
Abstract
In this study, loss of expression of the fragile site-encoded Wwox protein was found to contribute to radiation and cisplatin resistance of cells, responses that could be associated with cancer recurrence and poor outcome. WWOX gene deletions occur in a variety of human cancer types, and reduced Wwox protein expression can be detected early during cancer development. We found that Wwox loss is followed by mild chromosome instability in genomes of mouse embryo fibroblast cells from Wwox-knockout mice. Human and mouse cells deficient for Wwox also exhibit significantly enhanced survival of ionizing radiation and bleomycin treatment, agents that induce double-strand breaks (DSBs). Cancer cells that survive radiation recur more rapidly in a xenograft model of irradiated breast cancer cells; Wwox-deficient cells exhibited significantly shorter tumor latencies vs Wwox-expressing cells. This Wwox effect has important consequences in human disease: in a cohort of cancer patients treated with radiation, Wwox deficiency significantly correlated with shorter overall survival times. In examining mechanisms underlying Wwox-dependent survival differences, we found that Wwox-deficient cells exhibit enhanced homology directed repair (HDR) and decreased non-homologous end-joining (NHEJ) repair, suggesting that Wwox contributes to DNA DSB repair pathway choice. Upon silencing of Rad51, a protein critical for HDR, Wwox-deficient cells were resensitized to radiation. We also demonstrated interaction of Wwox with Brca1, a driver of HDR, and show via immunofluorescent detection of repair proteins at ionizing radiation-induced DNA damage foci that Wwox expression suppresses DSB repair at the end-resection step of HDR. We propose a genome caretaker function for WWOX, in which Brca1–Wwox interaction supports NHEJ as the dominant DSB repair pathway in Wwox-sufficient cells. Taken together, the experimental results suggest that reduced Wwox expression, a common occurrence in cancers, dysregulates DSB repair, enhancing efficiency of likely mutagenic repair, and enabling radiation and cisplatin treatment resistance.
Similar content being viewed by others


Introduction
The FRA16D locus at 16q23 is a common fragile site that forms gaps or breaks upon replication stress and is altered in many human cancers, showing up to 52% loss of heterozygosity in breast tumors.1 The 1.2 Mb WWOX gene is located within this fragile region,2 and reduced Wwox protein expression has been correlated with breast cancer progression and poor prognosis.1 Mouse models confirm a tumor suppressor function for Wwox, as complete knockout mice on various genetic backgrounds was postnatal lethal.1, 3 In addition, Wwox haploinsufficient mice, with a mammary tumor-susceptible C3H background, exhibit increased mammary gland tumorigenicity4 and re-expression of Wwox decreases tumor burden in ovarian and breast cancer xenografts.5, 6
Recently, FRA16D has been identified as the most active common fragile site in epithelial cells7 and large cancer copy number alteration studies identified WWOX as the third most commonly deleted gene across human cancers.8, 9 Despite many lines of evidence that suggest a role for loss of Wwox in the progression of cancer,10 our understanding of Wwox tumor-suppressive function is incomplete. Wwox protein exhibits distinct structural domains, two adjacent N-terminal WW protein-binding domains and a short-chain dehydrogenase/reductase (SDR) domain in its central region believed to have a role in steroid hormone metabolism2, 11 and cellular respiration.12 The first WW domain binds proteins with a PPxY or PPxL motif and many interactors have been identified, including p73, Ap2γ, ErbB-4, Smad3 and ATM,5, 13, 14, 15, 16 associated with induction of apoptosis, regulation of proliferation, interaction with extracellular matrix and checkpoint activation. Wwox functional interaction with these proteins has provided thus far the main clues for the role of Wwox in the prevention of cancer development.
In this study, we further examined the role of Wwox deficiency in cancer progression, specifically in the context of DNA damage. Our initial observation that Wwox-knockout mouse embryonic fibroblasts (MEFs) showed increased allele copy number gains and losses relative to wild-type MEFs led us to hypothesize that Wwox protein exhibits a genome caretaker function, suppressing mutations. We show that loss of Wwox expression significantly alters usage of all four double-strand break (DSB) repair pathways, suggesting a role for Wwox in pathway choice. Enhanced homology directed repair (HDR) in Wwox-deficient cells permits significantly increased resistance to cisplatin and radiation treatment-associated cell killing, a finding with clinical implications. Finally, we delineate a caretaker function for Wwox in DSB repair pathway choice, and propose its dependence on interaction with Brca1 protein.
