
Abstract
Celiac disease (CD) is caused by loss of tolerance toward gluten and related cereal products. The delivery of gliadin peptides (GP) to HLA-DR-positive late endosomes (LE) of enterocytes is required for antigen presentation and tolerance generation. We hypothesized that anti-gliadin antibodies in CD serum modify gliadin transport into LE within enterocytes. CD and control duodenal biopsies were incubated with digests of gluten as well as with serum of CD patients. Lissamin-labeled GP AA31–43 and AA56–68 were endocytozed by Caco-2 cells with serum of CD- or control patients. Colocalization of gliadin with the LE marker LAMP-2 and cathepsin D was determined and quantified on immunofluorescence and immunoelectron microscopical level. Up to 13% of internalized gliadin was located in LE of CD biopsies incubated with CD serum compared with less than 4% in CD biopsies without CD serum as well as in control biopsies. In Caco-2 cells, the colocalization coefficient of GP AA31–43 and LE was 0.82 with CD serum, 0.42 with control serum, and 0.48 with culture medium. Incubation with CD serum can direct GP AA31–43 into LE of enterocytes which is required for antigen presentation.
Similar content being viewed by others

Main
Celiac disease (CD) is a multiorgan disease with autoimmune characteristics affecting approximately 1% of the population. It is caused by the lack of tolerance induction toward gluten and related cereal products in HLA-DQ2- and DQ8-positive individuals. Glutamine residues of these cereal storage proteins being gliadin for wheat are deamidated to glutamic acid by tissue-transglutaminase type 2, the autoantigen of CD. This increases its binding capacity to HLA-DQ2/8 (1). “Toxic” gliadin peptides (GP) have shown to be presented to mucosal T cells by HLA-DQ2/8 on dendritic cells of the lamina propria (2,3).
GP AA31–49 and AA57–73 were found to exacerbate CD in vivo and in vitro (4). GPs are transported through enterocytes to the lamina propria and are found in plasma (5,6). The toxic GP AA31–43 provokes both adaptive and innate immune responses with upregulation of various mediators, including IL-15, cyclooxygenase-2, CD83, and CD25 (7). MHC class I chain-related gene A plays a key role in the damage mediated by intraepithelial T lymphocytes involving the natural killer cell receptor NKG2D (8). Unlike GP AA31–43/49, the immunodominant GP AA57–68 has the ability to stimulate gluten-sensitive T cells (7,9).
GPs are crossing the intestinal barrier through transcytosis of the epithelial layer (10,11). While GP AA57–68 reaches late endosomes (LE), GP AA31–49 bypasses LE within enterocytes (12). Most of endocytosed proteins are transported from EE to LE and to lysosomes (13). The localization of exogeneous antigens in LE is a prerequisite for MHC II-mediated antigen presentation to CD4+ T lymphocytes (14). Therefore, GP AA31–49 escapes antigen presentation and is unable to induce T lymphocytes. We have recently shown that a conjugation of GP AA31–49 to cholera toxin B can direct this peptide to LE (12). Routing food antigens to LE of enterocytes seems to be essential for the generation of oral tolerance (15).
As polyvalent ligands have also been found to be processed in LE (16), we addressed the question whether gliadin-specific antibodies from CD serum could direct the toxic GP AA31–43 into HLA DR-positive LE which is important for antigen presentation (14).
PATIENTS AND METHODS
Patients.
Duodenal biopsy specimens from 14 patients were examined. Eight patients had a recently diagnosed CD. None of them had been on a gluten-free diet. The CD patients were 9.24 ± 2.08 y old, two were males, six females. The control group consisted of six patients. Two of them suffered from Crohn's disease (both 13.7 y old, one male, one female) and four had diseases without bowel affection (16.95 ± 8.61 y old, one male, three females).
After the biopsies were extracted endoscopically, two specimens of each patient were incubated with Frazer's Fraction III (FF) (12) with or without 20% of CD serum for 60 min. FFIII was diluted 1:20 and pooled serum was obtained from six patients with an IgA- and IgG-anti-gliadin antibody titer of 121 ± 63 and 108 ± 80 U/mL, respectively.
The study was approved by the ethics committee of the Justus-Liebig-University Giessen (reference number 157/09), and written informed consent was obtained from all participants.
