
Abstract
Permissive hypercapnia because of reduced tidal volume is associated with improved survival in lung injury, whereas therapeutic hypercapnia—deliberate elevation of arterial Pco2—protects against in vivo reperfusion injury and injury produced by severe lung stretch. No published studies to date have examined the effects of CO2 on in vivo models of neonatal lung injury. We used an established in vivo rabbit model of surfactant depletion to investigate whether therapeutic hypercapnia would improve oxygenation and protect against ventilator-induced lung injury. Animals were randomized to injurious (tidal volume, 12 mL/kg; positive end-expiratory pressure, 0 cm H2O) or protective ventilatory strategy (tidal volume, 5 mL/kg; positive end-expiratory pressure, 12.5 cm H2O), and to receive either control conditions or therapeutic hypercapnia (fraction of inspired CO2, 0.12). Oxygenation (alveolar-arterial O2 difference, arterial Po2), lung injury (alveolar-capillary protein leak, impairment of static compliance), and selected bronchoalveolar lavage and plasma cytokines (IL-8, growth-related oncogene, monocyte chemoattractant protein-1, and tumor necrosis factor-α) were measured. Injurious ventilation resulted in a large alveolar-arterial O2 gradient, elevated peak airway pressure, increased protein leak, and impaired lung compliance. Therapeutic hypercapnia did not affect any of these outcomes. Tumor necrosis factor-α was not increased by mechanical stretch in any of the groups. Therapeutic hypercapnia abolished the stretch-induced increase in bronchoalveolar lavage monocyte chemoattractant protein-1, but did not affect any of the other mediators studied. Therapeutic hypercapnia may attenuate the impairment in oxygenation and inhibit certain cytokines. Because hypercapnia inhibits certain cytokines but does not alter lung injury, the pathogenic role of these cytokines in lung injury is questionable.
Similar content being viewed by others
Main
Mechanical ventilation is central to neonatal and pediatric critical care. Limiting Vt during mechanical ventilation in the setting of acute lung injury results in several important effects, including elevation of Paco2(1, 2), alteration of lung inflammatory events (3), decreased duration of mechanical ventilation (4), and improved survival (5). Although the associations among these effects are clear, the mechanistic relationships are uncertain. Lung stretch is associated with a complex series of pulmonary and systemic events, including neutrophil recruitment (6) and release of inflammatory prostanoids (7, 8) and cytokines (9–11) as well as morphologic injury (6, 12).
Alterations in CO2 tension may be important in this paradigm for several reasons (13). First, changes in Paco2 are inextricably linked to tidal stretch, such that reduced stretch increases Paco2 and increased stretch lowers Paco2. The impact of reduced Vt on Paco2 can be abrogated by inversely adjusting ventilatory frequency to maintain overall alveolar ventilation. Nonetheless, some residual elevation of Paco2 is usual (2, 3, 14), resulting in the original term “permissive hypercapnia”(2). Indeed, this clinical approach, originally reported in a case series by Wung et al.(1), has been extended in a randomized controlled trial to neonates with hypoxemic respiratory failure (4), with improvement in short-term respiratory outcome. In any case, when stretch is altered in patients, so too is Paco2. Second, alteration of Paco2 has important effects on gas exchange (15, 16) because hypocapnia worsens oxygenation whereas hypercapnia may enhance tissue oxygenation. Third, elevated CO2 can exert direct antiinflammatory effects in lung injury (17–19). Antioxidant effects of hypercapnia or acidosis have also been demonstrated in cell culture (20), and because many of these inflammatory processes may be operational in stretch-induced injury, their modulation by CO2 suggests that elevation of Paco2 could have protective effects in VALI. Fourth, deliberate elevation of CO2—“therapeutic hypercapnia”—protects against primary and secondary ischemia-reperfusion-induced lung injury (17–19), and protective effects have been reported for systemic ischemic injury (21). In addition, two recent studies reported that hypercapnic acidosis protects against stretch-induced lung injury ex vivo(22) and in vivo(23). However, the clinical role of CO2 alterations in neonatal practice is unclear (24, 25), and the efficacy of hypercapnic acidosis in in vivo models of lung injury associated with surfactant-depletion lung injury is not known.
We used an established in vivo rabbit model (saline lung lavage) of surfactant depletion and ventilator-induced lung injury, and used clinically relevant Vt. Our primary aim was to investigate whether therapeutic hypercapnia would improve oxygenation and protect against ventilator-induced lung injury.
