
Abstract
Epigenetics is defined as mitotically and meiotically heritable changes in gene expression that do not involve a change in the DNA sequence. Two major areas of epigenetics—DNA methylation and histone modifications—are known to have profound effects on controlling gene expression. DNA methylation is involved in normal cellular control of expression, and aberrant hypermethylation can lead to silencing of tumor-suppressor genes in carcinogenesis. Histone modifications control the accessibility of the chromatin and transcriptional activities inside a cell. MicroRNAs (miRNAs) are small RNA molecules, ∼22 nucleotides long that can negatively control their target gene expression posttranscriptionally. There are currently more than 460 human miRNAs known, and the total number is predicted to be much larger. Recently, the expression of miRNAs has been definitively linked to cancer development, and miRNA profiles can be used to classify human cancers. miRNAs are encoded in our genome and are generally transcribed by RNA polymerase II. Despite the growing evidence for their importance in normal physiology, little is known about the regulation of miRNA expression. In this review, we will examine the relationship between miRNAs and epigenetics. We examine the effects of miRNAs on epigenetic machinery, and the control of miRNA expression by epigenetic mechanisms. Epigenetics is defined as heritable changes in gene expression that do not involve a change in DNA sequence.
Similar content being viewed by others

Main
Epigenetics is defined as heritable changes in gene expression without a change in the DNA sequence itself (1). DNA cytosine methylation and histone modifications are two important mechanisms in the area of epigenetics that have profound roles in gene regulation, development, and carcinogenesis (2–5).
DNA methylation is a normal process used by mammalian cells in maintaining a normal expression pattern; it is involved in the regulation of imprinted gene expression and X-chromosome inactivation, among others (6–8). DNA methylation occurs almost exclusively on a cytosine in a CpG dinucleotide, and is achieved by the addition of a methyl group to the 5 position of a cytosine ring mediated by DNMTs (Fig. 1). CpG sites are roughly 80% depleted in the genome, and are asymmetrically distributed into CpG poor regions and dense regions called CpG “islands” (9,10), which are often located in the promoter regions of roughly half of all the protein-coding genes. The majority of the genome is rather CpG-poor due to the mutagenicity of a methylated cytosine; a methylated cytosine can undergo spontaneous deamination to become a guanine (Fig. 2) (1, 11). CpG islands normally remain unmethylated, whereas the sporadic CpG sites in the rest of the genome are normally methylated. There is a gradual reversal of this pattern during aging that leads to sporadic methylation in the CpG islands and a global loss of methylation, but this change is particularly pronounced during carcinogenesis (Fig. 3) (1,12). Methylation of CpG islands in promoter regions is often associated with gene silencing, and aberrant DNA methylation occurs in most cancers, leading to the silencing of some tumor suppressor genes (Fig. 2) (2,12). There are three major enzymes involved in establishing and maintaining DNA methylation patterns: DNMT 3A and 3B are de novo methyltransferases, and DNMT1 is the maintenance DNMT that ensures that methylation patterns are copied faithfully throughout each cell division (3). They cooperate with each other to establish and maintain the cellular DNA methylation patterns (Fig. 4) (1).

Mechanism of DNA methylation. DNA methylation involves the addition of a methyl group onto the 5 position of a cytosine residue, mediated by the enzymes DNMTs. DNA methylation happens almost exclusively on cytosines in front of a guanine in a CpG dinucleotide.

CpG sites in the genome are highly unevenly distributed. CpG islands can be found in the promoter regions of roughly half of the genes and normally remain unmethylated. When they become aberrantly hypermethylated, as can happen in many cancers, they lead to the silencing of downstream genes.

CpG islands normally remain unmethylated, whereas the sporadic CpG sites located in the rest of the genome often are methylated. With aging, there is a gradual reversal of this phenomenon. During carcinogenesis, this change is much more dramatic, leading to a global hypomethylation and hypermethylation of CpG islands. The results are chromosomal instability and silencing of some important tumor-suppressor genes.

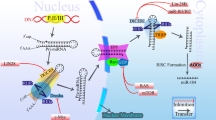
Biogenesis of miRNAs. miRNAs are endogenously encoded in the genome. They are generally transcribed by RNA polymerase II (PolII) into the primary miRNAs (pri-miRNAs), which therefore have 5′ caps and poly-A tails. The pri-miRNAs are processed by RNase III Drosha with its partner DGCR8 (DiGeorge syndrome chromosomal region 8; also known as Pasha in invertebrates) into the precursor miRNAs (pre-miRNAs), which are then exported by the nuclear export factor Exportin 5 and its cofactor RAN-GTP into the cytoplasm. It is in the cytoplasm that the pre-miRNAs are further processed by another RNase III Dicer into the mature miRNAs. The mature miRNA is incorporated into the RISC complex and negatively regulates its target mRNA by one of two ways. When it binds its target with complete complementarity, it leads to the target mRNA cleavage. On the other hand, it can also bind to its target with incomplete complementarity, and leads to translational repression of the latter by a yet poorly understood mechanism.
