
Abstract
Reducing body myopathy is a rare X-linked myopathy characterized by the presence of reducing bodies. The causative gene has been identified as FHL1. We presented with the clinical, muscle magnetic resonance imaging and genetic features of 6 unrelated Chinese patients with reducing body myopathy. We divided the patients into 2 groups according to their age at onset. In addition to limb muscle weakness, pronounced axial muscle involvement was a striking feature common to both groups. Muscle magnetic resonance imaging revealed fatty infiltration predominantly in the postero-medial muscles of the thigh and the soleus muscle of the calf, sparing the gluteus and sartorius muscles. Muscle pathology demonstrated the muscle fibres with reducing bodies distributed in small groups. Genetic analysis revealed FHL1 hemizygote variants in the 6 patients, including 4 novel and 2 reported variants. These variants were located in the LIM2 domain of FHL1 in 4 patients, but 2 located in the LIM4 domain. To the best of our knowledge, this is the first report of reducing body myopathy in the Chinese population. Our findings expand the genetic spectrum of reducing body myopathy.
Similar content being viewed by others


Introduction
Four-and-a-half LIM domain protein 1 (FHL1) is highly expressed in skeletal and cardiac muscle, and plays an important role in protein synthesis, cellular integrity and the intracellular Ras/mitogen-activated protein kinase pathway. Mutations in the FHL1 gene have recently been identified as the causative gene in multiple muscle diseases, including reducing body myopathy (RBM) [1,2,3,4,5,6,7], X-linked myopathy with postural muscle atrophy [8], X-linked dominant scapuloperoneal myopathy [9], distal myopathy with hypertrophic cardiomyopathy [10], Uruguay facio-cardio-musculo-skeletal syndrome with skeletal muscular hypertrophy and cardiomyopathy [11], muscular dystrophy with facial dysmorphology and pulmonary artery hypoplasia [12], isolated hypertrophic cardiomyopathy [13, 14], and Emery-Dreifuss muscular dystrophy with rigid spine [15].
RBM is characterized pathologically by the presence of intracytoplasmic inclusion bodies that reduce menadione nitroblue tetrazolium (NBT) in the absence of the substrate α-glycerophosphate in a menadione-linked α-glycerophosphate dehydrogenase (menadione-NBT) reaction [16]. RBM is a rare disease [1,2,3,4,5,6,7]. RBM showed wide range of clinical phenotypes, including X-linked scapuloperoneal myopathy [17], rigid spine syndrome with hypertrophic cardiomyopathy [18], progressive myopathy with rigid spine and joint contractures [19, 20], distal myopathy with asymmetric involvement [6] and X-linked scapulo-axio-peroneal myopathy [21]. The onset age of RBM is from early childhood to adulthood [4, 6]. Pathological examination is of great value in recognizing RBM and requires a menadione-NBT staining. Muscle magnetic resonance imaging (MRI) has revealed a recognizable pattern of muscle involvement in several patients with RBM, which might be helpful in facilitating diagnosis [22]. However, the muscle MRI findings still need to be verified with additional cases, considering the small sample sizes in those studies [5, 22].
To date, RBM has been reported in various ethnic populations, but not in Chinese. Herein, we present the clinical, pathological, muscle MRI, and genetic characteristics of 6 unrelated Chinese RBM patients.
Materials and methods
Patients
In the present study, six patients from unrelated families were recruited and referred to Peking University First Hospital between 2013 and 2017. These patients were diagnosed with RBM based on myopathological and genetic findings. The following clinical variables of the patients were collected retrospectively: age, sex, initial symptoms, motor function, joint contracture, spine deformities and cardiac involvement. The routine laboratory tests included an electrocardiogram (ECG), electromyography (EMG), and serum creatine kinase (CK) assays.
All clinical materials used in this study were obtained for diagnostic purposes with the informed consent of the patients or their guardians. The study was approved by the Ethics Committee of the Peking University First Hospital.
Muscle biopsy
Muscle biopsies were performed on all 6 patients after written informed consent was obtained. Serial frozen sections (8 μm) were stained with haematoxylin-eosin (H&E), modified Gomori trichrome (mGT) and a battery of histochemical methods. Menadione-NBT staining was also performed to detect reducing bodies (RBs). Immunohistochemical staining was performed with primary antibodies against dystrophin (the N-terminus, C-terminus and rod domain), sarcoglycan (α, β, and γ), desmin and dysferlin. Muscle specimens were also examined using routine ultrastructural techniques.