METHODS
Male New Zealand White rabbits (Charles River Inc., Quebec, Canada), 2.5 to 3.0 kg, were used in all experiments. All experimental work conformed to the guidelines of the Canadian Council for Animal Care, and was approved by the Animal Care Committee at the Research Institute, Hospital for Sick Children.
Anesthesia and surgical dissection.
Premedication was with ketamine (85 mg/kg, intramuscularly). Anesthesia was induced with pentobarbital sodium (15 to 25 mg/kg, i.v.). Incremental bolus doses of pentobarbital (5 mg/kg) were administered as required. Sterile technique was used during all manipulations. A tracheotomy was performed, and a 4-mm (internal diameter) ETT was inserted to a depth of 1 cm and secured in place. Pancuronium (1 mg, i.v.) was administered after depth of anesthesia was confirmed by absence of response to paw compression. Pilot experiments established that anesthetized animals did not exhibit escape behavior in the absence of neuromuscular blockade. The lungs were ventilated using a small animal ventilator (model 683; Harvard Apparatus, MA, U.S.A.) with Fio2 1.0, rate 22 breaths/min, Vt 9 mL/kg, and 1 cm H2O PEEP. Under aseptic conditions the right carotid artery was cannulated for arterial pressure measurement and blood sampling. Anesthesia was maintained with pentobarbital (3–6 mg·kg−1·h−1) and muscle relaxation with pancuronium (0.1–0.2 mg·kg−1·h−1), and lactated Ringer's crystalloid (10–20 mL·kg−1·h−1) was administered by continuous infusion into bilateral marginal ear vein cannulas. Body temperature was maintained (39.0° ± 1.0°C) by use of a heating pack, and confirmed with an indwelling rectal temperature probe.
Saline lavage.
Surfactant depletion was induced by lavaging the lungs with warmed sterile saline 0.9% (15 mL/kg, temperature 37°C), using a modification of previously described techniques (26, 27). Saline was infused into the trachea from a height of 30 cm. The abdomen and chest were massaged for 30 s to assist intrapulmonary distribution. The effluent was then drained by gravity. After the first lavage, ventilation was recommenced but with PEEP set to 5 cm H2O with the other ventilatory variables remaining as before. This lavage procedure was repeated until Pao2 was less than 100 mm Hg. After this target Pao2 was achieved, ventilation was continued for 30 min, and Pao2 was measured again. If the Pao2 had increased to greater than 100 mm Hg by the end of this period, additional lavage was performed.
Lung recruitment.
In rabbits in which the Pao2 was sustained at less than 100 mm Hg at 30 min after lavage, a recruitment maneuver was performed. CPAP at 25–30 cm H2O was administered for 30 s. After this, the ETT was clamped and reconnected to the ventilator circuit with PEEP set at 12.5 cm H2O. The ETT was unclamped during expiration, and ventilation was continued for 10 min. Pao2 measurement was repeated, and recruitment was considered successful when Pao2 was greater than 200 mm Hg. If Pao2 was less than 200 mm Hg, the recruitment maneuver was repeated. If at this stage Pao2 was less than 200 mm Hg, the animal was excluded from further study.
Group allocation.
Animals were then randomly allocated to one of four treatment groups, defined by the type of ventilation strategy (protective versus injurious) and Fico2, as follows:
-
Normocapnia with injurious ventilation (CONinj: Fico2 0.00; Fio2 0.75; balance N2)
-
Therapeutic hypercapnia with injurious ventilation (THinj: Fico2 0.12; Fio2 0.75; balance N2)
-
Normocapnia with protective ventilation (CONpro: Fico2 0.00; Fio2 0.75; balance N2)
-
Therapeutic hypercapnia with protective ventilation (THpro: Fico2 0.12; Fio2 0.75; balance N2)
In addition, Evans blue dye (10 mg/kg, i.v.) was administered after randomization.
Ventilatory strategy.
The injurious ventilatory strategy consisted of: Vt 12 mL/kg, PEEP 0 cm H2O, and a rate of 19 breaths/min. The protective ventilatory strategy consisted of: Vt 5 mL/kg, with PEEP 12.5 cm H2O and rate 52 breaths/min. Ventilation was continued for 2.5 h (150 min), at which time the animals were killed by exsanguination under anesthesia.
Physiologic variables.