Histone modifications, especially the posttranslational modifications of amino-terminal tail domains, are also important epigenetic mechanisms in controlling gene expression (1). Certain histone modifications, such as histone acetylation, are associated with active gene transcription, whereas others such as the methylation of histone H3 lysine 9 (H3K9) is an indicator of condensed and inactive chromatin (1,13,14).
The relationships between DNA methylation and histone modifications have recently become clearer, although much is still to be learned. It is now believed the two mechanisms cooperate in controlling gene expression. For example, methylation of histone H3 lysine 9 can be triggered by DNA methylation (15–17). DNA methyltransferases have also been shown to interact with histone deacetylases (HDAC), histone methyltransferases, and methyl-cytosine-binding proteins in a complex network (18–20).
The association between abnormal epigenetic changes such as DNA hypermethylation and human diseases, including cancer, has become increasingly clear (1). In contrast to genetic changes that cause cancer, epigenetic modifications of gene expression are more general and usually involve more than one gene. Many drugs have been found to have effects in reversing the abnormal epigenetic changes that occur in carcinogenesis, and they can be divided into two classes—DNA methylation inhibitors and HDAC inhibitors (1,21). These agents hold great promises in improving the treatment of cancer.
miRNAs ARE ENDOGENOUS SMALL RNA MOLECULES THAT CAN CONTROL GENE EXPRESSION
miRNAs are ∼22 nucleotides-long RNA molecules encoded in the genome that can have a profound effect in controlling gene expression. They are transcribed by RNA polymerase II (Pol II) into primary miRNAs (22,23), and are then processed in the nucleus by the RNase III Drosha and DGCR8 (microprocessor complex) into the precursor miRNAs. Precursor miRNAs are structured as imperfect stem-loops, and they are exported into the cytoplasm by Exportin-5. The precursor miRNAs are further processed in the cytoplasm by another RNase III Dicer into the final functional mature miRNAs (24–27).
miRNAs bind to their target mRNAs and down-regulate their stabilities and/or translation. When binding to its target mRNA with complete complementarity, the miRNA can lead to degradation of the target. MiRNAs can also bind to their targets with incomplete complementarity, often in the 3′ UTR regions, and this leads to the translational suppression of their target genes by a mechanism that has yet to be completely elucidated (24–26). Each miRNA is predicted to have many targets, and each mRNA may be regulated by more than one miRNA (28–30). Currently, there are more than 460 human miRNAs known (31).
miRNAs have recently been shown to be definitely linked to cancer, and they can act as either oncogenes (Table 1) or tumor-suppressor genes (Table 2) in carcinogenesis. For example, miR-15a and miR-16-1 can target the anti-apoptotic BCL2, and they are often down-regulated in chronic lymphocytic leukemia (32,33). miR-21 is found to be antiapoptotic, and it is up-regulated in glioblastomas and breast cancers (34–37). Lu et al. (38) showed that the expression profiles of miRNAs are able to classify human cancers.
miRNA CONTROL OF EPIGENETIC MECHANISMS
Small interfering RNAs (siRNAs), often considered to be closely related to miRNAs, have been shown to be involved in both DNA methylation and histone modifications. The processing pathways of siRNAs and miRNAs share many of the enzymes involved in the RNA interference (RNAi) pathway (39). Recent evidence also suggest that they affect histone modifications. Maison et al. (40) showed that RNase treatment can abolish the localization of methylated H3 lysine 9 and HP1 to pericentromeric chromatin. Fukagawa et al. (41) showed that Dicer-related RNAi machinery is necessary for the formation of heterochromatin structure. Because siRNAs and miRNAs are closely related, miRNAs could also play important roles in controlling DNA methylation and histone modifications.
miRNAs can be involved in establishing DNA methylation. miR-165 and miR-166 have been shown to be required for the methylation at the PHABULOSA (PHB) gene in Arabidopsis. They interact with the newly processed PHB mRNA to change the chromatin of the template PHB gene (42). This presents an exciting new mechanism by which miRNAs can control gene expression in addition to the RNAi pathway. Similar findings in mammalian cells have yet to be shown. In addition, key DNA methylation enzymes DNMT1, 3a, and 3b all are predicted to be potential targets of miRNAs (28), although it remains to be experimentally determined whether the DNMTs can indeed be regulated by miRNAs.
