
Abstract
Estrogen receptor α (ERα) is a master driver of a vast majority of breast cancers. Breast cancer cells often develop resistance to endocrine therapy via restoration of the ERα activity through survival pathways. Thus identifying the epigenetic activator of ERα that can be targeted to block ERα gene expression is a critical topic of endocrine therapy. Here, integrative genomic analysis identified MYST3 as a potential oncogene target that is frequently amplified in breast cancer. MYST3 is involved in histone acetylation via its histone acetyltransferase domain (HAT) and, as a result, activates gene expression by altering chromatin structure. We found that MYST3 was amplified in 11% and/or overexpressed in 15% of breast tumors, and overexpression of MYST3 correlated with worse clinical outcome in estrogen receptor+ (ER+) breast cancers. Interestingly, MYST3 depletion drastically inhibited proliferation in MYST3-high, ER+ breast cancer cells, but not in benign breast epithelial cells or in MYST3-low breast cancer cells. Importantly, we discovered that knocking down MYST3 resulted in profound reduction of ERα expression, while ectopic expression of MYST3 had the reversed effect. Chromatin immunoprecipitation revealed that MYST3 binds to the proximal promoter region of ERα gene, and inactivating mutations in its HAT domain abolished its ability to regulate ERα, suggesting MYST3 functioning as a histone acetyltransferase that activates ERα promoter. Furthermore, MYST3 inhibition with inducible MYST3 shRNAs potently attenuated breast tumor growth in mice. Together, this study identifies the first histone acetyltransferase that activates ERα expression which may be potentially targeted to block ERα at transcriptional level.
Similar content being viewed by others


Introduction
Targeting estrogen receptor α (ERα) has significantly improved the outcomes for patients with breast cancer. Many studies have suggested that restoration of ERα activity has been a key mechanism of breast cancer cell survival and resistance to endocrine therapy.1 Although many endocrine agents have been developed to target ERα ligand binding domain, posttranslational modifications of ER and its key coregulators (that is, phosphorylation) by survival pathways have been shown to restore ERα activity and render resistance to various endocrine therapies.2, 3 New therapeutic approaches that can eradicate ERα expression at transcriptional level will apparently complement the current endocrine therapies in the management of breast cancer. In particular, identifying the epigenetic activator of ERα expression that can be targeted to block ERα gene expression is a critical yet underexplored area of endocrine therapy. Histone acetylation and deacetylation are the processes by which the lysine residues at the N terminus of histone proteins are acetylated or deacetylated, which are catalyzed by histone acetyltransferase (HAT) and histone deacetylase, respectively. As a consequence, histone acetylation enhances gene transcription while histone deacetylation represses gene transcription. Although histone deacetylase that represses ERα promoter has been identified and are being targeted to modulate ERα expression during endocrine therapy (such as histone deacetylase 1), to date, the specific histone acetyltransferase that activate ERα promoter remains unclear.1, 4
In this study we have applied an integrative analysis to discover key drug targets from the complex cancer signaling network by combining genomic/pharmacological information with cancer-gene concept signatures (ConSig).5 This analysis has revealed a new and potentially druggable target, MYST3, which is frequently amplified in 11% and/or overexpressed in 15% of breast cancers. MYST3, also known as KAT6A and MOZ (monocytic leukemic zinc-finger), is a HAT enzyme that controls fundamental cellular processes, including gene transcription and maintenance of normal hematopoietic stem cell.6 Deregulation of the MYST3 HAT activity or aberrant expression of MYST3 has been associated with the development of disease, particularly acute myeloid leukemia.7 MYST3 interacts with transcriptional factors, such as p53 and RUNX1, to activate gene transcription by acetylating histones near target gene promoters.8 Fusions of MYST3 with CREB-binding protein (CBP), p300, TIF2 or NCOA3, resulting from chromosome translocation in acute myeloid leukemia lead to aberrant transcriptional activity.9, 10, 11 MYST3 acetylates histones via its HAT domain.12 However, its role in breast cancer remains unclear. Here, we discovered that MYST3 has a role in breast cancer development as novel histone acetyltransferase that activates ERα promoter and targeting MYST3 may serve as a novel therapeutic strategy to block ERα gene expression in MYST3-high breast cancers.