Systemic mean arterial pressure, peak airway pressures, and rectal temperature were recorded from a standard monitor (Spacelabs Monitor model 90303B; Spacelabs Medical Products Ltd., Mississauga, Ontario, Canada) at baseline and every 30 min after randomization.
Blood sampling protocol.
Arterial blood samples were measured for pH, Pco2, and Po2, using an ABL-300 blood gas analyzer (Radiometer, Copenhagen, Denmark).
Lung mechanics.
In all experiments, static inflation compliance was measured at baseline and at completion of the study. Static inflation compliance was determined by injection of incremental 5-mL volumes of 100% O2 and measurement of pressure attained 4 s after each injection until a total volume of 60 mL was injected. Airway pressure was recorded using a pressure transducer (MP45; Validyne Engineering Corporation, Northridge, CA, U.S.A.) connected to the side port of the ETT adapter, and recorded by an online PC IBM-compatible computer (IPC, Pentium microprocessor, 75 Hz, MS DOS 6.0), using analog-to-digital conversion at a sampling rate of 250 Hz (DT2802A; Data Translocation, Marlboro, MA, U.S.A.) and the software package ANADAT/LABDAT (McGill University, Montreal, Quebec, Canada). An inspiratory pressure-volume curve was then constructed.
Bronchoalveolar lavage.
Alveolar-capillary protein permeability was performed using a modification of previously described methodology (28, 29). At the end of the experiment, sternotomy was performed, and the lungs excised and lavaged with 20 mL of saline (0.9%, at 37°C), and 3 mL of BAL fluid was collected. This fluid was divided into two 1.5-mL aliquots and centrifuged. One aliquot was then snap-frozen in liquid nitrogen and stored at −70°C for subsequent cytokine analysis. In the second, the concentration of Evans blue dye in the supernatant was then determined spectrophotometrically by measurement of its absorbance at 620 nm.
Cytokine assay.
Analysis of serum and BAL fluid IL-8, GRO, MCP-1, and TNF-α was carried out in duplicate and in a blinded fashion. All were measured with previously described rabbit specific immunoassays (30, 31).
Statistical analysis.
Baseline variables and lung permeability were compared with one-way ANOVA. Serial variables were compared using 2-way repeated-measures ANOVA, followed, if significant, by one-way ANOVA within groups over time or among groups at each time. Student-Newman-Keuls or Tukey's tests were used for post hoc comparisons. Significance was set at p < 0.05. A-aO2 gradient was calculated using the following formula (32):

Results are expressed as mean ± SEM. Data were analyzed using SigmaStat (Version 2.0; Jandel Corporation, San Rafael, CA, U.S.A.).
RESULTS
Seven animals failed baseline exclusion criteria, and were not randomized. After in vivo surfactant depletion, 28 animals were randomized to one of the following groups:
-
Normocapnia with injurious ventilation (high Vt, no PEEP; CONinj; n = 8)
-
Therapeutic hypercapnia with injurious ventilation (high Vt, no PEEP; THinj; n = 8)
-
Normocapnia with protective ventilation (low Vt, PEEP; CONpro; n = 6)
-
Therapeutic hypercapnia with protective ventilation (low Vt, PEEP; THpro; n = 6)
Baseline variables.
The baseline variables (animal weight, temperature, airway pressure, Hb, and base excess) were comparable in all four experimental groups.
Mortality.
Two animals in the THinj group died near the end of the protocol; data from these animals up to the time of death were recorded and used in the analysis.
Arterial Pco2 and acid base.
Paco2 and arterial pH were comparable in all groups at baseline (Fig. 1). After surfactant depletion, randomization, and alteration of Fico2 and ventilatory strategy, Paco2 became most elevated in THinj (Fig. 1A). The final rank order of Paco2 was THinj > THpro > CONinj > CONpro (p < 0.05;Fig. 1A). Arterial pH decreased in all groups except CONpro after institution of the experimental protocol, with the final rank order of pH THinj < CONinj < THpro < CONpro (p < 0.05;Fig. 1B). There were no differences in plasma HCO3− among the groups at baseline. Through the experiment, HCO3− decreased in CONinj, remained unchanged in THinj, and increased in both protective ventilation (CONpro and THpro) groups (data not shown). These changes likely represent a combination of buffering response (renal retention of bicarbonate with hypercapnia) and lactic acidosis in the injury groups.

A, rank order of final Paco2 was THinj > THpro > CONinj > CONpro, *p < 0.05. B, rank order of final pH was THinj < CONinj < THpro < CONpro, *p < 0.05.