In addition, miRNAs may regulate chromatin structure by regulating key histone modifiers. miR-140, which is cartilage-specific, can target histone deacetylase 4 in mice (43). Costa et al. (44) suggested that miRNAs may be involved in meiotic silencing of unsynapsed chromatin in mice. Taken together, miRNAs can be considered important players in the epigenetic control of gene expression.
EPIGENETIC CONTROL OF miRNA EXPRESSION
Since their initial discovery, miRNAs had been assumed to be transcribed by RNA polymerase III (Pol III) due to their small sizes (45), yet the biogenesis of miRNAs has only been elucidated in recent years. Lee et al. (27) showed that miRNAs are transcribed from long primary transcripts in 2002, and two years later miRNAs were proven to be generally transcribed by Pol II (22,23). We are now only beginning to understand how miRNA expression is regulated. Because miRNAs are generally transcribed by Pol II, they can be spatially and temporally regulated (25). In addition to negatively regulating their target mRNAs, miRNAs themselves can be regulated by other factors. c-myc has been shown to activate transcription of the miR-17-92 cluster, which has a role in tumor neovascularization (46,47). NF-kB can induce the expression of miR-146a, which can then down-regulate IRAK1 and TRAF6 and thus acts as a component in a negative feedback loop that controls TLR signaling. Fazi et al. (48) showed that the transcription factors NFI-A and C/EBPalpha compete for binding to the miR-223 promoter, leading to low and up-regulated expression of miR-223, respectively. In addition, miR-223 participates in its own feedback loop and favors the C/EBPalpha binding by repressing the NFI-A translation (48). Despite mounting evidence for the importance of miRNAs, the regulation of their expression is still poorly understood.
An exciting new discovery by Saito et al. (49) showed that epigenetic mechanisms, such as DNA methylation and histone modifications, can affect the expressions of miRNAs. In particular, miR-127 was found to be remarkably up-regulated in cancer cell lines after the treatment with 5-Aza-CdR, a potent DNA methylation inhibitor, and 4-phenylbutyric acid (PBA), a histone deacetylase inhibitor (1). Together, 5-Aza-CdR and PBA lead to reduced DNA methylation levels and more open chromatin structures, and therefore induce the re-expression of genes that had been silenced epigenetically (Fig. 5) (1). The finding that miR-127, among many other miRNAs, can be expressed after treatment with 5-Aza-CdR and PBA, suggests that epigenetic mechanisms can control the expression of miRNAs. Scott et al. (50) also showed that treatment of breast cancer cell line SKBr3 with HDAC inhibitor LAQ824 led to a rapid change in miRNA expression profile.

The expression of miRNAs can be controlled by epigenetic mechanisms. Epigenetic mechanisms such as DNA methylation and histone modifications can contribute to the transcriptional control of miRNA expression. In the case of miR-127, methylation of the CpG sites and deacetylation of the histones around its promoter region contribute to its silencing in tumor cell lines. Treatment with 5′-Aza-CdR and PBA leads to reduced DNA methylation and increased histone acetylation, allowing the miRNA to be expressed. The gray circle depicts a nucleosome with histone tails. Open circles on the DNA strand represent unmethylated CpG sites, and the filled circles methylated CpG sites. Octagons on the histone tails represent acetyl groups.
Many miRNAs are located in the introns of protein-coding genes (25). It is believed that such miRNAs are co-regulated with their host genes (51). However, it is possible that these miRNAs can have their own promoters. The finding that CpG islands within introns can act as promoters suggests that perhaps intronic miRNAs that have CpG islands upstream within the same intron could be transcribed from their own promoters that are regulated by DNA methylation (52,53).
Much still needs to be learned before a definite role for epigenetic mechanisms in controlling the expression of miRNAs can be established. Knowing that epigenetics can control the expression of many protein-coding genes, and that miRNAs are also generally transcribed by Pol II, it is reasonable to hypothesize that epigenetics can play fundamental roles in controlling the miRNA expression.