Results and Discussion
MYST3 is amplified and overexpressed in breast cancer
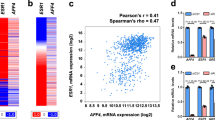
To systematically reveal new therapeutic targets, we performed integrative analysis of the genomic data set from The Cancer Genome Atlas (TCGA).13 We first identified all human genes that are upregulated by recurrent genomic amplification in 10% of breast cancer genomes. The candidate genes were benchmarked by the level of correlation between gene expression and copy number data, and then, by a cancer ConSig score that prioritizes biologically important genes.5 Druggable genes were selected according to a drug-target database compiled from multiple sources.14, 15, 16 This analysis not only revealed known amplified targets as lead candidates, such as ERBB2, WHSC1L1, PTK2, YWHAZ, FGFR1,17, 18, 19 but also identified a novel druggable target, MYST3, which is amplified in at least 11% of breast tumors (Figure 1a). Notably, MYST3 was amplified in tumors only, not in their adjacent normal breast tissues or in blood (Figure 1b). Furthermore, MYST3 is overexpressed in about 15% of breast tumors; a higher frequency (22%) was detected in the Luminal B subtype (HER2-) (Figure 1c). In addition, MYST3’s gene amplification highly correlated with its RNA overexpression (Figure 1d). Interestingly, survival analyses using the Metabric gene expression data set,20 suggested that high MYST3 levels particularly predicted worse clinical outcome for ER+/HER2− patients, the most common subtype of breast cancer. In addition, MYST3 showed prognostic effect in both systematically untreated (n=203) or hormone treated patients (n=362) (Figure 1e). We assessed the protein levels of MYST3 in breast cancer cell lines. MYST3 is highly expressed in multiple ER+ breast cancer cell lines, such as CAMA1, T47D and ZR75-1 (Figure 1f). Among these, MDAMB134VI, CAMA1 and ZR75-1 harbor MYST3 amplifications (Figure 1b). These results together indicate that MYST3 is frequently amplified and overexpressed in breast cancer, and imply that MYST3 might have a role in breast cancer development.

MYST3 is amplified and overexpressed in breast cancer. (a) MYST3 is nominated as a potential druggable oncogene target in breast cancer by integrative bioinformatics analysis. We used the Affymetrix SNP 6.0 copy number and Agilent 4502A gene expression data sets for invasive breast cancer from TCGA (http://cancergenome.nih.gov/). Normalized ‘level 3’ data segmented by the CBS algorithm were applied in the analysis.13 First, genes that are recurrently amplified in more than 10% of invasive breast tumors were nominated and their expressions correlated with copy number using Spearman’s correlation statistics (R>0.5 as cutoff). The druggability of these genes was predicted according to several drug-target databases.14, 15, 16 Then, all candidates were ranked by Pearson's correlation coefficient, and by the concept signature (ConSig) score that we have developed.5 This score prioritizes biologically important genes underlying cancer by accessing their strength of association with molecular concepts characteristic of cancer genes (see http://consig.cagenome.org, release 2). The overexpression cutoff is based on median+1 × MAD (median absolute deviation). MAD is calculated using the R with default constant (1.4826). (b) Amplification of MYST3 locus in breast cancer cell lines (data from Broad-Novartis Cancer Cell Line Encyclopedia), breast tumors, and blood (data from TCGA). (c) MYST3 is amplified and overexpressed in breast tumors based on TCGA breast cancer copy number and RNAseq data sets. Luminal B subtype has the highest overexpression rate. (d) The correlation of MYST3 overexpression in breast tumors based on the TCGA RNAseq data set, with its corresponding gene copy number. R value is based on Pearson’s correlation. The relative copy number was estimated by dividing the tumor signal intensities with signal intensities from the linear combination of all normal samples that are most similar to the tumor, which were then transformed with log2. (e) MYST3-high predicts worse survival outcome for ER+/HER2− breast patients. Both systematically untreated (n=203) or hormone treated (n=362) ER+/HER2− breast cancer patients showed worse clinical outcome in MYST3-high groups. Survival analyses results based on the cutoff of median+MAD (data from METABRIC provided by Molecular Taxonomy of Breast Cancer International Consortium (Illumina HT-12 v3)). MAD is calculated using the R with default constant (1.4826). Normalized gene expression data matrixes were used for survival analysis. The available probe for MYST3 in the Illumina HT-12 v3 array is ILMN_2095840. Patients were divided into two groups (MYST3-high and the rest), according to the cutoff based on median+1 × MAD. MAD is calculated using the R with default constant (1.4826). Kaplan–Meier analyses were carried out using the R survival package. Follow-up time was limited to a maximum of 10 years. P-values were calculated according to the log-rank test. (f) MYST3 protein expression in a subset of breast cancer cells. MCF7, T47D, CAMA1, ZR75-B and ZR75-1 cells were obtained from Dr Rachel Schiff. SK-BR-3, BT474, HCC1569, HCC1954, BT549 and MDA-MB-231 were obtained from American Type Culture Collection (ATCC). LY2 was cultured as described.40 SUM225CWN was purchased from Asterand Bioscience (Detroit, MI, USA). MCF7, ZR75-1, ZR75-B, BT474, T47D, HCC1569, HCC1954 and BT549 cells were cultured in RPMI 1640 (Cellgro, Manassas, VA, USA) with 10% fetal bovine serum and 200 mg/ml l-glutamine (Life Technologies, Carlsbad, CA, USA). CAMA1, SK-BR-3 and MDA-MB-231 cells were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum and 200 mg/ml l-glutamine (Life Technologies). MCF10A cells were cultured as described.41 The protein levels were determined with western blot. Cells were homogenized in RIPA Lysis Buffer (50 mm Tris·HCl pH8.0, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS), supplemented with HALT (Thermo Fisher Scientific). Forty micrograms of protein extracts were heated in sample buffer at 70 °C, separated by Tris-Acetate protein gels (NuPAGE Novex 3-8% Gels, Life Technologies), and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membranes were blocked for one hour and incubated overnight at 4 °C with either anti-MYST3 rabbit polyclonal (1:2000, Active Motif, #39867), or with anti-alpha Tubulin monoclonal (1:5000, Sigma, #T5168) antibodies. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (Life Technologies) and the signals were visualized by the enhanced chemiluminescence system (Perkin Elmer, Waltham, MA, USA) as per the manufacturer’s instructions.