Genetic analysis
Genomic DNA was extracted from peripheral blood samples or frozen muscle samples. Targeted next-generation sequencing was used to find the mutation associated with the inherited myopathy, and the mutation was verified by Sanger sequencing using specific primers. The mutations were also examined in available family members (patients 3–6) to determine the segregation.
Western blot analysis of muscle
Frozen muscle specimens were examined using standard immunoblotting techniques with a rabbit polyclonal antibody to FHL1 obtained from Proteintech Group (10991-1-AP).
Muscle MRI
Patients 1, 2, 5 and 6 underwent bilateral thigh muscle MRI, and Patients 4 and 5 underwent bilateral calf muscle MRI at 1.5/3.0 T with conventional T1-weighted spin echo, T2-weighted fast spin echo and short time inversion recovery (STIR) sequences. Scans were assessed for muscle bulk (atrophy/hypertrophy) and for signal intensity (fatty infiltration and an oedema-like signal) within different muscles. We scored the MRI signals for the gluteus maximus muscle, the medial, lateral and posterior muscles of the thighs, and the calf muscles. According to the scope and degree of muscle involvement on the T1WI sequence, we divided the muscle fatty infiltration into 6 grades (0–5 points) using the modified Mercuri score [23, 24]. We calculated the average fatty infiltration score of the thigh muscle in 4 patients and the calf muscle in 2 patients to compare the degree of fatty infiltration in different muscle groups. The muscle oedema was divided into six grades (0–5 points) according to muscle involvement on the STIR sequence [25]. Patient 3 underwent cervical MRI.
Statistical analysis
Statistical analysis was carried out using the SPSS Base 18.0 software, and comparisons between groups were assessed using Student’s t test. Statistical significance was set at P < 0.05.
Results
Clinical features
The clinical data of these patients (five male and one female) are summarized in Table 1. According to their onset age, these patients were divided into two groups, the late-onset group (Patients 1–3) and the early-onset group (Patients 4–6).
Patient 1 is a 47-year-old man who had experienced exercise-related myalgia and fatigue in the legs since the age of 26 years. At the age of 41, he noted persistent lower limb muscle weakness leading to a waddling gait. At the age of 42, he suffered from muscle stiffness in the neck, trunk and lower limbs, with difficulty bending the spine. A physical examination at the age of 45 showed a rigid spine and bilateral contracture of the Achilles tendon. The muscle strength (Medical Research Council) was normal in the upper limbs but 4/5 in the proximal and 5/5 in the distal lower limbs bilaterally. Because of his rigid cervical spine, neck extension and neck flexion were difficult. No muscle atrophy or hypertrophy could be observed. Serum CK was 781 IU/L (normal range: 75–195 IU/L). EMG revealed neither myogenic nor neurogenic changes in the examined upper and lower limb muscles. The ECG and echocardiography showed no remarkable findings. His maternal uncle and granduncle had similar symptoms. Unfortunately, the detailed information of the affected family members was unavailable.
Patient 2 is a 32-year-old man who had experienced difficulty climbing stairs since the age of 24. At age 27, he was not able to squat down or climb stairs without assistance. At age 28, he developed proximal upper limb weakness. He was wheelchair-bound at age 30. The physical examination revealed a rigid spine and winging of the scapulae. Neck extension and neck flexion were difficult for him. He presented with symmetrical limb weakness of 3–5/5 in the upper limbs and 0–3/5 in the lower limbs. Atrophy of the thigh muscles and hypertrophy of the calves was observed. Serum CK was 528 IU/L. EMG revealed a myopathic pattern. The ECG revealed sinus bradycardia. Echocardiography revealed decreased systolic function of the left ventricle with a 45% ejection fraction. No family member exhibited similar symptoms.
Patient 3 is a 22-year-old man who had experienced difficulties arising from a bent spine since the age of 11. At 14 years of age, he presented with difficulty running. At the age of 19, he had difficulty climbing stairs, squatting down and raising his upper arms. At the age of 21, he could not walk more than 10 metres. Physical examination revealed a rigid spine, and the movement of his neck was limited. Symmetrical proximal muscle weakness (MRC 3/5) in all four limbs, as well as foot extensor weakness (MRC3/5), was observed. Hypertrophy of the bilateral sternocleidomastoid muscles and atrophy of the bilateral biceps brachii were seen. His serum CK level was between 651 and 1063 IU/L. EMG showed a myopathic pattern. ECG disclosed no abnormality. No one in his family shared similar symptoms.