Results
Wwox-knockout MEFs exhibit chromosomal alterations and shared CNVs
To determine if Wwox contributes to genome stability, karyotype and copy number variation (CNV) analyses were carried out using Wwox-knockout and -wild-type MEF cell lines. Karyotype analysis of three cell lines (two knockout and one wild type, established from Wwox-knockout mouse models described previously3, 17) showed near tetraploidy with chromosomal losses and gains (Supplementary Figure 1). Both knockout MEFs exhibited two structural abnormalities not present in the wild-type cell line: del(7)(A1B4) and del(4)(C4). Loss of the distal arm of chromosome 4 encompasses the murine Cdkn2a gene, encoding p16, a locus frequently deleted in human cancers and in cultured cells.18 Hemizygous del(7)A1B4 encompasses 48.5 Mb and >650 genes.
CNVs, defined as allele gains or losses spanning >10 kb in size, were assessed in DNAs of MEFs from two distinct Wwox-knockout mouse models and compared with DNAs from corresponding wild-type littermates (cell line pairs, KO5/WT4 from Wwox-knockout17 and Wwox5/Wwox3 from a different Wwox-knockout mouse model3). CNVs were detected through array comparative genomic hybridization. Three distinct deletions were shared by the two knockout MEF lines at chromosome locations 1 H6, 4 B3 and 8 C2 (Tables 1a and b).
The karyotype and CNV results suggest that absence of Wwox protein is associated with mild genome instability, likely stemming from endogenous DNA damage, prompting our hypothesis that Wwox participates in protecting the genome from damage. We first considered whether Wwox loss leads to uninduced genome damage (i.e., damage not due to exposure to exogenous cytotoxic agent), but the comet assay, which detects DSBs as comet tails, and immunofluorescence assays for DSB markers, 53BP1 and γH2AX, did not reveal differences in untreated Wwox-deficient and -expressing cells (Supplementary Figure 2).
Wwox-deficient cells exhibit increased survival of ionizing radiation-induced DSBs
To determine if Wwox expression may affect repair of induced DSBs, we investigated the effects of ionizing radiation (IR) on various Wwox-deficient vs -sufficient cells. Early-passage MEFs were exposed to increasing IR doses up to 10 Gy, and plated for clonogenicity to quantify cell survival and proliferation. We observed a significant difference (P<0.01) in survival at doses 7.7 Gy and above (Figure 1a), with knockout lines KO3 and KO5 surviving 10-fold better than wild-type cell lines WT4 and WT7. Similarly, survival curves for transformed breast epithelial cell line, MCF10A clones stably transduced with Wwox silencing shRNAs, shWWOXA and shWWOXB,15 demonstrated increased survival at 7.7 Gy and above (P<0.05) vs the Wwox-sufficient Scr RNA-transfected cells (Figure 1b). To determine if Wwox loss has a similar effect in cancer cells, we used MDA MB-231 breast adenocarcinoma cells lacking endogenous Wwox expression, which had been stably transduced with a doxycycline-inducible Wwox expression vector.15 Wwox-induced cells that were treated with doxycycline for 24 h and were positive for Wwox are denoted as 231/Wwox-pos throughout the manuscript, whereas these Wwox-deficient cells untreated with doxycycline are referred to as 231/Wwox-neg. Clonogenicity assays of these cells showed that induced Wwox expression led to reduced survival at 5.1 Gy (Figure 1c; P<0.05), the highest dose at which 231/Wwox-pos cells formed colonies. The shWWOXA and shWWOXB cells were also treated with the radiomimetic, bleomycin, a glycopeptide antitumor antibiotic that induces DSBs, and strikingly significant differences in survival were observed; the Wwox-silenced cells survived bleomycin treatments 25-fold better at 1 and 1.5 μg/ml bleomycin and 100-fold better than Wwox-expressing MCF10A cells at 3 μg/ml bleomycin (P<0.01) (Figure 1d). To confirm that enhanced survival was because of the absence of Wwox, we established clones KO3A and KO5F from the knockout MEF cell lines KO3 and KO5, which were doxycycline inducible for the expression of Wwox. Upon Wwox induction, both clones exhibited significantly decreased survival (P<0.05) at 7.7 Gy and above (Figures 1e and f), confirming that Wwox expression sensitizes cells to radiation.