Depletion of MYST3 remarkably inhibits cell proliferation in ER+/ MYST3-high breast cancer cells
To explore the role of MYST3 in breast cancer, we knocked down the expression of MYST3 using two different MYST3 siRNAs, respectively. As compared with the negative control, each of these two siRNAs significantly attenuated the expression of MYST3, at both mRNA and protein levels (Figures 2c and 3b). To examine the effect of MYST3 inhibition in ER+ breast cancer cells, we selected two ER+ cell lines overexpressing MYST3—CAMA1 and T47D—with or without MYST3 amplification, respectively. Intriguingly, depletion of MYST3 with these siRNAs drastically inhibited cell growth in MYST3-high, ER+ breast cancer cell lines (T47D and CAMA1), but not in MYST3-low cells (MCF7 and SK-BR-3; Figure 2a). Furthermore, in breast epithelial cells, MCF10A, no significant inhibitory effects were observed (Supplementary Figure 1B). To confirm the inhibitory effect of MYST3 depletion, we generated lentiviruses inducible expressing MYST3 shRNAs. We found that stable infections with these viruses can efficiently knockdown MYST3 in T47D and CAMA1 cells (Figure 2c). Accordingly, we observed a profound reduction in anchorage-dependent and anchorage-independent growth in these cells with the expression of inducible shRNAs, in a clonogenic assay (Figure 2b), and a soft agar assay, respectively (Figure 2d). Together, these data demonstrate that targeting MYST3 can attenuate the growth of MYST3-high/ER+ cancer cells.

Knocking down MYST3 inhibits cell growth in ER+, MYST3-high breast cancer cells. (a) Clonogenic assay: MYST3-high (T47D and CAMA1) and MYST3-low (MCF7 and SKBR3) breast cancer cells were plated at a density of 500 cells/well in a 6-well plate, and treated with two MYST3-specific siRNAs (siRNA1: 5′-GCGCUAUACUAAUCCAAUA-3′ and siRNA2: 5′-GGAGUUGAGUGUUAAAGAU-3′) and negative control siRNA (Qiagen, Valencia, CA, USA) to knockdown MYST3. siRNAs were transfected using Lipofectamine RNAi MAX Reagent (Life Technologies) according to the manufacturer’s protocol. Transfected cells were incubated for 2–3 weeks. The colonies were stained with 0.5% crystal violet in 50% methanol and counted. Statistical data are presented as mean±s.d. Three experiments were performed. (b) MYST3-high (T47D and CAMA1) and MYST3-low (MCF7 and SKBR3) breast cancer cells were infected with shRNAs against MYST3 (shMYST3-1: 5′-TTGGAGTTGAGTGTTAAAGAT-3′; shMYST3-2: 5′-CGGCGCTATACTAATCCAATA-3′) and non-silencing controls (5′-GCGAAAGATGATAAGCTAA-3′). Control and shRNA oligos were annealed and cloned into the inducible lentiviral vector pLKO-Tet-On (Addgene plasmid 21915). Lentiviral vector mixed with packing plasmid (psPAX2) and envelope plasmid (pMD2g) were transfected into 293T cells using Lipofectamine 3000 (Life Technologies). Viral supernatants were collected after 48 h. The stable lines expressing shMYST3 were selected with Puromycin (Life Technologies). Doxycycline (Dox) (Sigma-Aldrich, St. Louis, MO) was used for the induction of shMYST3 for 2–3 weeks. Statistical data are presented as mean±s.d, P-values were calculated based on two-tailed student’s t-test. Three experiments were performed. (c) Protein and mRNA levels of MYST3 in T47D cells after induction of shMYST3 by 500 ng/ml Dox. The protein levels were determined with western blot. Total RNA from all samples was isolated with the RNeasy Mini Plus Kit (Qiagen) according to the manufacturer’s protocol. Complementary DNA was synthesized from 1μg total RNA, using a Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Indianapolis, IN) in the presence of random primers. All oligonucleotide primers used in this study were synthesized by Integrated DNA Technologies (Coralville, IA, USA). MYST3 qPCR primers are: forward: 5′- ATAATCCTGGGCGAATAGCACT-3′; reverse: 5′- CTGCCTCCGAATAATGCAGAC-3′. Quantative Real-Time PCR (qRT–PCR) was performed on Applied Biosystems ViiA 7 (Life Technologies). The relative expression level of each target gene was determined using the comparative threshold cycle (Ct) method and normalized to a housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Statistical data are presented as mean±s.d. P-values were calculated based on two-tailed student’s t-test. Three experiments were performed. (d) Soft agar assay: relative colony formation ability of MYST3-high T47D breast cancer cells after treatment with inducible MYST3 shRNAs. The soft agar assay was performed as described previously. Briefly, T47D cells (500 cells/well) were plated in 24-well plates in culture medium containing 0.35% agar. The cells were treated with by 500 ng/ml Dox for 4 weeks. The top layer of T47D colonies was stained with p-iodonitrotetrazolium (1 mg/ml), and counted. Data are presented as mean±s.d. Three experiments were performed. P-values were calculated based on two-tailed student’s t-test.