Patient 4 is an 8-year-old girl. The pregnancy and delivery were uneventful, and her motor development was normal. At the age of 4, she experienced difficulty walking on her toes. The disease rapidly deteriorated her condition, and she lost ambulation at age 5. A physical examination at age 6 showed she had a rigid spine, contracture of the Achilles tendon and the knee joint, and strephexopodia. She showed no dropped head at that time, and her neck flexor was MRC grade 4/5. Muscle weakness was more marked on the left (MRC 2/5) than on the right side (MRC 4/5) in her proximal upper limbs. However, weakness was more symmetrical and severe in the lower limbs, with an MRC of 1–3/5. Her CK level was 819 IU/L. EMG showed a myopathic pattern. ECG showed sinus arrhythmia and ST-segment depression in II and aVF lead. No family member exhibited similar signs.
Patient 5 is a 5-year-old boy. The pregnancy and achievement of motor milestones were normal until age 3, when he began to fall frequently and had difficulty getting up from a squatting position. A physical examination showed a rigid spine and contracture of the Achilles tendon. He had no dropped head, and his neck flexor was MRC grade 2/5. He had mild symmetrical distal weakness of the lower limbs (MRC3/5). Neither muscle atrophy nor hypertrophy was observed. The serum CK level was 520 IU/L. EMG showed a myopathic pattern, and an NCV examination was normal. No family member exhibited similar signs.
Patient 6 is a 4-year-old boy. He was born following a normal pregnancy and delivery. He achieved normal motor milestones until the age of 21 months, when he began to fall down frequently. 2 months later, he had difficulty raising his upper arms, lifting up his head, squatting or maintaining a sitting position without assistance. He was wheelchair-bound at the age of 27 months. Physical examination revealed a floppy child with a dropped head. The muscle strength was 0/5 in the proximal muscles and 3/5 in the distal muscles for all extremities. The serum CK level was between 630 and 1141 IU/L. EMG showed a myopathic pattern. ECG was normal. No one in his family history shared similar symptoms.
None of these patients had signs of impaired intelligence, ptosis, facial weakness, respiratory insufficiency, dyspnoea, or repeated pneumonias.
Muscle pathology
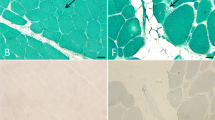
Muscle biopsies showed mild to marked variation in fibre size with endomysium proliferation of the connective tissue. Muscle fibres containing RBs were demonstrated on menadione-NBT staining in all the patients, although with varying degrees. These abnormal fibres showed intracytoplasmic inclusions on H&E and mGT staining (Fig. 1). It is noteworthy that the fibres with RBs were distributed focally in all the patients (Fig. 1). Additionally, necrotic and regenerative fibres appeared in all the patients, and rimmed vacuoles were observed in Patients 3, 4, 5 and 6. Swirled fibres were observed in Patient 5, and RBs were also found in the swirled fibres. Immunohistochemical staining showed the accumulation of desmin and dysferlin in these abnormal fibres.

Muscle pathology of Patient 1 (a–c) and Patient 6 (d–f). Intracytoplasmic inclusions (a, b, d, e) and rimmed vacuoles (a) are seen on H&E (a, d) and mGT staining (b, e). Reducing bodies are positive on melanodine–NBT staining (c, f). The myofibers with intracytoplasmic inclusions or reducing bodies are multifocally distributed
Genetic analysis
All patients were shown to harbour an FHL1 gene variant through targeted next-generation sequencing, followed by confirmation via Sanger sequencing (Table 1). Patient 1 and 4 carried the c.672 C > G (p.C224W) and c.369 C > G (p.123 H > Q) missense mutations of the FHL1 gene, respectively, both have been previously reported [1, 2, 5, 8]. In Patients 2, 3, 5 and 6, the following four novel FHL1 variants were found: c.446_448delACT (p.Y149del), c.448 T > C (p.C150R), c.745 T > C (p.249 C > R), and c.386 G > A(p.C129Y), respectively. All of these variants were located in regions highly conserved among species. Family segregation analysis showed P4 and P6 had de novo mutation, while P3 and P5’ healthy mothers had the same heterozygous mutation as the probands.