Antibodies.
The primary antibodies and their characteristics are described elsewhere (12). For electron microscopic experiments, gold-conjugated goat serum (Jackson Immunoresearch, Suffolk, Great Britain) was used in dilutions of 1:2.5 (6 nm gold particles) and 1:5 (12 nm gold particles) for anti-mouse antibodies and 1:40 (6 nm gold particles) and 1:50 (12 nm gold particles) for anti-rabbit antibodies. For fluorescence microscopy, Alexa-488-conjugated donkey anti-mouse (dilution 1:200) and donkey anti-goat antibodies (dilution 1:100) (Molecular Probes, Darmstadt, Germany) as well as EEA1 (dilution 1:100) (Santa Cruz, Heidelberg, Germany) were used. The samples were incubated with secondary antibodies for 45 min in the dark at room temperature. To characterize the labeling specificity of the gliadin antibodies (R5, WB6/8 and α-Gliadin) on electron microscopical level, we analyzed at least 12 randomly chosen electron micrographs from four different specimen with mitochondria and nuclei as negatively and early endosomes (EE) as positively labeled organelles.
Immunoelectron microscopy of duodenal biopsies.
Duodenal biopsies were sectioned and labeled using the technique of Tokuyasu as described previously (15). Microscopic evaluation was done with magnifications between 13.300- and 33.900-fold. For quantitative analysis of subcellular labeling distribution, 100 gold particles (12 nm) were counted by a meander-shaped pass through each grid. Their ultrastructural localizations were listed. LE were characterized by positive staining with cathepsin D (dilution 1:40; Dako, Hamburg, Germany) and LAMP-2 antibodies (dilution 1:5; BD Pharmingen, NJ, USA). Vacuoles without staining by these antibodies were taken as EE.
Immunofluorescence microscopy of Caco-2 cells.
Cultivation of cells, incubation of peptides and antibodies, and microscopical evaluation have recently been described in detail (17). Cells were incubated with GP at 37°C for 20 min, washed, and fixed immediately or after 180 min.
Statistical analysis.
Statistical analyses were performed by using SPSS 12.0 (IBM, Chicago, IL, USA). The calculation of significance levels was done using the Mann-Whitney U-test. p values less than 0.05 were defined as significant.
RESULTS
Visualization of gliadin localization sites within enterocytes on ultrastructural level.
Duodenal biopsies were incubated with gliadin, and thin frozen sections were stained with antibodies against gliadin and LE markers. Two monoclonal (R5, WB6/8) and one polyclonal antibody (α-gliadin) against gliadin were used to determine the subcellular distribution of gliadin within enterocytes; 1% of gliadin-related gold particles were counted in mitochondria, 5% in nuclei, and 17% in the cytosol (Table 1). These compartments are considered to be unspecific for gliadin labeling considering the large volume of nuclei and cytosol. Thus, labeling densities as the number of gold particles per μm2 representing a determination for antibody specificity were low for R5 on mitochondria and nuclei, compared with EE as a positive control with 0.8, 0.1, and 39.1, respectively. Incubation experiments, omitting the primary antibody, yielded in no significant labeling.
Detection of endocytosed gliadin in LE of enterocytes from CD biopsies after incubation with CD serum.
Our experiments showed that the gliadin content of LE could be increased significantly, if FFIII incubation of CD biopsies was done in the presence of CD serum. Without CD serum, the endocytosed gliadin only reached EE but not LE in CD biopsies (Fig. 1A). However, when CD serum was added to the incubation media of CD biopsies, an increased colocalization of gliadin within LE was observed (Fig. 1B-D). The use of gliadin with different antibodies (R5 and WB 6/8 as monoclonal and p-α-Gliadin as polyclonal antibodies) revealed comparable results in all eight CD patients. This result was similar to the effect of cholera toxin B conjugation with GP AA 31–49 observed ex vivo and in vitro (12). In contrast to LE, the amount of endocytosed gliadin within EE could not be enhanced by CD serum. About 50% of intracellular gliadin was found in EE of enterocytes from patients with CD, either with or without incubation with CD serum (Table 1).