MYST3 regulates ERα expression. (a) Association of ERα expression and protein level with MYST3 overexpression status in breast tumors. MYST3 expression analyses results based on TCGA RNAseq data using the cutoff of median+MAD (MAD is calculated using the R with default constant (1.4826)). MYST3-high: n=148; MYST3-low: n=954. The unit of Y-axis is transcript level of ERα (log2 of transformed upper quartile normalized read count). To estimate the correlation of ERα protein level with MYST3 expression in breast tumors, we analyzed the reverse phase protein array (RPPA) data set from TCGA. MYST3-high: n=86; MYST3-low: n=491. P-values were calculated based on student’s t-test. (b) T47D and CAMA1 cells were transfected with negative control siRNA or two MYST3 siRNAs. The mRNA levels of ERα were determined with qRT–PCR (data are presented as mean±s.d, three experiments were performed), and the protein levels with western blot. Polycolonal against ERα (1:2000, Santa Cruz, #sc8002) was used to detect ERα. (c) Inducible ectopic expression of MYST3 elevated ERα levels in a dose-dependent manner. The full-length cDNA of MYST3 was purchased from Harvard plasmid deposit (Clone ID: HsCD00399202), and then was introduced into an inducible lentiviral destination vector by Gateway LR Clonase II enzyme (Life Technologies). Total protein of MCF7 cells overexpressing Flag-tagged MYST3 or its mutants were collected after a 3-day incubation with different doses of doxycycline (50–1000 ng/ml Dox). (d) T47D cells were used in the ChIP assay. ChIP was carried out by following the protocol of EZ-ChIP kit (17-371, Upstate, Temecula, CA, USA). In general, formaldehyde (final concentration: 1 %) was added to growth media to crosslink the protein to the DNA. After quenched the residual formaldehyde, cells were collected and lysed in SDS lysis buffer. Cell lysate were sonicated with three pulses of 45 s to achieve fragment sizes of around 500 bp by sonic dismembrator (Thermo Fisher Scientific). The anti-MYST3 rabbit polyclonal antibody (39867, Active Motif), anti-acetyl-Histone H3 antibody (06-599, Millipore, Darmstadt, Germany) and anti-acetyl-Histone H3(Lys9) monoclonal antibody (9649S, Cell Signaling, Danvers, MA, USA) were used to immunoprecipitate the respective crosslinked protein/DNA. qRT–PCR and primers was carried out as described23 and primer sequences are provided in Supplementary Table 1. The primers (+135, and +528) are the promoter regions of ESR1. Normal IgG was used as negative control. Data are presented as mean±s.d. Three experiments were performed. P-values were calculated based on two-tailed student’s t-test. *P<0.05; **P<0.01. (e) Restoration ERα expression in stably inducible MYST3 shRNAs cells partially recovered the cell growth. The full-length cDNA of ERα was purchased from Harvard plasmid deposit (Clone ID: HsCD00376961), and then was introduced into an inducible lentiviral destination vector by Gateway LR Clonase II enzyme. Inducibly expressing ERα and MYST3 shRNAs in T47D cells were induced with doxycycline. The protein levels were determined with western blot. Statistical data of clonogenic assay are presented as mean±s.d. Five experiments were performed. P-values were calculated based on two-tailed student’s t-test.