Western blot analysis of muscle specimens
An immunoblot analysis showed that the amount of FHL1 protein was significantly reduced in patient muscles compared to normal controls after normalization to tubulin (Fig. 2a, c, P = 0.0042). Patients with early-onset disease seemed to have less FHL1 than did patients with late-onset disease (Fig. 2b).

Immunoblot analysis of FHL1. a Western blot analyses of FHL-1 protein levels in four normal control subjects and five patients. b Quantification of FHL-1 protein levels (relative to tubulin) in each group (n = 3, *P < 0.05). c The amount of FHL1 in biopsied muscle from all five patients was reduced compared to levels in four normal controls after normalization tubulin (**P < 0.01)
Muscle MRI
At the thigh level, Patients 1, 2 and 5 showed similar patterns of muscle involvement (Fig. 3). A predominant fatty infiltration of the postero-medial thigh muscles on T1WI images was the main alteration, while the STIR sequence revealed mild oedema-like signals. The muscles most seriously affected by fatty infiltration were the adductor magnus (average score 2.5), the semimembranosus (average score 2.25), and the long head of the biceps femoris (average score 2.25), followed by the short head of the biceps femoris (average score 2), vastus medialis (average score 1.75), vastus lateralis (average score 1.75) and vastus intermedius (average score 1.5) muscles. The gluteus maximus was selectively spared, with an average score of 0.25. The sartorius muscle was also mildly affected (average score 1) (Supplementay Fig. 1). The thigh muscle MRI of Patient 6 showed severe, uniform oedema on STIR and very mild fatty infiltration on T1W1(Fig. 3). The score of the oedema-like signal was 2 in the rectus femoris, vastus lateralis, vastus intermedius, vastus medialis, sartorius, adductor magnus, adductor longus, semimembranosus, semitendinosus and biceps femoris muscles. However, the gluteus maximus was relatively well preserved on STIR.

Muscle MRI scans of Patient 1 (a, b, h), Patient 2 (c), Patient 3 (i), Patient 4 (f), Patient 5 (g), and Patient 6 (d, e). Thigh muscles MRI showed that predominant fatty infiltration of postero-medial thigh muscles on T1WI (b, c) was the primary change, while the STIR sequence revealed mild oedema-like signals (h). Patient 1 also had hypertrophy of semitendinosus. The gluteus maximus was selectively spared in all four patients (a). Sartorius muscles was also relatively spared (b, c). The thigh muscle MRI scan of Patient 6 showed severe, uniform oedema on the STIR sequence (d), with very mild fatty infiltration on the T1 sequence (e). The T1WI sequence of calf MRI showed that the soleus muscle was the most seriously fatty infiltrated muscle in Patients 4 and 5 (f, g). Cervical MRI of Patient 3 showed severe fatty infiltration of the paraspinal and most cervical muscles were seen, whereas the infrahyoid muscles were relatively well preserved (i)
At the calf level, Patient 4 and Patient 5 (Fig. 3) showed similar changes, although those in Patient 5 were much milder. T1WI revealed that the soleus muscle was the most seriously fatty-infiltrated muscle (average score 3) (Supplementay Fig. 2). The other affected muscles included the extensor digitorum longus (average score 2), medial head of the gastrocnemius (average score 1.5), tibialis anterior (average score 1), tibialis posterior (average score 1), lateral head of the gastrocnemius (average score 1), flexor hallucis longus (average score 1), flexor digitorum longus (average score 1), peroneus longus (average score 1) and brevis (average score 1) muscles. The popliteus (average score 0.5) was relatively spared Supplementay Fig. 2). On the STIR sequence, mild oedema-like signals in the medial and posterior compartment of the calf muscles were seen.
Only Patient 3 had a cervical MRI scan, in which showed severe fatty infiltration of the paraspinal and most cervical muscles (score 3–4), whereas the infrahyoid muscles were relatively well preserved (Fig. 3).
Discussion
In this study, all patients presented with progressive muscle weakness, especially pronounced axial muscle involvement. Muscle biopsies indicated the presence of fibres with RBs. The above clinical findings supported the diagnosis of RBM. Finally, the identification of FHL1 variants in these 6 cases confirmed the diagnosis of FHL1-related RBM. To the best of our knowledge, this is the first report of Chinese patients with FHL1-related RBM.