Human and mouse Wwox-deficient cells exhibit increased survival to DSBs. Wwox-deficient cell lines are represented in red or orange, Wwox-sufficient cell lines are represented in blue or green. (a) Graph depicting % survival of MEF cell lines, Wwox knockout (KO3, KO5) and wild type (WT4, WT7), following IR treatment. (b) Graph depicting % survival in the transformed breast epithelial MCF10 cells and Wwox stably silenced MCF10A clones, shWWOXA and shWWOXB, following increasing IR doses. (c) Graph depicting % survival in doxycycline-inducible MDA MB-231 cells following IR treatment. (d) Graph depicting % survival in MCF10A cells and shWWOXA and shWWOXB clones following bleomycin treatment. (e) Graph depicting % survival of doxycycline-inducible KO3 clone, 2A2, with and without doxycycline after IR treatment. (f) Graph depicting % survival of doxycycline-inducible KO5 clone, 2F2, with and without doxycycline after various IR doses. (a–f) Embedded western blots (WBs) demonstrate Wwox expression in the cell lines. Experiments were performed in triplicate. Error bars denote 1 s.d.; lower standard error bars are abbreviated due to log scale.
Wwox expression inhibits growth of irradiated cancer cells in vivo
To determine if the IR resistance of Wwox-deficient cells persists in vivo, Wwox-induced (231/Wwox-pos) and Wwox-deficient (231/Wwox-neg) breast cancer cells were tested for tumor growth in immunocompromised mice. The rationale was that despite irradiating the same number of cells for the two groups, more 231/Wwox-neg cells would survive IR, forming tumors more quickly than 231/Wwox-pos cells (Figure 2a). Both groups of cells were exposed to 5 Gy IR, immediately harvested and injected (1 × 107 cells per mouse) subcutaneously into the flanks of athymic nude mice. Control mice from each group were unexposed to IR and exhibited mean tumor latencies (days from injection to first sign of tumor) that were not different: 11 days for 231/Wwox-neg cells and 14 days for 231/Wwox-pos cells. For mice receiving irradiated cells, 7/8 mice injected with 231/Wwox-neg cells formed tumors, whereas 6/8 mice injected with 231/Wwox-pos cells formed tumors. Of the tumor-bearing mice, those injected with 231/Wwox-neg cells had significantly shorter tumor latencies (mean 17 days) vs mice receiving 231/Wwox-pos cells (mean 28 days) (Figure 2b; P<0.05).

Wwox expression inhibits growth of irradiated cancer cells in vivo in mouse and human models. Wwox-deficient cells are depicted as red and Wwox-sufficient cells are depicted as blue. (a) Illustration of experimental rationale: on injection of equal cell numbers, it is expected that more Wwox-deficient cells will survive IR and therefore form earlier tumors. (b) Box plot depicting tumor latency (days from injection to sign of first tumor) of mice which developed tumors following injection of Wwox-deficient or Wwox-expressing IR and non-IR-treated 231/Wwox cells. Tumors developing in mice injected with 231/Wwox-neg cells exhibited significantly shorter latencies (P<0.05) following exposure to 5 Gy IR. (c) Kaplan–Meier plot of all brain cancer patients indicating no significant difference in overall survival (months) of brain cancer patients stratified by Wwox expression. (d) Kaplan–Meier plot of only brain cancer patients treated with radiation, demonstrating a significantly shorter survival (P<0.01) of Wwox-deficient brain cancer patients compared with Wwox-sufficient brain cancer patients upon treatment with radiation.