MYST3 regulates the gene expression of ERα
MYST3 has been shown to epigenetically regulate the expression of genes, such as HOXA9, via modification of the chromatin structure.21 In fact, patient data set analyses showed that ERα expression level is significantly higher in MYST3-high tumors at both mRNA and protein levels (Figure 3a). We thus hypothesized that MYST3 may regulate ERα expression by modifying the ERα promoter region via its HAT activity. To clarify this, we determined ERα expression after MYST3 knockdown. Both mRNA and protein levels of ERα were remarkably reduced after depleting MYST3 with its siRNAs (Figure 3b). We also confirmed that ERα expression could be reduced with our inducible MYST3 shRNAs (data not shown). To exclude off-target effect, we electroporated T47D cell with siRNAs and siMYST3-2 resistant MYST3 plasmids. The ERα expression was restored in T47D cells expressing siMYST3-2 resistant MYST3 (Supplementary Figure 2A). Also overexpressing MYST3 partially rescued cell growth in siMYST3-2 cells (Supplementary Figure 2B). In addition, inducible overexpression of MYST3 in MCF7 cells, which are MYST3-low, resulted in a dose-dependent increase of ERα expression (Figure 3c). We also observed similar dose-dependent increase of ERα in LY2 cells in response to MYST3 induction (Supplementary Figure 3). To examine whether the HAT domain of MYST3 is involved in this ERα upregulation, we generated MYST3 mutants MYST3G657E and MYST3C543G/G657E, which are HAT activity deficient22 and found that these mutants cannot upregulate ERα as the wild-type does (Figure 3c). Together, these data demonstrate that MYST3 can promote ERα expression at transcriptional level via its HAT activity.
To assess how MYST3 regulates the expression of ERα, we performed a ChIP assay to determine whether MYST3 binds to the promoter regions of ESR1 gene.23 Indeed, MYST3 occupancy was significantly enhanced at the proximal site of ESR1 promoter (6.4 and 5.83-fold enrichment, Figure 3d), indicating that MYST3 is recruited to the promoter regions of ESR1. Noticeably, the promoter region of ERα also showed the enrichment of H3ac and H3K9ac marks (Figure 3d), suggesting that MYST3 may regulate the expression of ERα by directly binding to its promoter region via acetylation of H3. Furthermore, we restored the ERα expression in T47D cells carrying inducible MYST3 shRNAs. Cell growth of these MYST3-depleted cells were partially recovered after ERα re-expression using an inducible lentiviral expression system (Figure 3e), suggesting that ERα is important for MYST3-mediated cancer cell growth.
MYST3 knocking down attenuates tumor growth in a xenograft tumor model
To determine the therapeutic potential of targeting MYST3 in vivo, we employed a tumor xenograft mouse model. Among the two ER+ cell line models that we have studied in vitro (T47D and CAMA1), only T47D cells are known to be transplantable in nude mice. We thus injected T47D-shMYST3 cells (T47D cells with inducible stable expression of MYST3 shRNA) into the mammary fat pad of the nude mice to establish a breast tumor xenograft model. After tumors reached 150 mm3, we fed the mice with doxcycline to induce the expressison of MYST3 shRNA. MYST3 shRNA was able to effectively knockdown MYST3 in vivo and ERα levels were reduced in the tumors with MYST3 knockdown (Figure 4b). Notably, depletion of MYST3 expression in T47D cells led to significant tumor regression in these mice, resulting in prolonged progression-free survival (Figure 4a). This data strongly suggests that targeting MYST3 may be an attractive strategy to treat the disease.

The therapeutic effect of knocking down MYST3 in a xenograft tumor model. All animal works have been approved by BCM Institutional Animal Care and Use Committee. 1 × 107 T47D cells Dox dependently expressing shMYST3 were transplanted bilaterally to the mammary fat pad of 4–6 weeks female athymic nude mice (Harlan Sprague–Dawley) supplemented with 17β-estradiol pellets (Sigma-Aldrich). Xenograft tumors of the T47D models are successfully engrafted in 16 mice which were randomized into±doxycycline (Dox) (8 mice per group). Briefly when tumors reached 150 mm3, 0.2 mg/ml Dox was administered in drinking water to turn on shRNA expression. Animals were euthanized after 33 days of treatment and tumors were collected. (a) MYST3 depletion suppresses tumor growth in vivo. The growth of the xenograft tumors was measured three times per week and tumor volume was calculated using the formula 1/2(length × width2). Data are presented as mean±s.d. ***P<0.001. Kaplan–Meier analyses were carried out using the R survival package. P-values were calculated according to the log-rank test. **P<0.01. (b) The protein levels of MYST3 in the collected tumors treated with Dox were decreased. The expression levels of MYST3 and ERα were determined with Western blot. The density of bands was analyzed with ImageJ.