On the basis of the clinical features of this patient series, all the patients could be classified as having progressive myopathy with a rigid spine and joint contractures. Our series had a disease onset ranging from 2 to 26 years of age, and could be divided into a late-onset group, in which the disease had a juvenile or early-adulthood onset (after 10 years of age), and an early-onset group, with symptoms starting in early childhood (before 10 of age). We found that the early-onset group exhibited a rapid progressive course, which was consistent with other reports [1, 4, 5, 16, 26], while the late-onset group had a relatively benign and slow progression [4]. However, both groups shared one common feature, axial muscle weakness, as indicated by a rigid spine in five patients and a dropped head in the other patient. A rigid spine has also been noted in 4/9 patients in a French study [4] and in some German patients [1]. All of these data indicate that axial muscle involvement could suggest the possibility of RBM. Respiratory failure had been reported in the patients with RBM in previous studies [1, 7], but none of our patients showed symptoms of respiratory involvement at the diagnosis. Obviously, respiratory function evaluation and follow-up of our patients are needed in the future. While most of our patients had predominantly limb girdle muscle weakness, one patient showed only distal muscle weakness in the lower limbs, indicating the heterogeneity of the clinical manifestations of RBM [6]. Although RBM is X-linked, females can also be affected [3]. Among the 38 patients reported by others to date, 26 were female patients [1, 4]. In our patient series, only one patient was female.
The muscle biopsies revealed intracytoplasmic inclusions on H&E and mGT staining. RBs were identified by menadione-NBT staining. The myofibres with RBs were focally distributed, which has also been found in Japanese patients [2], although the precise mechanism responsible for this distribution remains unclear. The numbers of RBs and the severity of the pathologic abnormalities observed in each biopsied muscle did not seem to correlate with either an early or late onset. In addition, it was not uncommon to find rimmed vacuoles, as well as desmin abnormally aggregating in the fibres. All of these findings confirmed that RBM also has features of myofibrillar myopathy [4, 27].
To date, all the mutations detected in the reported RBM patients have been located in the second LIM domain of FHL1. In the present study, four patients (P2, 3, 4 and 6) were found to harbour mutations in the LIM2 domain, including three novel mutations (c.386 G > A, c.446_448del and c.448 T > C) and one reported mutation (c.369 C > G) [1, 2, 5] (Table 1). P4 and P6 had more severe phenotypes than did P2 and P3; this finding was consistent with the notion that mutations at positions H123 and C132 result in a more severe phenotype than those in residues more distal to the LIM2 domain central core [1, 28]. However, Fujii reported a 32-year-old female patient harbouring a mutation at C126 near the central core, who had slowly worsening muscle weakness and remained ambulatory 4 years after onset, suggesting that the clinical course of RBM patients is not defined solely by the localization of the mutation but may involve other factors [5, 6]. It is worth noting that in this study, Patients 1 and 5, who both had the mildest phenotypes in their corresponding groups, were found to have mutations located in the LIM4 domain, a novel (c.745 T > C) and a reported (c.672 C > G) mutation, respectively (Table 1). The c.672 C > G mutation has been reported to cause X-linked myopathy with postural muscle atrophy (XMPMA), which has been categorized as an RB-negative subgroup of FHL1-related myopathy [8, 29]. c.745 T > C was located near a reported mutation, c.736 C > T/p.H246Y, which also has been reported to cause XMPMA [30]. A mutation in the LIM4 domain only decreases the expression of the FHL1A and FHL1B proteins, while the FHL1C protein is retained. Mutations in the LIM2 domain affect all three splice forms of FHL1. This may partially explain the difference in the clinical manifestations. Our findings expand the mutational spectrum of the FHL1 gene.
The western blot analysis revealed reduced levels of FHL1 protein in our patients compared to normal controls. Patients in the early-onset group seemed to have less FHL1 than did patients in the late-onset group, but we found no correlation between clinical severity and amount of FHL1 protein among the members within a group. Western blot analyses of other RBM cases in previous studies showed variable levels of FHL1, ranging from decreased to normal or even increased levels [2, 31]. This variation could be explained by the possibility that the mutant FHL1 protein forms insoluble aggregates, which are therefore not detectable in the immunoblot [5]. Since RBM cases exhibit asymmetric muscle involvement and focal pathologic changes in the same muscle specimen, the decrease or increase in the level of FHL1 may depend upon the degree to which the biopsied portion of the muscle is affected. We should also consider the degree of protein degradation/turnover [19].