CLINICAL SIGNIFICANCE AND FUTURE DIRECTIONS
Epigenetics and miRNAs are two important subjects of study that warrant significant growth in their fields in the future, and the relationship between epigenetics and miRNA is just beginning to be understood (Fig. 6). Some miRNAs have been found to play important roles in carcinogenesis. These miRNAs can serve as therapeutic targets in future cancer therapies. For example, knockdown of the oncogenic miRNA miR-21 can trigger apoptosis in cultured glioblastoma cells (36). Other examples of oncogenic miRNAs exist. MiR-372 and 373 are up-regulated in testicular germ cell tumors (54). MiR-155 is overexpressed in B-cell lymphomas and breast cancers (34,55,56). These, and other oncogenic miRNAs, can all serve as important targets in cancer therapy; knocking down of these miRNAs may stunt the cancer growth. On the other hand, restoring tumor-suppressor miRNAs can also be a powerful approach in treating cancer. The finding that epigenetic drugs 5-Aza-CdR and PBA are able to lead to the up-regulation of miR-127, which can down-regulate BCL6, is especially exciting (49). It demonstrates that epigenetics drugs may exert their antitumor effects on two fronts: they not only turn on the tumor-suppressor genes that were aberrantly silenced epigenetically, but they also turn on tumor-suppressor miRNAs that down-regulate target oncogenic mRNAs. 5-Azacytidine and 5-Aza-CdR, both DNA methylation inhibitors, have been approved by the FDA to treat myelodysplasia (21). Knowing that these drugs can affect the expression of miRNAs helps us further understand the mechanisms of action of these agents. More studies are needed in these areas to further illuminate the therapeutic potential of epigenetic modifiers and miRNAs.

The interplay of epigenetics and miRNAs. Recent evidence has shone light on the relationship between miRNAs and epigenetics. miRNAs can affect the epigenetic mechanisms by targeting key enzymes involved in establishing epigenetic memory. For example, miR-140 has been shown to target HDAC4. It is likely that miRNAs can target other epigenetic players such as DNMTs. On the other hand, epigenetics can control the expression of miRNAs. DNMTs and HDACs can affect the expression of some miRNAs, and DNA methylation and histone modifications have been shown to control the expression of miR-127. Epigenetic drugs such as 5-Aza-CdR and PBA can reverse the changes done by DNMTs and HDACs, and this adds a new layer of understanding to the pharmacological actions of these drugs.
Abbreviations
- 5-Aza-CdR:
-
5-Aza-2′-deoxycytidine
- DNMT:
-
DNA methyltransferase
- miR and miRNA:
-
microRNA
- PBA:
-
4-phenylbutyric acid
References
Egger G, Liang G, Aparicio A, Jones PA 2004 Epigenetics in human disease and prospects for epigenetic therapy. Nature 429: 457–463
Jones PA, Baylin SB 2002 The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428
Klose RJ, Bird AP 2006 Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 31: 89–97
Baylin SB, Ohm JE 2006 Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction?. Nat Rev Cancer 6: 107–116
Jones PA, Laird PW 1999 Cancer epigenetics comes of age. Nat Genet 21: 163–167
Jones PA, Takai D 2001 The role of DNA methylation in mammalian epigenetics. Science 293: 1068–1070
Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H 2004 Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900–903
Csankovszki G, Nagy A, Jaenisch R 2001 Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol 153: 773–784
Takai D, Jones PA 2003 The CpG island searcher: a new WWW resource. In Silico Biol 3: 235–240
Takai D, Jones PA 2002 Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 99: 3740–3745
Gardiner-Garden M, Frommer M 1987 CpG islands in vertebrate genomes. J Mol Biol 196: 261–282
Bird A 2002 DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21
Peters AH, Mermoud JE, O'Carroll D, Pagani M, Schweizer D, Brockdorff N, Jenuwein T 2002 Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet 30: 77–80
Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL 2001 Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107: 727–738
Johnson L, Cao X, Jacobsen S 2002 Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12: 1360–1367
Soppe WJ, Jasencakova Z, Houben A, Kakutani T, Meister A, Huang MS, Jacobsen SE, Schubert I, Fransz PF 2002 DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO J 21: 6549–6559
Tariq M, Saze H, Probst AV, Lichota J, Habu Y, Paszkowski J 2003 Erasure of CpG methylation in Arabidopsis alters patterns of histone H3 methylation in heterochromatin. Proc Natl Acad Sci U S A 100: 8823–8827
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A 1998 Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393: 386–389
Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T 2003 The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278: 4035–4040
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T 2000 DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24: 88–91
Yoo CB, Jones PA 2006 Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 5: 37–50
Cai X, Hagedorn CH, Cullen BR 2004 Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10: 1957–1966
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN 2004 MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: 4051–4060
Esquela-Kerscher A, Slack FJ 2006 Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 6: 259–269
Kim VN, Nam JW 2006 Genomics of microRNA. Trends Genet 22: 165–173
Meltzer PS 2005 Cancer genomics: small RNAs with big impacts. Nature 435: 745–746
Lee Y, Jeon K, Lee JT, Kim S, Kim VN 2002 MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21: 4663–4670
Rajewsky N 2006 microRNA target predictions in animals. Nat Genet 38: S8–S13
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003 Prediction of mammalian microRNA targets. Cell 115: 787–798
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM 2005 Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773