In this study, we identified MYST3 as a novel acetyltransferase that positively regulates ERα promoter. MYST3 is amplified and overexpressed in a significant portion of breast cancers and its overexpression correlates with poor survival in ER+ patients. MYST3 depletion remarkably inhibits breast cancer cell growth both in vitro and in vivo. Here, we not only show that the MYST3 gene is significantly amplified, but that it is also overexpressed in 15% of breast cancers, including ER+ cancers. Of note, during the course of this study, MYST3 has also recently reported as a potential oncogene for luminal breast cancer.24 In that study, knocking down MYST3 in ER+ breast cancer, SUM-52, resulted in attenuated cell growth; this inhibitory effect is consistent with our observations in T47D and CAMA1 cells. Additionally, they showed that MYST3 is required for the growth of tumor initiating cells. However, our study is the first one to report MYST3 as a novel acetyltransferase that activates ERα promoter. We also evaluated the therapeutic effect of MYST3 inhibition in multiple cell line models as well as using inducible shRNA system in nude mice (Figure 4a). Our study, along with that report,24 strongly supports that MYST3 acts as a key oncogenic driver for luminal breast tumor development. Of note, besides its role in ER+ tumors, knocking down MYST3 in MYST3-high/ER- cancer cells renders a modest inhibitory effect (Supplementary Figure 1C), although it is not as dramatic as the effect in ER+ cells. This implies that MYST3 may have ERα-independent functions as well.
MYST3 is localized in chromosome 8p11-p12. This locus is amplified in 12–15% of breast cancers.25 Of note, other oncogenes including growth factor receptor FGFR1 are also identified to be amplified in this interval and contribute to breast cell growth and transforming.26, 27 Thus, it is plausible that MYST3 cooperates with these oncogenes to promote the tumorigenesis. In response to MYST3 depletion, we observe the significant changes in cell proliferation (BrdU incorporated) but not apoptosis (Supplementary Figure 1D,E). Thus, MYST3 depletion-mediated tumor suppression is caused at least partially by decreased cell proliferation. Additionally, recent reports suggest that reducing levels of MYST3 may lead to cellular senescence in stem cells and mouse embryonic fibroblasts by enhancing the expression of p16 tumor suppressor.28, 29 It will be interesting to investigate whether MYST3 reduction induces cellular senescence in cancer cells and whether this cellular senescence contributes the tumor suppression.
Importantly, our study reveals a novel mechanism for epigenetic regulation of ER expression. ERα expression is regulated at several levels and its aberrant expression has been implicated in breast cancer development. Transcriptional factors, such as GATA3, FOXO3A and FOXM1, are participated in the upregulation of ERα expression.4 In addition, promoter DNA hypermethylation also contributes to the aberrant ERα expression observed in breast cancer.30 Recently, it has been reported that histone methyltransferase WHSC1 is also a regulator to facilitate ERα gene expression,31 which has been evaluated as a therapeutic targets. Here, we provide solid evidence that MYST3 is another epigenetic activator of ERα expression in breast cancer. As far as we know it is the first acetyltransferase reported to regulate ERα promoter. We observed that knocking down MYST3 attenuates ERα expression at both mRNA and protein levels (Figure 3b), while elevating MYST3 increases ERα expression, as verified in two MYST3-low breast cancer cells, MCF7 and LY2 (Figure 3c; Supplementary Figure 3). Our ChIP analysis revealed that MYST3 is recruited and bound to the ERα promoter. This promoter region also showed enrichment of H3ac and H3K9ac marks (Figure 3d). MYST3 has been reported to regulate H3K9 acetylation at HOX gene loci and their expression.32 Thus, our data support a model in which MYST3 may regulate ERα gene expression via direct binding to ERα promoter, enhancing histone acetylation at this locus, and activating ERα gene expression by altering the chromatin structure.6 In fact, MYST3 mutants MYST3G657E and MYST3C543G/G657E, which are HAT activity deficient, lose their ability to regulate ERα expression (Figure 3c). This further supports our model in which MYST3 may regulate the ERα promoter via histone modification at this locus. Currently, it is not clear how MYST3 recognizes and binds to the ERα promoter region. It is possible that other cofactors are involved in directing MYST3 specifically to the ERα promoter region. We also could not turn on ERα expression in two MYST3-low/ER- breast cancer cells, MDA-MB-231 and SK-BR-3 (data not shown). Epigenetic status of ERα promoter region may prevent the binding of MYST3. Furthermore, many MYST family proteins, such as MYST1/MOF,33 MYST2/HBO134 and TIP60,35 as well as other histone acetyltransferases, p300/CBP,36 have been reported to function as ERα coactivators to regulate the expression of ERα target genes. p300/CBP also acetylates ERα protein at different regions with distinct functions.37, 38 It is possible that MYST3 might involve these processes to affect estrogen receptor function besides it acts at the level of activation of ESR1 gene expression.