Gliadin localization in enterocytes of CD patients. (A) Without CD serum: gliadin within EE (cathepsin D negative) but not in LE. (B-D) With CD serum: gliadin present in LE. Gliadin (R5, WB6/8, α-gliadin: large gold, arrows). LE (cathepsin D, LAMP-2: small gold, arrowheads). Bars, 0.1 μm.
No enrichment of gliadin within LE within control enterocytes by CD serum.
To examine the specificity of the enhanced endocytosis of gliadin into LE in CD serum-treated biopsies of CD patients, we compared the amount of gliadin within LE of enterocytes, originating from duodenal biopsies of healthy controls and CD patients. Four healthy control biopsies showed a low uptake of gliadin in LE with and without CD serum (Fig. 2A, B) which is comparable to CD biopsies without treatment with CD serum and biopsies of two patients with Crohn's disease. The use of three different antibody combinations, R5 with cathepsin D, WB 6/8 with cathepsin D, and α-Gliadin with LAMP-2 yielded in similar results. The labeling amount of gliadin within EE was independent of the treatment with or without CD serum (data not shown). These data support previous results, indicating a lack of gliadin transport into LE in control enterocytes (10–12).

Lack of gliadin within LE with (A) or without (B) CD serum in control biopsies. Gliadin (R5, WB6/8: large gold). LE (cathepsin D+, LAMP-2+: small gold, arrowheads). BL, basolateral membrane, Mi, mitochondrium. Bars, 0.1 μm.
Gliadin ratios within LE of CD and control biopsies treated with and without CD serum.
The quantitative analysis of the distribution of intracellular gliadin label was performed through meander-shaped counting of 100 gold particles in the epithelial layer through each grid in relation to their ultrastructural localizations (Fig. 3). LE within enterocytes of duodenal biopsies, which were obtained from eight CD patients, contained 13% of intracellular gliadin when incubated with CD compared with 3% in CD biopsies without CD serum incubation. Low amounts of gliadin in LE were also found in four healthy control biopsies with (3%) and without (4%) CD serum as well as in biopsies from two patients with Crohn's disease, again, independently of the incubation with (4%) or without CD serum (3%) (values are median). The amount of gliadin, reaching LE of CD enterocytes mediated with CD serum, was four times higher than the amount of gliadin in LE found in control biopsies with or without CD serum and CD biopsies without CD serum. Although the anti-gliadin antibodies used have different binding properties, the values of the WB 6/8 and polyclonal-α-gliadin antibodies confirmed the result obtained by using the R5 antibody (data not shown). In specimens of the two patients with Crohn's disease, 4% of intracellular gliadin was found in LE by using the R5 and p-α-Gliadin antibodies, but only 2% when detected with WB 6/8 antibody (data not shown). As WB 6/8, but not R5 is able to recognize deamidated GP (12), WB 6/8 staining indicated that the majority of LE gliadin has not been deamidated.

Quantitation of gliadin (R5) in LE (cathepsin D) within enterocytes on electron micrsocopical level. CD biopsies incubated with CD serum had a significant increase of gliadin in LE compared with CD biopsies incubated without CD serum as well as control biopsies with and without CD serum. *p ≤ 0.05.
Redirection of GP AA31–43 to LE in Caco-2 cells through CD serum.
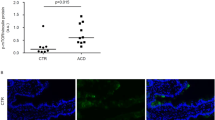
The synthetic peptide GP AA31–43 was incubated in Caco-2 cells in the presence of CD serum and control serum. Figure 4 demonstrates representative examples of the immunfluorescence evaluation with control serum (Fig. 4A-C) and with CD serum (Fig. 4D-F). Figure 4A and D shows the isolated signal of the Lissamin-rhodamine-labeled GP AA31–43, which is localized in vacuoles around the nonstained nuclei. Figure 4B and E visualizes the labeled LAMP-2, a marker for LE. The merged images seen in Figure 4C and F revealed possible overlapping of both colors. After incubation of Caco-2 cells with control serum, no colocalizations of both color signals could be observed, as indicated by the absence of yellow color in Figure 4C. This suggests a lack of GP AA31–43 transport into LAMP-2-positive LE. In contrast to control serum, CD serum markedly enhanced GP AA31–43 transport into LE (Fig. 4F).