Next, we sought a human cancer database with expression and clinical data for cancers treated with radiation to determine if loss of Wwox enabled tumor cell resistance to radiation and decreased overall patient survival in a human model. As we did not find publicly available breast cancer databases with sufficient clinical data or patients treated with radiation, we examined brain cancers via the Repository of Molecular Brain Neoplasia Data (REMBRANDT).19 Stratification of patients by Wwox expression did not predict overall survival in a large cohort of brain cancer patients (Figure 3c); however, in cancers treated with radiation, reduced Wwox expression correlated significantly with decreased overall survival vs Wwox normal cancers (Figure 3d), suggesting that loss of Wwox facilitates resistance to radiation therapy, disease recurrence and shorter overall survival. Collectively, results of the xenograft model and the brain cancer assessment confirm that Wwox loss supports radiation resistance in vivo and that Wwox deficiency provides a survival advantage to cancer cells carrying DSBs.

Wwox-deficient cells exhibit increased homologous recombination, which is responsible for enhanced IR survival. In bar charts, Wwox-deficient cells are depicted as black and Wwox-sufficient cells are depicted as white. Western blot (WBs) below bar charts demonstrate transfection efficiency and Wwox expression. (a) Bar graph indicating % of cells positive for GFP as indication of relative HR, with corresponding immunoblots of U87 cells carrying HR reporter, DR-GFP, and transfected with siCtrl, siWwox and siWwox+myc-Wwox plasmid resistant to silencing. (b) Graph indicating % GFP-pos HeLa cells DR-GFP transfected with empty vector or Myc-Wwox. (c) Bar graph indicating relative mRNA as a measure of C-NHEJ in H1299 cells carrying pHW1 and transfected with siCtrl and siWwox. (d) Bar graph depicting fraction of GFP-pos cells in H1299 EJ2 cells indicating relative Alt-NHEJ efficiencies following transfection with siCtrl, siWwox+myc-Wwox and siWwox. (e) Bar graph indicating % of GFP-pos cells as a measure of SSA in HeLA Sa26 cells following transfection with empty vector and myc-Wwox. (f) Schematic of the four DSB repair pathways: NHEJ, HDR, SSA and Alt-NHEJ and corresponding changes in efficiency dependent on Wwox expression (right). (g) Line graph indicating % survival of MEFs with and without Rad51 silencing (denoted as R51 in embedded WBs) following exposure to designated IR doses. Solid lines indicate survival of MEFs with normal Rad51 expression, whereas dashed lines indicate Rad51-silenced cells. (h) Line graph depicting % survival of 231/Wwox cells with and without doxycycline following exposure to various levels of cisplatin for 2 h. (i) Line graph depicting % survival of MEFs following exposure to various levels of cisplatin for 2 h. (a–e and g–i) Data from three independent experiments were subjected to two-tailed Student’s t-test.
Wwox expression regulates DSB repair pathway choice
To elucidate the mechanism that underlies Wwox deficiency associated radiation resistance, we hypothesized that loss of Wwox could enhance DSB repair. Although there are four DSB repair pathways, two pathways predominate: non-homologous end-joining (NHEJ), which bluntly re-ligates DSB ends independent of sequence homology, or HDR, which prevails in S and G2 cell cycle phases and uses sister chromatids as templates for accurate DSB repair.20 To assess the HDR pathway in the presence or absence of Wwox, we used the direct repeat-green fluorescent protein (DR-GFP) reporter construct21 that has been stably integrated into HeLa cells22 and U87 cells.23 Repair of the DR-GFP construct via HDR is measured as the fraction of GFP-positive cells. Wwox-sufficient U87 cells transfected with siWwox and I-SceI revealed a 2.5-fold increase in HDR (P<0.01) vs control cells transfected with siCtrl and I-SceI (Figure 3a). To confirm Wwox specificity, we performed a rescue experiment in the U87 DR-GFP cells by transfecting siWwox (directed toward Wwox 5′-untranslated region) simultaneously with a full-length Wwox expression plasmid, myc-Wwox.13 When Wwox expression levels were restored in the presence of siWwox, we observed decreased HDR, similar to HDR efficiency of Wwox-sufficient cells (Figure 3a). As further confirmation that Wwox deficiency increases HDR efficiency, transfection of endogenous Wwox-deficient HeLa DR-GFP cells with myc-Wwox resulted in a significant decrease (P<0.05) in HDR (Figure 3b). Thus, the DR-GFP assays consistently showed that Wwox expression suppressed HDR.