Finally, we confirmed that an MRI scan of muscles reveals a recognizable pattern of muscle involvement that might be helpful in diagnosing RBM [22, 32]. However, the fatty infiltration pattern in RBM is associated with not only disease severity but also disease course. Sparing of the gluteal muscles was found in all patients. The predominant involvement of the soleus muscle at the calf level was observed in late-onset patients. The predominant involvement of the adductor magnus and of postero-medial muscles symmetrically at the thigh level appeared in 2 patients, as reported by others [22, 32]. One finding unique to our study was that one patient’s gracilis muscle was more affected than the sartorius muscle. Severe fatty infiltration of the paraspinal and oblique abdominal muscles with the rectus abdominus muscles relatively well preserved has been found in other late-onset RBM patients [5]. Our early-onset patients showed a uniform oedema or oedema with fatty infiltration on thigh MRI scans; this finding is unique from those of other reported RMB patients.
In conclusion, our findings expand the ethnic origin and genetic spectrum of RBM. The patients could be divided into late- and early-onset groups. Novel FHL1 mutations found in this study expand the mutational spectrum associated with RBM.
Data availability
Supplementary information is available at Jounal of human genetics’s website.
References
Schessl J, Taratuto AL, Sewry C, Battini R, Chin SS, Maiti B, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132:452–64.
Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology. 2009;72:375–6.
Schessl J, Columbus A, Hu Y, Zou Y, Voit T, Goebel HH, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics. 2010;41:43–6.
Malfatti E, Olivé M, Taratuto AL, Richard P, Brochier G, Bitoun M, et al. Skeletal muscle biopsy analysis in reducing body myopathy and other FHL1-related disorders. J Neuropathol Exp Neurol. 2013;72:833–45.
Schreckenbach T, Henn W, Kress W, Roos A, Maschke M, Feiden W, et al. Novel FHL1 mutation in a family with reducing body myopathy. Muscle Nerve. 2013;47:127–34.
Fujii T, Hayashi S, Kawamura N, Higuchi MA, Tsugawa J, Ohyagi Y, et al. A case of adult-onset reducing body myopathy presenting a novel clinical feature, asymmetrical involvement of the sternocleidomastoid and trapezius muscles. J Neurol Sci. 2014;343:206–10.
Sabatelli P, Castagnaro S, Tagliavini F, Chrisam M, Sardone F, Demay L, et al. Aggresome-autophagy involvement in a sarcopenic patient with rigid spine syndrome and a p.C150R mutation in FHL1 gene. Front Aging Neurosci. 2014;6:215.
Windpassinger C, Schoser B, Straub V, Noor A, Lohberger B, Farra N, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet. 2008;82:88–9.
Quinzii CM, Vu TH, Min KC, Tanji K, Barral S, Grewal RP, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–13.
D’Arcy C, Kanellakis V, Forbes R, Wilding B, McGrath M, Howell K, et al. X-linked recessive distal myopathy with hypertrophic cardiomyopathy caused by a novel mutation in the FHL1 gene. Child Neurol. 2015;30:1211–7.
Xue Y, Schoser B, Rao AR, Quadrelli R, Vaglio A, Rupp V, et al. Exome sequencing identified a splice site mutation in FHL1 that causes uruguay syndrome, an X-linked disorder with skeletal muscle hypertrophy and premature cardiac death. Circ Cardiovasc Genet. 2016;9:130–5.
Pen AE, Nyegaard M, Fang M, Jiang H, Christensen R, Mølgaard H, et al. A novel single nucleotide splice site mutation in FHL1 confirms an Emery-Dreifuss plus phenotype with pulmonary artery hypoplasia and facial dysmorphology. Eur J Med Genet. 2015;58:222–9.
Hartmannova H, Kubanek M, Sramko M, Piherova L, Noskova L, Hodanova K, et al. Isolated X-linked hypertrophic cardiomyopathy caused by a novel mutation of the four-and-a-half LIM domain 1 gene. Circ Cardiovasc Genet. 2013;6:543–51.
Gossios TD, Lopes LR, Elliott PM. Left ventricular hypertrophy caused by a novel nonsense mutation in FHL1. Eur J Med Genet. 2013;56:251–5.
Gueneau L, Bertrand AT, Jais JP, Salih MA, Stojkovic T, Wehnert M, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–53.
Brooke MH, Neville HE. Reducing body myopathy. Neurology. 1972;22:829–40.