Redirection of gliadin peptide AA31–43 into LE of Caco-2 cells. No colocalization with control serum (A-C) in contrast to CD serum (D-F). Endocytosed gliadin peptide AA31–43 is marked with red and LAMP-2 with green fluorochroms. Note the numerous colocalization sites in F (yellow dots). Bars, 100 μm.
Quantitative evaluation of GP AA31–43 transport into LE of Caco-2 cells comparing the incubation with CD serum, control serum, and culture medium.
Incubation with control serum did not change the amount of endocytosed GP AA31–43 in comparison to cell culture medium (DMEM) alone neither in EE (0.81 and 0.75, respectively) nor in LE (0.46 and 0.48, respectively) (Fig. 5). The colocalization coefficients with GP AA31–43 and the EE marker EEA1 were significantly higher after treatment with CD serum (0.93), indicating its increased uptake into EE. The colocalization coefficients of this peptide with the LE marker LAMP-2 were low after incubation with medium alone (0.48) as well as with control serum (0.46), but could be doubled after incubation with CD serum (0.81). Thus, the absence of GP AA31–43 in LE could be enhanced by using CD serum (Fig. 5). This treatment delivered toxic GP into LE and may influence its presence in EE through recycling. The transport into LE seemed to be completed within 20 min, as longer incubation times for up to 180 min did not significantly change colocalization coefficients (data not shown). The immunodominant GP AA57–68 showed high colocalization fractions in EE (0.83 with control and 0.92 with CD serum, p ≥ 0.05) and LE (0.67 with control and 0.82 with CD serum, p ≥ 0.05), independently of incubation with CD serum and control serum. Thus, it reaches LE without any treatment, supporting previous data (12); its translocation into LE observed with CD serum tended to be enhanced.

GP AA31–43 uptake into EE is not affected by control serum (Co-S) and culture medium (DMEM). CD serum (CD-S) significantly increases translocation into EE and almost twice as much into LE in contrast to Co-S and DMEM. *p ≤ 0.05.
DISCUSSION
Gliadin is partly digested by gastric and pancreatic proteases and peptidases. GP can be endocytosed by enterocytes and partially processed in endosomes until it reaches the lamina propria and the circulation (18). The toxic GP AA31–49 is poorly degraded in CD enterocytes; the immunodominant GP AA57–68, however, is totally degraded during epithelial transport (18). Both peptides segregate along the endosomal pathway and GP AA31–49 bypasses LE (12). This cellular compartment is thought to play a key role in HLA class II antigen-mediated CD4+ T-cell stimulation induced by exogenous antigen presentation of intestinal epithelial cells (14,19,20). HLA class II expression and CD4+ T cells are required to generate oral tolerance (21). A prerequisite for enterocytes to induce tolerogenic effects is the presence of HLA class II antigens in LE, which belong to the HLA-DR type (22). HLA-DQ antigens are more strongly expressed on dendritic cells in the lamina propria required for the toxic reaction (3). In experiments with mice, it was shown that BALB/c mice, which generated oral tolerance toward ovalbumin, took up this food antigen very efficiently into LE of enterocytes in contrast to SCID mice that were unable to initiate such a tolerance reaction (15,23). Gliadin processing and presentation through enterocytes seems to be crucial for the understanding of CD pathogenesis, as CD is caused by a tolerance failure toward gliadin. In our study, we observed the toxic GP AA31–43 only in EE, but not in LE of enterocytes, while the immunogenic GP AA57–68 accumulated in this compartment within 20 min. Interestingly, Luciani et al. (24) described GP AA31–43 localized in LE of T-84 and Caco-2 cells without any treatment. These findings might result from long incubation times and the biotinylation of this peptide applied in their study.