As HDR and NHEJ have both been implicated in resistance to radiation,24, 25 we next assessed the contribution of Wwox to NHEJ efficiency using 293/HW1 cells. Following induction of DSBs and silencing of Wwox, NHEJ is measured by real-time PCR using a probe that spans the junction of the break sites.26 In 293/HW1 cells, silencing Wwox significantly decreased NHEJ by 50% (P<0.05) (Figure 3c), indicating that, unlike HDR, Wwox expression enhanced NHEJ.
As Wwox expression significantly altered the efficiencies of the predominant DSB repair pathways HDR and NHEJ, we also sought to determine the consequences of Wwox loss on the less common, more error-prone pathways: alternative NHEJ (Alt-NHEJ) and single-strand annealing (SSA). Both are mutagenic and result in deletions initiated by 5′ to 3′ resection from the DSB site up to microhomologous regions.27 Alt-NHEJ efficiency was assessed in H1299 EJ2 cells28 via restoration of GFP fluorescence following I-SceI transfection. We observed a marked, statistically significant (P<0.0001) decrease in Alt-NHEJ (Figure 3d) upon transfection with siWwox and I-SceI. This effect was confirmed in a rescue experiment where siWwox and myc-Wwox simultaneously co-transfected with I-SceI restored Wwox expression and significantly increased Alt-NHEJ (P<0.001), indicating that Wwox expression significantly enhanced Alt-NHEJ. To evaluate the effects of Wwox on SSA repair, Wwox-deficient HeLa Sa26 cells29 were transfected with I-SceI plus either empty vector or myc-Wwox (Figure 3e) and % GFP-positive cells were assessed. Upon Wwox expression, SSA in HeLa Sa26 cells was markedly repressed (P<0.01) to less than one-third, demonstrating that Wwox expression significantly repressed SSA. Altogether, the data indicate that Wwox expression significantly alters repair efficiency for all four DSB repair pathways (summarized in Figure 3f) such that Wwox expression enhances NHEJ and Alt-NHEJ, but impairs HDR and SSA.
Enhanced HDR in Wwox-deficient cells results in resistance to radiation and cisplatin
Although HDR and SSA repair are both elevated in Wwox-deficient cells, we hypothesized that HDR was responsible for the observed radiation resistance. To verify that Wwox-deficient cells survive IR-induced DSBs because of enhanced HDR, we inhibited HDR via transient knockdown of Rad51, a critical protein for HDR-specific strand invasion. As expected, knockout MEFs transfected with siCtrl RNAs (solid lines, Figure 3g) exhibited significantly increased survival (P<0.001) compared with wild-type MEFs, but following Rad51 depletion (dashed lines), knockout MEFs had survival curves similar to wild-type cells. Thus, HDR inhibition significantly decreased survival after radiation (P<0.001) only for Wwox-knockout MEFs, confirming that increased HDR underlies the radiation resistance of Wwox-deficient cells.
As alterations in DSB repair pathway efficiencies would affect responses to DNA-damaging chemotherapeutic agents, we next investigated the effect of Wwox expression on sensitivity to cisplatin, a crosslinking chemotherapeutic agent that causes DSBs. We hypothesized that Wwox-deficient cells would be more resistant to cisplatin vs Wwox-sufficient cells because their enhanced HDR would resolve DSBs induced by cisplatin more efficiently. Indeed, treatment of 231/Wwox-neg cells with various doses of cisplatin revealed enhanced survival to cisplatin treatment at 10 μm in 231/Wwox-neg vs 231/Wwox-pos cells (P<0.01) and at higher doses (Figure 3h). Similarly, knockout MEFs survive cisplatin at 25 μm (P<0.001) and higher concentrations vs WT4 MEF (Figure 3i). Thus, the dysregulation of DSB repair pathways and subsequent enhanced HDR associated with Wwox deficiency results in cellular resistance to the commonly used breast cancer therapeutic agents: radiation and cisplatin. Given that cellular loss of Wwox encourages therapy resistance, we next sought clues to the mechanism involved in Wwox protein suppression of HDR.