miRBase::Sequences. Sanger Institute, Wellcome Trust. Available at: http://microrna.sanger.ac.uk/sequences/index.shtml
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM 2002 Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99: 15524–15529
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM 2005 miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102: 13944–13949
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM 2005 MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65: 7065–7070
Ciafre SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM, Farace MG 2005 Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun 334: 1351–1358
Chan JA, Krichevsky AM, Kosik KS 2005 MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65: 6029–6033
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY 2007 miR-21-mediated tumor growth. Oncogene [Epub ahead of print]
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR 2005 MicroRNA expression profiles classify human cancers. Nature 435: 834–838
He L, Hannon GJ 2004 MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522–531
Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G 2002 Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30: 329–334
Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, Nakayama T, Oshimura M 2004 Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat Cell Biol 6: 784–791
Bao N, Lye KW, Barton MK 2004 MicroRNA binding sites in Arabidopsis class III HD-ZIP mRNAs are required for methylation of the template chromosome. Dev Cell 7: 653–662
Tuddenham L, Wheeler G, Ntounia-Fousara S, Waters J, Hajihosseini MK, Clark I, Dalmay T 2006 The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett 580: 4214–4217
Costa Y, Speed RM, Gautier P, Semple CA, Maratou K, Turner JM, Cooke HJ 2006 Mouse MAELSTROM: the link between meiotic silencing of unsynapsed chromatin and microRNA pathway?. Hum Mol Genet 15: 2324–2334
Lee RC, Feinbaum RL, Ambros V 1993 The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854
O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT 2005 c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843
Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A 2006 Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38: 1060–1065
Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I 2005 A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123: 819–831
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA 2006 Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9: 435–443
Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC 2006 Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 66: 1277–1281
Ying SY, Lin SL 2005 Intronic microRNAs. Biochem Biophys Res Commun 326: 515–520
Lyle R, Watanabe D, te Vruchte D, Lerchner W, Smrzka OW, Wutz A, Schageman J, Hahner L, Davies C, Barlow DP 2000 The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat Genet 25: 19–21
Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP 1997 Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 389: 745–749
Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, Nojima H, Looijenga LH, Agami R 2006 A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 124: 1169–1181
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE 2005 Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 102: 3627–3632
Tam W, Dahlberg JE 2006 miR-155/BIC as an oncogenic microRNA. Genes Chromosomes Cancer 45: 211–212
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM 2004 Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101: 2999–3004
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T 2004 Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64: 3753–3756
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ 2005 RAS is regulated by the let-7 microRNA family. Cell 120: 635–647
Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, Harris CC 2006 Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9: 189–198
Michael MZ, van Holst SM, Pellekaan NG, Young GP, James RJ 2003 Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 1: 882–891
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM 2005 A microRNA polycistron as a potential human oncogene. Nature 435: 828–833
Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T 2005 A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 65: 9628–9632
Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A 2004 High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer 39: 167–169
Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A 2005 BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol 207: 243–249
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM 2006 A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103: 2257–2261
Taganov KD, Boldin MP, Chang KJ, Baltimore D 2006 NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 103: 12481–12486
Acknowledgements
The authors thank Dr. Yoshimasa Saito and Jeffrey Friedman for their critical reading and critique of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chuang, J., Jones, P. Epigenetics and MicroRNAs. Pediatr Res 61, 24–29 (2007). https://doi.org/10.1203/pdr.0b013e3180457684
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/pdr.0b013e3180457684
This article is cited by
-
Elevation of hsa-miR-7-5p level mediated by CtBP1-p300-AP1 complex targets ATXN1 to trigger NF-κB-dependent inflammation response
Journal of Molecular Medicine (2023)
-
Phytotoxic Responses and Plant Tolerance Mechanisms to Cadmium Toxicity
Journal of Soil Science and Plant Nutrition (2023)
-
Identification of Metabolic Syndrome-Related miRNA–mRNA Regulatory Networks and Key Genes Based on Bioinformatics Analysis
Biochemical Genetics (2023)
-
Unravelling blood-based epigenetic mechanisms: the impact of hsa-miR-146a and histone H3 acetylation in lead-induced inflammation among occupational workers
International Archives of Occupational and Environmental Health (2023)
-
miRNA expression patterns in blood leukocytes and milk somatic cells of goats infected with small ruminant lentivirus (SRLV)
Scientific Reports (2022)