To generate oral tolerance toward gliadin in the context of primary prevention toward CD, our study was designed to test the potential of guiding GP into LE so that they can be presented by HLA-DR antigens in the basolateral membrane. Different ways to modulate the intracellular transport, for example by adding a signal sequence, have been reported (20). Cholera toxin B subunit acts as an efficient transporter, using the ganglioside GM-1 receptor for internalization and tolerance induction (25). A conjugate of the cholera toxin B subunit and GP AA31–49 is transported effectively into LE (12). Our experiments demonstrated that there is an additional mechanism to redirect GP AA31–43 to LE of enterocytes similar to GP AA31–49 conjugated by cholera toxin B. Mellman et al. (26) found that small amounts of monovalent antibodies entered mouse macrophages, reached EE, and are subsequently recycled to the cell membranes. With polyvalent immune complexes, both ligand and receptor are degraded after being delivered to lysosomes (16). The “neonatal” Fc receptor (FcRn) mediates the postnatal transfer of IgG from colostrum and milk (27); FcRn is also expressed in adult intestinal epithelial cells and Caco-2 cells (28). Furthermore, the polymeric Ig receptor (pIgR) is involved in the transepithelial IgA transport. Although pIgR mainly functions in basolateral to apical transcytosis, apically uncleaved pIgR can take up pIgA from the luminal site (29). Unspecific secretory IgA is able to enhance probiotic adhesion on Caco-2 cells. On this background, we examined the hypothesis that sera from CD patients containing specific anti-gliadin IgG and IgA might be able to redirect gliadin and more specifically GP AA31–43 into LE of enterocytes (Fig. 6). In fact, we observed ex vivo and in vitro that CD serum yielded the anticipated effect which neither could be induced by cell culture medium nor by serum from healthy control patients. This is most likely due to high titer of gliadin specific IgA and IgG in the serum from CD patients. Isolation of gliadin antibodies from CD sera would be required to finally prove this hypothesis. Moreover, our experiments showed that translocation of GP AA57–68 into LE, which was much stronger than for GP AA31–43 under native conditions (which is in line with Matysiak-Budnik et al. (18)), was also slightly enhanced by CD serum.

Polyvalent effect of anti-gliadin Igs on intracellular processing of toxic GP. (A) Transcytosis of GP without CD serum through EE to the paracellular space bypassing LE. (B) The pathway of GP in the presence of CD serum, GP enters HLA-DR-positive LE and can be presented on the basolateral surface.
Enterocytes are able to suppress the proliferation of CD4+ and CD8+ lymphocytes by expressing the immunosuppressive cytokines IL-10 and TGF-β, which can also induce T2-helper cells and regulatory T cells for generation and maintenance of oral tolerance (30,31). Two mechanisms are important for the development of immune tolerance: (1) deletion and anergy of CD4+ T cells and (2) induction of regulatory CD4+-T cells. We now speculate that toxic GP, such as GP AA31–43, are delivered to LE of the infant's intestinal epithelium mediated by gliadin specific IgA and IgG. Eventually, these GP can be presented on the cellular surface, resulting in the induction of Foxp3+ Treg lymphocytes (32).
Breastfeeding may reduce the risk of developing infections, allergies, and CD; the underlying mechanisms, however, are still unclear (33). Protection is suggested to be mainly deriving from IgA antibodies, lactoferrin, and oligosaccharides, which are present in maternal milk. Timing for the introduction of gluten in the infants diet can reduce the risk for CD significantly, especially when breastfeeding is continued during that time and only small amounts of gluten are given (34,35). Because no valid follow-up studies have been done so far, the question still remains whether CD is simply delayed or whether the overall risk is reduced. Any ingredient of breast milk could be involved in this immune mechanism. Besides other antigens, human milk contains gliadin derived from the mother's diet and specific IgA antibodies against gliadin. The anti-gliadin IgA concentration was found to be the highest in colostrum compared with milk and serum (36,37).
In conclusion, toxic GP such as GP AA31–43 does not reach LE of enterocytes. This might be one reason for the failure in generating a physiological tolerance toward gliadin and its related cereal-storage proteins. Our experiments demonstrated that CD serum was able to transfer gliadin and specifically GP AA31–43 ex vivo and in vitro into LE of enterocytes, which is essential for their presentation to regulatory CD4+-T cells at the basolateral membrane. This translocation process is enhanced by gliadin antibodies from CD serum. It remains to be investigated whether this mechanism would probably explain the protective effect of breastfeeding in individuals with a genetic predisposition to CD.