Analyses of available patient data sets show that high MYST3 levels predict a worse disease-free survival in ER+/HER2− breast cancer patients (Figure 1e). Furthermore, our animal studies showed that targeting MYST3 attenuates breast tumor growth. Notably, MYST3 depletion results in an inhibitory effect only in MYST3-high cells, but not in normal cells or MYST3-low cells (Supplementary Figure 1B). In line with our finding, MYST3 haploinsufficiency retards MYC-induced lymphoma development in a mouse model.29 As an enzyme, MYST3 is expected to be druggable and small molecular compounds capable of blocking the enzymatic activity of MYST3 may be developed in the future based on the available MYST3 crystal structure.39 All of these features make MYST3 an attractive cancer therapeutic target. In clinic, targeting ER with endocrine therapy is an effective treatment for ER+ breast cancer. However, de novo and acquired resistance to endocrine therapies are major challenges for treating ER+ breast cancer with a significant portion of ER+ breast tumors treated with endocrine therapy eventually relapsed.3 Interestingly, in our study, high MYST3 levels also predicted worse survival outcome for ER+/HER2− patients treated with endocrine therapy (Figure 1d). Thus, targeting MYST3 may be an alternative strategy to treat MYST3-high ER+/HER2− breast cancers in addition to endocrine therapy. Further targeting MYST3 not only blocks ERα pathways, but also blocks other MYST3-mediated oncogenic pathways. For example, recent genome-wide analysis suggested that IGF1R and IRS1 may also be potential targets for MYST3, although these targets are not yet validated.24 Activation of IGF1R or IRS1 has also been shown to contribute to therapeutic resistance in breast cancer. Overall, compared with hormone therapy targeting ERα alone, dual targeting of ERα and MYST3 may achieve a better therapeutic effect in MYST3-high ER+/HER2− breast tumors.
References
Ignatiadis M, Sotiriou C . Luminal breast cancer: from biology to treatment. Nat Rev Clin Oncol 2013; 10: 494–506.
Musgrove EA, Sutherland RL . Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 2009; 9: 631–643.
Osborne CK, Schiff R . Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011; 62: 233–247.
Thomas C, Gustafsson JA . The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer 2011; 11: 597–608.
Wang XS, Prensner JR, Chen G, Cao Q, Han B, Dhanasekaran SM et al. An integrative approach to reveal driver gene fusions from paired-end sequencing data in cancer. Nat Biotechnol 2009; 27: 1005–1011.
Yang XJ, Ullah M . MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 2007; 26: 5408–5419.
Avvakumov N, Cote J . The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007; 26: 5395–5407.
Bristow CA, Shore P . Transcriptional regulation of the human MIP-1alpha promoter by RUNX1 and MOZ. Nucleic Acids Res 2003; 31: 2735–2744.
Collins HM, Kindle KB, Matsuda S, Ryan C, Troke PJ, Kalkhoven E et al. MOZ-TIF2 alters cofactor recruitment and histone modification at the RARbeta2 promoter: differential effects of MOZ fusion proteins on CBP- and MOZ-dependent activators. J Biol Chem 2006; 281: 17124–17133.
Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet 1996; 14: 33–41.
Esteyries S, Perot C, Adelaide J, Imbert M, Lagarde A, Pautas C et al. NCOA3, a new fusion partner for MOZ/MYST3 in M5 acute myeloid leukemia. Leukemia 2008; 22: 663–665.
Carlson S, Glass KC . The MOZ histone acetyltransferase in epigenetic signaling and disease. J Cell Physiol 2014; 229: 1571–1574.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70.
Chen X, Ji ZL, Chen YZ . TTD: therapeutic target database. Nucleic Acids Res 2002; 30: 412–415.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S . The protein kinase complement of the human genome. Science 2002; 298: 1912–1934.
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006; 34: D668–D672.
Bernard-Pierrot I, Gruel N, Stransky N, Vincent-Salomon A, Reyal F, Raynal V et al. Characterization of the recurrent 8p11-12 amplicon identifies PPAPDC1B, a phosphatase protein, as a new therapeutic target in breast cancer. Cancer Res 2008; 68: 7165–7175.
Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS . A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 2010; 10: 59–64.
Agochiya M, Brunton VG, Owens DW, Parkinson EK, Paraskeva C, Keith WN et al. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene 1999; 18: 5646–5653.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486: 346–352.
Paggetti J, Largeot A, Aucagne R, Jacquel A, Lagrange B, Yang XJ et al. Crosstalk between leukemia-associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34+ cells. Oncogene 2010; 29: 5019–5031.
Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 2003; 3: 259–271.
Ellison-Zelski SJ, Solodin NM, Alarid ET . Repression of ESR1 through actions of estrogen receptor alpha and Sin3A at the proximal promoter. Mol Cell Biol 2009; 29: 4949–4958.
Turner-Ivey B, Guest ST, Irish JC, Kappler CS, Garrett-Mayer E, Wilson RC et al. KAT6A, a chromatin modifier from the 8p11-p12 amplicon is a candidate oncogene in luminal breast cancer. Neoplasia 2014; 16: 644–655.