Oxygenation.
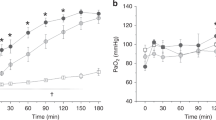
There was no difference in baseline Pao2 among groups (Table 1). After randomization and alteration of Fio2 (from 1.00 to 0.75) and ventilatory strategy, Pao2 fell significantly in all groups, but was significantly higher in CONpro and THpro compared with CONinj and THinj. Pao2 remained stable thereafter within all groups for the remainder of the experiment. Pao2 was higher in THproversus CONpro at 30 min (Table 1), indicating an effect of hypercapnia that is independent of the ventilation strategy. The rank order of Pao2 at 150 min was CONinj = THinj < CONpro = THpro (p < 0.05;Table 1). The A-aO2 gradient was comparable at baseline in all four groups (Fig. 2A). At the end of the experiment (150 min), the rank order of the A-aO2 gradient was CONinj = THinj < CONpro = THpro (p < 0.05;Fig. 2A).

A, baseline values for A-aO2 were similar among groups. The final A-aO2 was greater in both injurious (THinj, CONinj) vs both protective (CONpro, THpro) groups (*p < 0.05). B, represents BAL absorbance of Evans blue dye, reflecting microvascular leak. The rank order of final values was THinj = CONinj > CONpro = THpro (*p < 0.05).
Lung permeability.
Injurious ventilation worsened alveolar-capillary protein permeability as measured by BAL Evans blue dye concentration, compared with protective ventilation (Fig. 2B), producing a rank order of microvascular leakage of CONinj = THinj > CONpro = THpro (p < 0.05). Thus, lung permeability was not altered by CO2 tension.
Pulmonary mechanics.
Peak airway pressure (Paw) was comparable in all groups at baseline. The ΔPaw (Pawfinal − Paw30 min) was significantly greater in the two injurious ventilation groups (CONinj, THinj) versus the two protective groups (CONpro, THpro; p < 0.05;Table 1), demonstrating a significant impact of ventilatory strategy but no effect of therapeutic hypercapnia. Static inspiratory compliance was comparable in all groups at baseline (Fig. 3A). Static inspiratory compliance decreased significantly over the course of the experiment in both injurious ventilation groups (CONinj, THinj), but remained unchanged throughout the experiment in both protective ventilation groups (CONpro, THpro;Fig. 3B). Thus, the rank order of final static compliance was CONinj = THinj < CONpro = THpro (p < 0.05), demonstrating an absence of effect of CO2 tension on final static lung compliance.

A, static inflation compliance was comparable among all groups at baseline. B, rank order of final static compliance was THinj = CONinj < CONpro = THpro (*p < 0.05).
Cytokine measurements.
Cytokine measurements were obtained from a limited number of animals (n = 6 CONinj, THinj; n = 4 CONpro, THpro). No TNF-α was detected in the BAL from any samples from any of the groups, and there were only minimal levels of TNF-α in the plasma after injurious ventilation (Table 2). After injurious ventilation, therapeutic hypercapnia was associated with significantly lower BAL MCP-1 levels compared with CONinj and comparable to levels seen with protective ventilation (THpro, CONpro;Fig. 4A). Plasma MCP-1 (Fig. 4B) levels were significantly elevated in all groups. Mean levels of these cytokines were lower in both therapeutic hypercapnic groups (THinj, THpro) compared with CONinj, although this was not statistically significant. Plasma IL-8 levels increased significantly in all groups during the course of the experiment (Table 2). Plasma GRO levels were significantly elevated compared with baseline in CONinj, and were not significantly elevated in other groups (Table 2). Finally, there were no among-group differences in BAL GRO levels (Table 2).

A, BAL MCP-1 concentration was less in THinjvs CONinj, *p < 0.05; values were similar in the other groups. B, plasma MCP-1 increased with time in all groups, †p < 0.05, with no among-group differences.
DISCUSSION
The current study demonstrates that therapeutic hypercapnia did not protect against VALI in terms of oxygenation, vascular permeability, or lung mechanics in an in vivo surfactant-depletion model.
Ventilation-induced lung injury.
The model used in the current study has been used since the 1980s (6, 33), and is well characterized. The histologic injury in this model reflects the deterioration in oxygenation, as well as the impairment of compliance and microvascular leak (6, 33). The model is useful because the high Vt-low PEEP approach combines the effects of tidal stretch, as well as atelectasis, and thus may be a better model of clinical VALI than pure extreme stretch models given that extreme Vt are no longer used.
