsy [27, 28]. However, early-onset DEEs are unlikely to be caused solely by subtle genetic alterations that confer susceptibility; therefore, whether 15q11.2 rearrangements could in fact be involved in the etiology of early-onset seizures is unclear. Thus, the four CNVs in this study were classified as VUS, even though two of them (found in patients 9 and 11) occurred de novo.
Among the CNVs classified as VUS were two heterozygous deletions involved in genes that have never been associated with epilepsy; yet, based on their functions, we considered that they could be novel candidate genes: LRFN4 (11q13.2) in patient 6 and SYT10 (12p11.1) in patient 7. However, as parental analysis revealed that the two deletions were inherited from unaffected parents, a causal role in epilepsy cannot be attributed to either of these genes yet.
Discussion
We performed an SNP array analysis of 83 patients with early-onset DEE and detected rare CNVs in 12 cases. Among them, three cases with clearly pathogenic CNVs that encompassed known epilepsy genes were found.
The deletion detected in patient 1 included the 5q34 cluster of six genes encoding gamma-aminobutyric acid A (GABAA) receptors of which GABRB2, GABRA1 and GABRG2 have been previously associated with epileptic phenotypes. Mutations in GABRB2 have been reported to cause infantile or early childhood epileptic encephalopathy-2 (IECEE2, OMIM# 617829), characterized by seizures of several types and variable severities associated with global developmental delay and variable ID. Missense or loss-of-function mutations in GABRA1 are the cause of idiopathic generalized epilepsy or early infantile epileptic encephalopathy-19 (EIEE19, OMIM# 615744). Conversely, GABRG2 mutations have mostly been associated with familial FS 8 (FEB8, OMIM# 607681) or early infantile epileptic encephalopathy-74 (EIEE74, OMIM# 618396), in which the seizure phenotypes range from FS or childhood absence seizures to genetic epilepsy with febrile seizures plus (GEFS+) or Dravet syndrome [29]. In addition to the GABAA receptor gene cluster, the deletion in patient 1 also involved CYFIP2, a component of the WASP-family verprolin-homologous protein (WAVE) regulatory complex that is involved in actin dynamics [30], in which missense mutations have been recently reported to cause early infantile epileptic encephalopathy-65 (EIEE65, OMIM# 618008). While most cases with epileptic phenotypes are due to SNVs in these genes, to date, only three patients with deletions at 5q34 involving the GABAA receptor gene cluster have been previously described [27, 31, 32], with the shortest deletion (0.5 Mb) involving only GABRA6, GABRA1 and GABRG2 [27]. Collectively, the aforementioned three cases with 5q34 deletions and our patient 1 showed extremely similar clinical features: FS as the initial seizure type and mild to moderate ID. Nevertheless, due to the large deletion size (10 Mb), determining which of these gene(s) is responsible for the phenotype of patient 1 is difficult.
In patient 2, a deletion disrupting the PCDH19 gene, which encodes a calcium-dependent cell adhesion protein that is primarily expressed in the brain, was found. Over a hundred cases of PCDH19-related epilepsy described in the literature are caused by point mutations, whereas CNVs were reported in only 15 cases [33,34,35,36,37,38]. A correlation between the phenotype and the type of mutation does not appear to exist. Since a highly but not completely skewed X chromosome inactivation pattern (87%:13%) was observed in patient 2, the patient likely presents with mosaic expression of PCDH19-normal and deficient cells, which characterizes the unique inheritance seen in PCDH19-related epilepsy [39].
Finally, the duplication in patient 3 included the MECP2 gene, the cause of Rett syndrome (OMIM# 312750), which predominantly affects females with loss-of-function mutations. Conversely, MECP2 duplication syndrome affects only males, with a penetrance of 100%. Epilepsy is reported in 50% of subjects with MECP2 duplication syndrome [40]. Additionally, most affected patients have severe to profound ID, absence of speech (>70%), infantile hypotonia (75%), loss of ambulation (33%), recurrent infections (~75%) and mild facial dysmorphism [40]. Lugtenberg et al. [41] described a 0.1-Mb microduplication including MECP2 in two brothers with similar phenotypic features to that of patient 3, such as ID, speech impairment, and loss or delay of ambulation. One of the brothers had absence seizures like patient 3.