Abbreviations
- CD:
-
celiac disease
- EE:
-
early endosomes
- FF:
-
Frazer's Fraction III
- GP:
-
gliadin peptide
- LE:
-
late endosomes
- pIgR:
-
polymeric Ig receptor
References
Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Noren O, Roepstorff P, Lundin KE, Sjostrom H, Sollid LM 1998 Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 4: 713–717
Koning F 2005 Celiac disease: caught between a rock and a hard place. Gastroenterology 129: 1294–1301
Sollid LM 2002 Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2: 647–655
Sturgess R, Day P, Ellis HJ, Lundin KE, Gjertsen HA, Kontakou M, Ciclitira PJ 1994 Wheat peptide challenge in coeliac disease. Lancet 343: 758–761
Bethune MT, Ribka E, Khosla C, Sestak K 2008 Transepithelial transport and enzymatic detoxification of gluten in gluten-sensitive rhesus macaques. PLoS One 3: e1857
Mazumdar K, Alvarez X, Borda JT, Dufour J, Martin E, Bethune MT, Khosla C, Sestak K 2010 Visualization of transepithelial passage of the immunogenic 33-residue peptide from alpha-2 gliadin in gluten-sensitive macaques. PLoS ONE 5: e10228
Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, Picard J, Osman M, Quaratino S, Londei M 2003 Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362: 30–37
Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, Verkarre V, Fodil N, Bahram S, Cerf-Bensussan N, Caillat-Zucman S 2004 A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 21: 367–377
Camarca A, Anderson RP, Mamone G, Fierro O, Facchiano A, Costantini S, Zanzi D, Sidney J, Auricchio S, Sette A, Troncone R, Gianfrani C 2009 Intestinal T cell responses to gluten peptides are largely heterogeneous: implications for a peptide-based therapy in celiac disease. J Immunol 182: 4158–4166
Zimmer KP, Naim H, Weber P, Ellis HJ, Ciclitira PJ 1998 Targeting of gliadin peptides, CD8, alpha/beta-TCR, and gamma/delta-TCR to Golgi complexes and vacuoles within celiac disease enterocytes. FASEB J 12: 1349–1357
Zimmer KP, Poremba C, Weber P, Ciclitira PJ, Harms E 1995 Translocation of gliadin into HLA-DR antigen containing lysosomes in coeliac disease enterocytes. Gut 36: 703–709
Zimmer KP, Fischer I, Mothes T, Weissen-Plenz G, Schmitz M, Wieser H, Buning J, Lerch MM, Ciclitira PC, Weber P, Naim HY 2010 Endocytotic segregation of gliadin peptide 31–49 in enterocytes. Gut 59: 300–310
Fader CM, Colombo MI 2009 Autophagy and multivesicular bodies: two closely related partners. Cell Death Differ 16: 70–78
Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ 1991 Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature 349: 669–676
Zimmer KP, Buning J, Weber P, Kaiserlian D, Strobel S 2000 Modulation of antigen trafficking to MHC class II-positive late endosomes of enterocytes. Gastroenterology 118: 128–137
Mellman I, Plutner H 1984 Internalization and degradation of macrophage Fc receptors bound to polyvalent immune complexes. J Cell Biol 98: 1170–1177
Barone MV, Nanayakkara M, Paolella G, Maglio M, Vitale V, Troiano R, Ribecco MT, Lania G, Zanzi D, Santagata S, Auricchio R, Troncone R, Auricchio S 2010 Gliadin Peptide p31–43 localises to endocytic vesicles and interferes with their maturation. PLoS ONE 5: e12246
Matysiak-Budnik T, Candalh C, Dugave C, Namane A, Cellier C, Cerf-Bensussan N, Heyman M 2003 Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 125: 696–707
Hershberg RM, Mayer LF 2000 Antigen processing and presentation by intestinal epithelial cells - polarity and complexity. Immunol Today 21: 123–128
Rowell JF, Ruff AL, Guarnieri FG, Staveley-O'Carroll K, Lin X, Tang J, August JT, Siliciano RF 1995 Lysosome-associated membrane protein-1-mediated targeting of the HIV-1 envelope protein to an endosomal/lysosomal compartment enhances its presentation to MHC class II-restricted T cells. J Immunol 155: 1818–1828
Desvignes C, Bour H, Nicolas JF, Kaiserlian D 1996 Lack of oral tolerance but oral priming for contact sensitivity to dinitrofluorobenzene in major histocompatibility complex class II-deficient mice and in CD4+ T cell-depleted mice. Eur J Immunol 26: 1756–1761