CO2 tension and adult respiratory distress syndrome.
Limiting Vt during mechanical ventilation in the setting of acute lung injury results in elevation of Paco2(2). This clinical approach, originally reported in a case series by Wung et al.(1), has been extended in a randomized controlled trial to neonates with hypoxemic respiratory failure (4). CO2 has been traditionally considered to play a passive role in the improved outcome observed with protective ventilatory strategies, and was thus termed permissive hypercapnia (2). However, deliberate elevation of CO2—therapeutic hypercapnia—directly attenuated acute lung injury after free radical generation (19), as well as both primary and secondary ischemia-reperfusion–induced acute lung injury (17–19). In the clinical context, the original reports of deliberate addition of inspired CO2 were in patients with posttraumatic lung injury (34), in whom the elevated CO2 appeared to improve cardiac output and oxygenation.
CO2 and stretch-induced injury.
Many of the inflammatory processes described above play a pivotal role in the pathogenesis of VALI. This has led to the suggestion that therapeutic hypercapnia could have protective effects in VALI, a contention supported by recent studies that reported hypercapnic acidosis protects against lung injury induced by severe lung stretch, in both ex vivo(22) and in vivo(23) models. However, the data in the current study demonstrate that therapeutic hypercapnia does not attenuate indices of lung damage, such as lung permeability and lung compliance in this in vivo model of VALI.
Several important experimental differences exist that may help reconcile the contrasting results. The experimental model used by Broccard et al.(22) was an ex vivo isolated perfused lung preparation, which allows a unique opportunity to measure specific variables (e.g. capillary filtration coefficient, microvascular hydrostatic pressure), but which is devoid of the effects of important circulating blood components. The study by Sinclair et al. demonstrated protective effects of elevated CO2, but important differences include the fact that the Vt was extreme (25 mL/kg), the Fio2 was high (100% inspired O2), and surfactant depletion was not performed (23); these issues may restrict interpretation. Thus, differences between the current data and other data in the literature may reflect a variety of model characteristics, including saline lavage, lung volume derecruitment, extremes of Vt, and differences in Fio2 or Pao2. Finally, the model used in the current study is prone to development of atelectasis (26, 27), and it is possible that whereas CO2 may attenuate injury caused by tidal stretch—i.e. large Vt(22, 23) —CO2 may be less effective in ameliorating injury caused by atelectasis. This may be important with the increasingly recognized spectrum of atelectasis-associated lung injury (35).
CO2 and oxygenation.
Alteration of CO2 tension has important effects on gas exchange (16). In a canine oleic acid–induced lung injury model, hypocapnia worsened oxygenation (15), whereas in a bovine pneumonia model, hypercapnia improved Pao2(36). When the complete formula (32) for calculation of A-aO2 gradient was used with the current data, there was no effect of CO2. However, the complexity of in vivo models is such that important interactions, such as the obligate effect of increased cardiac output on intrapulmonary shunt (37), must be taken into account. The lower systemic arterial blood pressure observed in the CONinj group may reflect the complex nature of heart-lung interactions.
Cytokines and VALI.
An evolving body of evidence points to a link between high-stretch ventilation and the release of cytokines, such as TNF-α(8–10, 38, 39) and IL-1β(9, 10, 39). These findings are supported by clinical data demonstrating that high Vt is associated with elevated levels of BAL (3) and plasma IL-6 (3, 5) in patients with adult respiratory distress syndrome. However, three separate in vivo studies (40–42) have not been able to demonstrate stretch-induced production of TNF-α, challenging the contention that TNF-α is central to the pathogenesis of VALI. Our findings demonstrate no release of TNF-α into the BAL fluid, and minimal TNF-α release into the plasma with injurious ventilation. These findings cast further doubt on the likelihood of a central pathogenic role for this cytokine in VALI.
A number of studies have reported elevation of IL-8 (or of its functional analog, macrophage inflammatory protein-2) in a variety of different experimental models (40, 43, 44). In the current study, IL-8 is increased to a similar degree in all groups; however, the degree of injury is quite different between protective versus injurious ventilatory strategies. Taken together, these data suggest that although IL-8 is released in this model—and other models (40, 43, 44) —the cytokine is unlikely to be uniformly pathogenic in the development of VALI.