Among the rare CNVs detected in this study, two involved genes that have never been associated with epilepsy, namely, LRFN4 and SYT10 in patients 6 and 7, respectively. LRFN4 is a member of the synaptic adhesion-like molecule (SALM) family, which regulates synapse formation and is highly expressed in the brain [42]. Its pLI score from the ExAC database is 0.94, a high score suggestive of haploinsufficiency. Moreover, LRFN4 was reported to show significantly high expression in the hippocampus and cerebral cortex of epileptic rat models [43]. Downregulation of LRFN4 was also reported to inhibit status epilepticus [43]. SYT10 is a member of the synaptotagmin family, which plays an essential role in neuronal exocytosis [44]. In rats, Syt10 was shown to be strongly upregulated in the hippocampus of animals with status epilepticus induced by kainic acid [45]. SYT10 was also reported to be required for the protection of hippocampal neurons against excitotoxic cell death [46]. Additionally, another member of the same family, SYT2, was considered to be a novel candidate gene after being involved in a de novo 700-kb duplication found in a patient with epilepsy-aphasia syndrome and mild ID [31]. Since the deletions in patients 6 and 7 were inherited from unaffected parents, we sequenced the coding regions of LRFN4 and SYT10 to detect mutations in the remaining alleles of the respective patients, but no mutations were detected. Nevertheless, due to the possibilities of incomplete penetrance or a second hit in other genes, the pathogenicity of these two CNVs cannot be fully discarded. The discovery of additional EE patients with point mutations or CNVs involving LRFN4 or SYT10 might offer more significant clues about their hypothetical associations with epilepsy.
To date, few studies have examined the contributions of CNVs to early-onset DEEs. Allen et al. [47] reported that pathogenic CNVs accounted for 5.9% of 52 Irish infants with unexplained EEs under the age of 1 year or with unexplained refractory epilepsy with abnormal development. In the study of Ma et al. [48], pathogenic CNVs accounted for 3.4% of 116 Chinese patients with early-onset DEEs who were negative for SCN1A and KCNT1 mutations. Although our study and these previous studies differ in their inclusion criteria for their respective cohorts, the overall diagnostic yields for clearly pathogenic CNVs in the three studies are similar. It should be noted that the sample size might be relatively small to thoroughly evaluate the CNV frequency in early-onset DEEs, both in our study and in previous reports.
Our study showed that rare CNVs might play a causative role in the phenotypes of 14.4% of Japanese patients with early-onset DEEs, of which 3.6% definitely involve pathogenic CNVs. Notably, the genes found to be implicated with the three pathogenic CNVs were in fact included in the 109-gene panel that had been applied prior to the CNV analysis [17]. These findings emphasize the importance of CNVs in the genetic etiology of children with early-onset DEE, and also underline the limitation of the application of targeted next-generation sequencing as the main approach for the genetic diagnosis of epilepsy, at the risk of missing large duplications and deletions. The application of several genetic analysis tools, ranging from the detection of single nucleotide to larger variants, may help to establish accurate and early diagnosis. Consequently, more effective treatment planning may enable better neurological outcomes for these patients with early-onset DEE.
References
Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005−2009. Epilepsia. 2010;51:676–85.
Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), Accessed 23 Jan 2017. http://omim.org/.
Oguni H, Otsuki T, Kobayashi K, Inoue Y, Watanabe E, Sugai K, et al. Clinical analysis of catastrophic epilepsy in infancy and early childhood: results of the Far-East Asia Catastrophic Epilepsy (FACE) study. Brain Dev. 2013;35:786–92.
Sugai K. Current management of febrile seizures in Japan: an overview. Brain Dev. 2010;32:64–70.
Stafstrom CE. The incidence and prevalence of febrile seizures. In: Baram TZ, Shinnar S, editors. Febrile seizures. San Diego: Academic Press; 2002. p. 1−25.
Marsh ED, Brooks-Kayal AR, Porter BE. Seizures and antiepileptic drugs: does exposure alter normal brain development? Epilepsia. 2006;47:1999–2010.
Dulac O. Epileptic encephalopathy. Epilepsia. 2001;42:23–6.
Berg AT, Zelko FA, Levy SR, Testa FM. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes. Neurology. 2012;79:1384–91.
Speed D, O’Brien TJ, Palotie A, Shkura K, Marson AG, Balding DJ, et al. Describing the genetic architecture of epilepsy through heritability analysis. Brain. 2014;137:2680–9.
Hildebrand MS, Dahl HH, Damiano JA, Smith RJ, Scheffer IE, Berkovic SF. Recent advances in the molecular genetics of epilepsy. J Med Genet. 2013;50:271–9.
Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53:1387–98.
McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21.
Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97.
Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196–204.
Chen Y, Zhao L, Wang Y, Cao M, Gelowani V, Xu M, et al. SeqCNV: a novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinf. 2017;18:147.
Ishii A, Kang JQ, Schornak CC, Hernandez CC, Shen W, Watkins JC. et al. A de novo missense mutation of GABRB2 causes early myoclonic encephalopathy. J Med Genet. 2017;54:202–11.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–5.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–92.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8.
Kiedrowski LA, Raca G, Laffin JJ, Nisler BS, Leonhard K, McIntire E, et al. DNA methylation assay for X-chromosome inactivation in female human iPS cells. Stem Cell Rev. 2011;7:969–75.
Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol. 2000;18:233–4.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–45.