Dubois B, Goubier A, Joubert G, Kaiserlian D 2005 Oral tolerance and regulation of mucosal immunity. Cell Mol Life Sci 62: 1322–1332
Furrie E, Turner MW, Strobel S 1994 Failure of SCID mice to generate an oral tolerogen after a feed of ovalbumin: a role for a functioning gut-associated lymphoid system. Immunology 83: 562–567
Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello Mantovani M, Guido S, Cexus ON, Peake N, Londei M, Quaratino S, Maiuri L 2010 Lysosomal accumulation of gliadin p31–43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 59: 311–319
Sun JB, Xiao BG, Lindblad M, Li BL, Link H, Czerkinsky C, Holmgren J 2000 Oral administration of cholera toxin B subunit conjugated to myelin basic protein protects against experimental autoimmune encephalomyelitis by inducing transforming growth factor-beta-secreting cells and suppressing chemokine expression. Int Immunol 12: 1449–1457
Mellman I, Plutner H, Ukkonen P 1984 Internalization and rapid recycling of macrophage Fc receptors tagged with monovalent antireceptor antibody: possible role of a prelysosomal compartment. J Cell Biol 98: 1163–1169
Baker K, Qiao SW, Kuo T, Kobayashi K, Yoshida M, Lencer WI, Blumberg RS 2009 Immune and non-immune functions of the (not so) neonatal Fc receptor, FcRn. Semin Immunopathol 31: 223–236
Sato K, Nagai J, Mitsui N, Ryoko Y, Takano M 2009 Effects of endocytosis inhibitors on internalization of human IgG by Caco-2 human intestinal epithelial cells. Life Sci 85: 800–807
Breitfeld PP, Casanova JE, McKinnon WC, Mostov KE 1990 Deletions in the cytoplasmic domain of the polymeric immunoglobulin receptor differentially affect endocytotic rate and postendocytotic traffic. J Biol Chem 265: 13750–13757
Cruickshank SM, McVay LD, Baumgart DC, Felsburg PJ, Carding SR 2004 Colonic epithelial cell mediated suppression of CD4 T cell activation. Gut 53: 678–684
Galliaerde V, Desvignes C, Peyron E, Kaiserlian D 1995 Oral tolerance to haptens: intestinal epithelial cells from 2,4-dinitrochlorobenzene-fed mice inhibit hapten-specific T cell activation in vitro. Eur J Immunol 25: 1385–1390
Sun JB, Czerkinsky C, Holmgren J 2010 Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand J Immunol 71: 1–11
Akobeng AK, Ramanan AV, Buchan I, Heller RF 2006 Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies. Arch Dis Child 91: 39–43
Norris JM, Barriga K, Hoffenberg EJ, Taki I, Miao D, Haas JE, Emery LM, Sokol RJ, Erlich HA, Eisenbarth GS, Rewers M 2005 Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 293: 2343–2351
Ivarsson A, Hernell O, Stenlund H, Persson LA 2002 Breast-feeding protects against celiac disease. Am J Clin Nutr 75: 914–921
Ozkan T, Ozeke T, Meral A 2000 Gliadin-specific IgA antibodies in breast milk. J Int Med Res 28: 234–240
Rumbo M, Chirdo FG, Anon MC, Fossati CA 1998 Detection and characterization of antibodies specific to food antigens (gliadin, ovalbumin and beta-lactoglobulin) in human serum, saliva, colostrum and milk. Clin Exp Immunol 112: 453–458
Acknowledgements
We would like to thank Mrs B Goritzka, Mrs L Thiessen, Mr M Zoremba, and Mrs N Metz for excellent technical assistance, Mrs I Panzer for support in performing the experiments, Prof. P Ciclitira for providing the WB6/8 antibody, and Dr. R-H Bödeker for reviewing the study design.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lübbing, N., Barone, MV., Rudloff, S. et al. Correction of Gliadin Transport Within Enterocytes Through Celiac Disease Serum. Pediatr Res 70, 357–362 (2011). https://doi.org/10.1203/PDR.0b013e31822a31e7
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31822a31e7
This article is cited by
-
Endocytosis and transcytosis of gliadin peptides
Molecular and Cellular Pediatrics (2016)
-
Endocytosis in enterocytes
Wiener Medizinische Wochenschrift (2016)
-
Effect of anti-gliadin IgY antibody on epithelial intestinal integrity and inflammatory response induced by gliadin
BMC Immunology (2015)