Therapeutic hypercapnia and cytokines.
Although we detected minimal—or zero—levels of TNF-α resulting from high Vt in the current study, the issue with MCP-1 and GRO is less clear. However, the fact that therapeutic hypercapnia attenuated BAL MCP-1 levels and may have decreased plasma GRO, but did not attenuate the development of severe lung injury in this model, suggests that these cytokines may not be pathogenic in the current model, and casts doubt on an obligatory pathogenic role for these cytokines in VALI. Finally, these findings raise the possibility that in other disease processes in which a pathogenic role for cytokines has been clearly established (e.g. sepsis), therapeutic hypercapnia may, by suppressing cytokine production, provide some protection against injury.
Limitations of current findings.
There are several issues that limit extrapolation to other contexts. The systemic organ effects of therapeutic hypercapnia were not examined in the current experiments. It is possible that therapeutic hypercapnia-induced attenuation of cytokines may translate to improved systemic organ function; however, the possibility of increased mortality with therapeutic hypercapnia does not support this. It is possible that significant species—or model—variability exists, e.g. distribution of pulmonary xanthine oxidase content (45) or species susceptibility to capillary stress failure (46), that could limit application of the findings. However, the current model, or modifications thereof, has been widely studied and has played a major part in the evolution of the literature leading to the clinical recognition of VALI (6, 38, 47). In addition, although designed to represent surfactant depletion as observed in neonates, it is important to note that the animals used in the current study were all adult animals. Finally, the duration of effect may be important, because like virtually all in vivo experimental models, the current model uses experimental intervals that are orders of magnitude shorter than the duration of almost any relevant clinical illness. It is possible that with increased duration of injury, different results may have been observed.
CONCLUSIONS
The recognition that lower Vt—without substantial differences in Paco2—improves patient outcome from adult respiratory distress syndrome demonstrates the potential that exists to institute protective ventilatory strategies without resorting to significant hypercapnia. This may be critical, because there could be additional beneficial effects of hypercapnia. However, the opposite might be true, and management with what is now accepted permissive hypercapnia might counter the (now-proven) benefits of lessened lung stretch. This work demonstrates that deliberate hypercapnia does not appear to modulate the severity of ventilation-induced lung injury after surfactant depletion. In addition, we were unable to confirm a consistent effect of stretch on release of cytokines. These findings cast doubt on the potential for therapeutic hypercapnia to attenuate VALI, particularly in the context of atelectasis and severe hypoxemia. Additional experimental work will be needed before clinical testing of therapeutic hypercapnia on outcome can be undertaken.
Abbreviations
- A-aO2:
-
alveolar-arterial O2 gradient
- BAL:
-
bronchoalveolar lavage
- CONinj:
-
normocapnia, injurious ventilation
- CONpro:
-
normocapnia, protective ventilation
- CPAP:
-
continuous positive airway pressure
- ΔPaw:
-
delta airway pressure
- ETT:
-
endotracheal tube
- Fico2:
-
fraction of inspired carbon dioxide
- Fio2:
-
fraction of inspired oxygen
- GRO:
-
growth-related oncogene
- MCP-1:
-
monocyte chemoattractant protein-1
- Paco2:
-
partial pressure of arterial carbon dioxide
- Pao2:
-
partial pressure of arterial oxygen
- PEEP:
-
positive end-expiratory pressure
- THinj:
-
therapeutic hypercapnia, injurious ventilation
- THpro:
-
therapeutic hypercapnia, protective ventilation
- TNF-α:
-
tumor necrosis factor-α
- VALI:
-
ventilator-associated lung injury
- Vt:
-
tidal volume
References
Wung JT, James LS, Kilchevsky E, James E 1985 Management of infants with severe respiratory failure and persistence of the fetal circulation, without hyperventilation. Pediatrics 76: 488–494