Honda S, Hayashi S, Nakane T, Imoto I, Kurosawa K, Mizuno S, et al. The incidence of hypoplasia of the corpus callosum in patients with dup (X)(q28) involving MECP2 is associated with the location of distal breakpoints. Am J Med Genet A. 2012;158A:1292–303.
Burnside RD, Pasion R, Mikhail FM, Carroll AJ, Robin NH, Youngs EL, et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130:517–28.
Olson H, Shen Y, Avallone J, Sheidley BR, Pinsky R, Bergin AM, et al. Copy number variation plays an important role in clinical epilepsy. Ann Neurol. 2014;75:943–58.
de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32.
Kang JQ, Macdonald RL. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 2016;73:1009–16.
Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–9.
Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol. 2011;70:974–85.
Lee JH, Kim HJ, Yoon JM, Cheon EJ, Lim JW, Ko KO, et al. Interstitial deletion of 5q33.3q35.1 in a boy with severe mental retardation. Korean J Pedia. 2016;59:S19–24.
Depienne C, LeGuern E. Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females. Hum Mutat. 2011;32:E1959–75.
Higurashi N, Nakamura M, Sugai M, Ohfu M, Sakauchi M, Sugawara Y, et al. PCDH19-related female-limited epilepsy: further details regarding early clinical features and therapeutic efficacy. Epilepsy Res. 2013;106:191–9.
Vincent AK, Noor A, Janson A, Minassian BA, Ayub M, Vincent JB. et al. Identification of genomic deletions spanning the PCDH19 gene in two unrelated girls with intellectual disability and seizures. Clin Genet. 2012;82:540–5.
Breuillard D, Leunen D, Chemaly N, Auclair L, Pinard JM, Kaminska A, et al. Autism spectrum disorder phenotype and intellectual disability in females with epilepsy and PCDH-19 mutations. Epilepsy Behav. 2016;60:75–80.
Kurian M, Korff CM, Ranza E, Bernasconi A, Lübbig A, Nangia S, et al. Focal cortical malformations in children with early infantile epilepsy and PCDH19 mutations: case report. Dev Med Child Neurol. 2018;60:100–5.
Smith L, Singhal N, El Achkar CM, Truglio G, Rosen Sheidley B, Sullivan J, et al. PCDH19-related epilepsy is associated with a broad neurodevelopmental spectrum. Epilepsia. 2018;59:679–89.
Pederick DT, Richards KL, Piltz SG, Kumar R, Mincheva-Tasheva S, Mandelstam SA, et al. Abnormal cell sorting underlies the unique X-linked inheritance of PCDH19 epilepsy. Neuron. 2018;97:59–66.
Van Esch H. MECP2 duplication syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle: University of Washington; 1993−2019.
Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17:444–53.
Mah W, Ko J, Nam J, Han K, Chung WS, Kim E. Selected SALM (synaptic adhesion-like molecule) family proteins regulate synapse formation. J Neurosci. 2010;30:5559–68.
Li J, Chen L, Wang N, Jiang G, Wu Y, Zhang Y. Effect of synaptic adhesion-like molecule 3 on epileptic seizures: evidence from animal models. Epilepsy Behav. 2017;69:18–23.
Cao P, Maximov A, Südhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–11.
Babity JM, Armstrong JN, Plumier JC, Currie RW, Robertson HA. A novel seizure-induced synaptotagmine gene identified by differential display. Proc Natl Acad Sci USA. 1997;94:2638–41.
Woitecki AM, Müller JA, van Loo KM, Sowade RF, Becker AJ, Schoch S. Identification of synaptotagmin 10 as effector of NPAS4-mediated protection from excitotoxic neurodegeneration. J Neurosci. 2016;36:2561–70.
Allen NM, Conroy J, Shahwan A, Ennis S, Lynch B, Lynch SA, et al. Chromosomal microarray in unexplained severe early onset epilepsy—a single centre cohort. Eur J Paediatr Neurol. 2015;19:390–4.
Ma Y, Chen C, Wang Y, Wu L, He F, Chen C, et al. Analysis copy number variation of Chinese children in early-onset epileptic encephalopathies with unknown cause. Clin Genet. 2016;90:428–36.
Acknowledgements
We thank the patients and their families for their cooperation. This study was supported by Nanken-Kyoten, Tokyo Medical and Dental University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hirabayashi, K., Uehara, D.T., Abe, H. et al. Copy number variation analysis in 83 children with early-onset developmental and epileptic encephalopathy after targeted resequencing of a 109-epilepsy gene panel. J Hum Genet 64, 1097–1106 (2019). https://doi.org/10.1038/s10038-019-0661-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0661-x
This article is cited by
-
IRAK1 Duplication in MECP2 Duplication Syndrome Does Not Increase Canonical NF-κB–Induced Inflammation
Journal of Clinical Immunology (2023)