Chen DH, Raskind WH, Parson WW, Sonnen JA, Vu T, Zheng Y, et al. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci. 2010;296:22–9.
Knoblauch H, Geier C, Adams S, Budde B, Rudolph A, Zacharias U, et al. Contractures and hypertrophic eardiomyopathy in a novel FHLl mutation. Ann Neurol. 2010;67:136–40.
Shalaby S, Hayashi YK, Goto K, Ogawa M, Nonaka I, Noguchi S, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1). Neuromuscul Disord. 2008;18:959–61.
Selcen D, Bromberg MB, Chin SS, Engel AG. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology. 2011;77:1951–9.
Feldkirchner S, Walter MC, Muller S, Kubny C, Krause S, Kress W, et al. Proteomic characterization of aggregate components in an intrafamilial variable FHL1-associated myopathy. Neuromuscul Disord. 2013;23:418–26.
Astrea G, Schessl J, Clement E, Tosetti M, Mercuri E, Rutherford M, et al. Muscle MRI in FHL1-linked reducing body myopathy. Neuromuscul Disord. 2009;19:689–91.
Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F, et al. Muscle MRI in inherited neuromuscular disorders:past, present and future. J Magn Reson Imaging. 2007;25:433–40.
Nali M, Areehavala-Gomcza V, Cirak S, Glover A, Guglieri M, Feng L, et al. Muscle histology VS MRI in Duchenne muscular dystrophy. Neurology. 2011;76:346–53.
Stramare R, Beltrame V, Dal Borgo R, Gallimberti L, Frigo AC, Pegoraro E, et al. MRI in the assessment of muscular pathology: a comparison between limb-girdle muscular dystrophies, hyaline body myopathies and myotonic dystrophies. Radio Med. 2010;115:585–99.
Waddell LB, Tran J, Zheng XF, Bönnemann CG, Hu Y, Evesson FJ, et al. A study of FHL1, BAG3, MATR3, PTRF and TCAP in Australian muscular dystrophy patients. Neuromuscul Disord. 2011;21:776–8.
Selcen D. Myofibrillar myopathies. Curr Opin Neurol. 2008;21:585–9.
Cowling BS, Cottle DL, Wilding BR, D’Arcy CE, Mitchell CA, McGrath MJ. Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromusclar Disord. 2011;21:237–51.
Bertrand AT, Bonnemann CG, Bonne G. FHL1 myopathy consortium. 199th ENMC international workshop: FHL1 related myopathies, June 7-9, 2013, Naarden, The Netherlands. Neuromuscul Disord: NMD. 2014;24:453–62.
Schoser B, Goebel HH, Janisch I, Quasthoff S, Rother J, Bergmann M, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73:543–51.
Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–12.
Komagamine T, Kawai M, Kokubun N, Miyatake S, Ogata K, Hayashi YK, et al. Selective muscle involvement in a family affected by a second LIM domain mutation offhl1: an imaging study using computed tomography. J Neurol Sci. 2012;318:163–7.
Acknowledgements
We extend our sincere appreciation to the patients and their parents for their participation and enthusiastic support. We thank Ichizo Nishino (National Institute of Neuroscience, NCNP) for reviewing and commenting on the manuscript.
Funding
This work was financially supported by grants from the Ministry of Science and Technology of China (No. 2011ZX09307-001-07), the National Natural Science Foundation of China (No. 81541118) and the Beijing Municipal Science and Technology Commission (No. Z151100003915126).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hu, Z., Zhu, Y., Liu, X. et al. FHL1-related clinical, muscle MRI and genetic features in six Chinese patients with reducing body myopathy. J Hum Genet 64, 919–926 (2019). https://doi.org/10.1038/s10038-019-0627-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0627-z
This article is cited by
-
FHL1 promotes chikungunya and o’nyong-nyong virus infection and pathogenesis with implications for alphavirus vaccine design
Nature Communications (2023)
-
Identification of novel FHL1 mutations associated with X-linked scapuloperoneal myopathy in unrelated Chinese patients
Journal of Human Genetics (2023)
-
Clinical, pathological, and molecular genetic analysis of 7 Chinese patients with hereditary myopathy with early respiratory failure
Neurological Sciences (2022)
-
A Dominant C150Y Mutation in FHL1 Induces Structural Alterations in LIM2 Domain Causing Protein Aggregation In Human and Drosophila Indirect Flight Muscles
Journal of Molecular Neuroscience (2021)