Hickling KG, Henderson SJ, Jackson R 1990 Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med 16: 372–377
Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS 1999 Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 282: 54–61
Mariani G, Cifuentes J, Carlo WA 1999 Randomized trial of permissive hypercapnia in preterm infants. Pediatrics 104: 1082–1088
Acute Respiratory Distress Syndrome Network 2000 Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342: 1301–1308
Kawano T, Mori S, Cybulsky M, Burger R, Ballin A, Cutz E, Bryan AC 1987 Effect of granulocyte depletion in a ventilated surfactant-depleted lung. J Appl Physiol 62: 27–33
Edmonds JF, Berry E, Wyllie JH 1969 Release of prostaglandins caused by distension of the lungs. Br J Surg 56: 622–623
Imai Y, Kawano T, Miyasaka K, Takata M, Imai T, Okuyama K 1994 Inflammatory chemical mediators during conventional ventilation and during high frequency oscillatory ventilation. Am J Respir Crit Care Med 150: 1550–1554
von Bethmann AN, Brasch F, Nusing R, Vogt K, Volk HD, Muller KM, Wendel A, Uhlig S 1998 Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med 157: 263–272
Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS 1997 Injurious ventilatory strategies increase cytokines and c-fos mRNA expression in an isolated rat lung model. J Clin Invest 99: 944–952
Held HD, Boettcher S, Hamann L, Uhlig S 2001 Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med 163: 711–716
Webb HH, Tierney DF 1974 Experimantal pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures: protection by positive end-expiratory pressure. Am Rev Respir Dis 110: 556–565
Laffey JG, Kavanagh BP 2002 Hypocapnia. N Engl J Med 347: 43–53
Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Fihlo G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR 1998 Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338: 347–354
Domino KB, Lu Y, Eisenstein BL, Hlastala MP 1993 Hypocapnia worsens arterial blood oxygenation and increases Va/Q heterogeneity in canine pulmonary edema. Anesthesiology 78: 91–99
Hickling KG, Joyce C 1995 Permissive hypercapnia in ARDS and its effects on tissue oxygenation (Review). Acta Anaesthesiol Scand 107: 201–208
Laffey JG, Tanaka M, Engelberts D, Luo X, Yiang S, Tanswell TK, Post M, Lindsay T, Kavanagh BP 2000 Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am J Respir Crit Care Med 162: 2287–2294
Strand M, Ikegami M, Jobe AH 2003 Effects of high PCO2 on ventilated preterm lamb lungs. Pediatr Res 53: 468–472
Shibata K, Cregg N, Engelberts D, Takeuchi A, Fedorko L, Kavanagh BP 1998 Hypercapnic acidosis may attenuate acute lung injury by inhibition of endogenous xanthine oxidase. Am J Resp Crit Care Med 158: 1578–1584
Rehncrona S, Hauge HN, Siesjö BK 1989 Enhancement of iron-catalyzed free radical formation by acidosis in brain homogenates: difference in effect by lactic acid and CO2 . J Cereb Blood Flow Metab 9: 65–70
Vannucci RC, Towfighi J, Heitjan DF, Brucklacher RM 1995 Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics 95: 868–874
Broccard AF, Hotchkiss JR, Vannay C, Markert M, Sauty A, Feihl F, Schaller M 2001 Protective effects of hypercapnic acidosis on ventilator-induced lung injury. Am J Respir Crit Care Med 164: 802–806
Sinclair SE, Kregenow DA, Lamm WJ, Starr IR, Chi EY, Hlastala MP 2002 Hypercapnic acidosis is protective in an in vivo model of ventilator-induced lung injury. Am J Respir Crit Care Med 166: 403–408
Ambalavanan N, Carlo WA 2001 Hypocapnia and hypercapnia in respiratory management of newborn infants. Clin Perinatol 28: 517–531
Woodgate PG, Davies MW 2001 Permissive hypercapnia for the prevention of morbidity and mortality in mechanically ventilated newborn infants. PG - CD002061 Cochrane Database Syst Rev
Lachmann B, Robertson B, Vogel J 1980 In vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta Anaesthesiol Scand 24: 231–236
Murphy D, Cregg N, Tremblay L, Engelberts D, Laffey JG, Slutsky AS, Romaschin A, Kavanagh BP 2000 Adverse ventilator strategy causes pulmonary to systemic translocation of endotoxin. Am J Resp Crit Care Med 162: 27–33
Moxley MA, Baird TL, Corbett JA 2000 Adoptive transfer of acute lung injury. Am J Physiol Lung Cell Mol Physiol 279: L985–993