Yang ZQ, Albertson D, Ethier SP . Genomic organization of the 8p11-p12 amplicon in three breast cancer cell lines. Cancer Genet Cytogenet 2004; 155: 57–62.
Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J et al. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene 2009; 28: 1892–1903.
Yang ZQ, Streicher KL, Ray ME, Abrams J, Ethier SP . Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res 2006; 66: 11632–11643.
Perez-Campo FM, Costa G, Lie ALM, Stifani S, Kouskoff V, Lacaud G . MOZ-mediated repression of p16(INK) (4) (a) is critical for the self-renewal of neural and hematopoietic stem cells. Stem Cells 2014; 32: 1591–1601.
Sheikh BN, Phipson B, El-Saafin F, Vanyai HK, Downer NL, Bird MJ et al. MOZ (MYST3, KAT6A) inhibits senescence via the INK4A-ARF pathway. Oncogene 2015; 34: 5807–5820.
Hervouet E, Cartron PF, Jouvenot M, Delage-Mourroux R . Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013; 8: 237–245.
Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res 2014; 24: 809–819.
Voss AK, Collin C, Dixon MP, Thomas T . Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev Cell 2009; 17: 674–686.
Zhang X, Peng D, Xi Y, Yuan C, Sagum CA, Klein BJ et al. G9a-mediated methylation of ERalpha links the PHF20/MOF histone acetyltransferase complex to hormonal gene expression. Nat Commun 2016; 7: 10810.
Georgiakaki M, Chabbert-Buffet N, Dasen B, Meduri G, Wenk S, Rajhi L et al. Ligand-controlled interaction of histone acetyltransferase binding to ORC-1 (HBO1) with the N-terminal transactivating domain of progesterone receptor induces steroid receptor coactivator 1-dependent coactivation of transcription. Mol Endocrinol 2006; 20: 2122–2140.
Jeong KW, Kim K, Situ AJ, Ulmer TS, An W, Stallcup MR . Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol 2011; 18: 1358–1365.
Kim MY, Hsiao SJ, Kraus WL . A role for coactivators and histone acetylation in estrogen receptor alpha-mediated transcription initiation. EMBO J 2001; 20: 6084–6094.
Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL . Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol 2006; 20: 1479–1493.
Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem 2001; 276: 18375–18383.
The crystal structure of human MYST histone acetyltransferase 3 in complex with acetylcoenzyme A. Available at http://www.rcsborg/pdb/explore/exploredo?structureId=2OZU.
Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515–527.
Debnath J, Muthuswamy SK, Brugge JS . Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30: 256–268.
Acknowledgements
This work was supported by grants from the National Institutes of Health (KL, CA161513, CA155151; X-SW, CA181368 & CA183976), a Pilot Project Grant from the Dan L. Duncan Cancer Center at Baylor College of Medicine (KL), Susan G. Komen foundation PDF15333523 (XW), CDMRP W81XWH-13-1-0201(X-XC). This study was partly supported by the Susan G. Komen for the cure grants Promise grant PG12221410 (RS), NIH SPORE Grants P50 CA058183 (RS) and CA186784-01 (RS), and Cancer Center Grant P30CA125123 (RS), Breast Cancer Research Foundation (RS), Cancer Prevention Research Institute of Texas (RS, CPRIT RP140102) and SYL, CRPIT RP130310. We also thank Dr. Ana Rodríguez from the Research Core Team at Michael E. DeBakey Department of Surgery, Baylor College of Medicine for editing of this manuscript. The results published here are in part based upon data generated by TCGA (dbGaP accession: phs000178.v6.p6). This study also makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium (funding for that project was provided by Cancer Research UK and the British Columbia Cancer Agency Branch).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yu, L., Liang, Y., Cao, X. et al. Identification of MYST3 as a novel epigenetic activator of ERα frequently amplified in breast cancer. Oncogene 36, 2910–2918 (2017). https://doi.org/10.1038/onc.2016.433
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.433
This article is cited by
-
Matrix stiffness-induced upregulation of histone acetyltransferase KAT6A promotes hepatocellular carcinoma progression through regulating SOX2 expression
British Journal of Cancer (2022)
-
miR-339-3p inhibits cell growth and epithelial–mesenchymal transition in nasopharyngeal carcinoma by modulating the KAT6A/TRIM24 axis
International Journal of Clinical Oncology (2022)
-
Targeting Histone Modifications in Bone and Lung Metastatic Cancers
Current Osteoporosis Reports (2021)
-
Histone methyltransferases regulate the transcriptional expression of ERα and the proliferation of tamoxifen-resistant breast cancer cells
Breast Cancer Research and Treatment (2020)
-
Mechanisms of resistance to estrogen receptor modulators in ER+/HER2− advanced breast cancer
Cellular and Molecular Life Sciences (2020)

{kind=link}
{kind=link}
{kind=link}