Verbrugge SJ, Vazquez de Anda G, Gommers D, Neggers SJ, Sorm V, Bohm SH, Lachmann B 1998 Exogenous surfactant preserves lung function and reduces alveolar Evans blue dye influx in a rat model of ventilation-induced lung injury. Anesthesiology 89: 467–474
Kajikawa O, Johnson MC, Goodman RB, Frevert CW, Martin TR 205: 1997 A sensitive immunoassay to detect the alpha-chemokine GRO in rabbit blood and lung fluids. J Immunol Methods 135–143
Kajikawa O, Goodman RB, Johnson MC, Konishi K, Martin TR 1996 Sensitive and specific immunoassays to detect rabbit IL-8 and MCP-1: cytokines that mediate leukocyte recruitment to the lungs. J Immunol Methods 197: 19–29
Tsukimoto K, Arcos JP, Schaffartzik W, Wagner PD, West JB 1990 Effect of common dead space on VA/Q distribution in the dog. J Appl Physiol 68: 2488–2493
McCulloch PR, Forkert PG, Froese AB 1988 Lung volume maintenance prevents lung injury during high frequency oscillatory ventilation in surfactant-deficient rabbits. Am Rev Respir Dis 137: 1185–1192
Trimble C, Smith DE, Rosenthal MH, Fosburg RG 1971 Pathophysiologic role of hypocarbia in post-traumatic pulmonary insufficiency. Am J Surg 122: 633–638
Duggan M, McCaul CL, McNamara PJ, Engelberts D, Ackerley C, Kavanagh BP 2003 Atelectasis causes vascular leak and lethal right ventricular failure in uninjured rat lungs. Am J Respir Crit Care Med 167: 1633–1640
Keenan RJ, Todd TR, DeMajo W, Slutsky AS 1990 Effects of hypercarbia on arterial and alveolar oxygen tensions in a model of gram-negative pneumonia. J Appl Physiol 68: 1820–1825
Cheney FW, Colley PS 1980 The effect of cardiac output on arterial blood oxygenation. Anesthesiology 52: 496–503
Takata M, Abe J, Tanaka H, Kitano Y, Doi S, Kohsaka T, Miyasaka K 1997 Intraalveolar expression of tumor necrosis factor-alpha gene during conventional and high-frequency ventilation. Am J Respir Crit Care Med 156: 272–279
Chiumello D, Pristine G, Slutsky AS 1999 Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med 160: 109–116
Ricard JD, Dreyfuss D, Saumon G 2001 Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med 163: 1176–1180
Verbrugge SJ, Uhlig S, Neggers SJ, Martin C, Held HD, Haitsma JJ, Lachmann B 1999 Different ventilation strategies affect lung function but do not increase tumor necrosis factor-alpha and prostacyclin production in lavaged rat lungs in vivo. Anesthesiology 91: 1834–1843
Copland IB, Kavanagh BP, Engelberts D, McKerlie C, Belik J, Post M 2003 Early changes in lung gene expression due to high tidal volume. Am J Resp Crit Care Med (e-published ahead of print - http://ajrccm.atsjournals.org/cgi/reprint/200208-964OCv1)
Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD 1999 Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol 277: L167–173
Oudin S, Pugin J 2002 Role of MAP kinase activation in interleukin-8 production by human BEAS-2B bronchial epithelial cells submitted to cyclic stretch. Am J Respir Cell Mol Biol 27: 107–114
Parks DA, Granger DN 1986 Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl 548: 87–99
Mathieu-Costello O, Willford DC, Fu Z, Garden RM, West JB 1995 Pulmonary capillaries are more resistant to stress failure in dogs than in rabbits. J Appl Physiol 79: 908–917
Imai Y, Kawano T, Iwamoto S, Nakagawa S, Takata M, Miyasaka K 1999 Intratracheal anti-tumor necrosis factor-alpha antibody attenuates ventilator-induced lung injury in rabbits. J Appl Physiol 87: 510–515
Acknowledgements
The authors are grateful to Drs. A.C. Bryan and J. Lewis for their insightful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by The Canadian Institutes of Health Research.
Rights and permissions
About this article
Cite this article
Rai, S., Engelberts, D., Laffey, J. et al. Therapeutic Hypercapnia Is Not Protective in the in vivo Surfactant-Depleted Rabbit Lung. Pediatr Res 55, 42–49 (2004). https://doi.org/10.1203/01.PDR.0000098502.72182.55
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000098502.72182.55
This article is cited by
-
Effects of permissive hypercapnia on pulmonary and neurodevelopmental sequelae in extremely low birth weight infants: a meta-analysis
SpringerPlus (2016)
-
Hypercapnic acidosis in ventilator-induced lung injury
Intensive Care Medicine (2010)
-
Does Hypercapnia Ameliorate Hyperoxia-Induced Lung Injury in Neonatal Rats?
Lung (2010)