Cell cycle phase distribution in IR-treated Wwox-pos and -neg cells
As HDR is thought to occur mainly in S and G2 phases of the cell cycle where a sister chromatid is available as repair template,20 we considered whether cell cycle phase distribution and checkpoint activation differences might contribute to increased HDR and radiation resistance in Wwox-deficient cells. Flow cytometric analysis in 231/Wwox cells, regardless of induced Wwox expression, showed nearly identical cell cycle phase distributions (Figure 4a), with accumulation in G2/M by 10 h postirradiation (Figure 4b), although 231/Wwox-neg cells appeared to have stronger activation of pChk2 and pChk1 (Supplementary Figure 3). Similar experiments in early-passage (~p10) Wwox-knockout and -wild-type MEFs revealed differences for the wild-type and knockout cells in cell cycle phase distribution (Figure 4c) and checkpoint activation (Figure 4d), neither of which activities showed correlation with the responses of the cells to IR (Supplementary Figure 4). Results of the cell cycle analyses of MEFs and 231/Wwox cells demonstrated that the differences in cell cycle kinetics among various Wwox-pos and -neg cells did not account for the differences in responses to IR exposure.

Cell cycle characterization in Wwox MEFs and 231/Wwox cells after IR. (a) Stacked column graph depicting cell cycle distribution of 231/Wwox cells with and without doxycycline at designated time points after 10 Gy IR. Bottom panel depicts corresponding protein expression with darker shades of blue indicating increasing intensity. (b) Representative images of flow cytometric DNA content analysis in 231/Wwox cells with and without doxycycline at 0, 1, 10 and 24 h after 10 Gy IR. (c) Stacked column graph depicting cell cycle distribution in wild-type and knockout MEFs at designated time points after 10 Gy IR. Bottom panel depicts corresponding protein expression with darker shades of blue indicating increasing intensity of checkpoint proteins. (d) Representative flow cytometric analysis of DNA content in WT7, KO3 and KO5 at 0, 1, 10 and 24 h after 10 Gy IR. (a, b) The 231/Wwox cells were untreated or induced for Wwox expression by treatment with 1 μg/ml doxycycline for 24 h before IR treatment and harvesting for flow cytometry, as described.41
Brca1 and Wwox proteins form a complex
Given the multiple protein interactors for Wwox, we hypothesized that Wwox interaction with a protein(s) in the HDR pathway might underlie its role in DSB repair pathway choice. Brca1 was a candidate interactor as it contains a PPxL Wwox-binding motif at amino acid 981 and promotes HDR over NHEJ repair by initiating DSB end resection.30 To investigate a potential interaction between Wwox and Brca1, we performed GST pulldown experiments using GST fused to various Wwox constructs in human embryonic kidney 293T (HEK293T) cells transfected with hemagglutinin-Brca1 (HA-Brca1) (Figure 5a). A schematic of the Wwox domains is presented in Figure 5c. GST fused to wild-type full-length Wwox bound HA-Brca1 and was detected by anti-HA antibody (Figure 5a, lane 7). Similarly, GST fused to the WW domain fragment (WW1 and 2, lane 3) bound HA-Brca1; however, the WW domain fragment with mutated WW1 (W44F/P47A),31 which renders the binding domain non-functional, did not bind HA-Brca1 (lane 4), suggesting that Wwox interaction with Brca1 occurs through the WW1 domain. In support of this, a WW domain with mutated WW2 (Y85A/P88A in lane 5) pulls down HA-Brca1, indicating that WW2 is not essential for the interaction; finally, a GST-fused SDR fragment did not bind HA-Brca1 (lane 6) and does not contain either WW domain.

Brca1 and Wwox interact in a complex through Brca1 central region. (a) Western blot (WB) and corresponding Coomassie blue staining of GST pulldown using GST, and GST-Wwox WW1/WW2 fragment, GST-Wwox WW1/WW2 fragment with mutation in WW1 domain, GST-Wwox WW1/WW2 fragment with mutation in WW2 domain, GST-Wwox SDR fragment and GST-Wwox (full-length) to IP HA-Brca1 following transfection in HEK293T cells. GST-Wwox with WW1 mutation and GST-Wwox SDR fragment do not IP HA-Brca1, suggesting that Wwox interacts with Brca1 through its first WW domain. (b) WB of HEK293T cells transfected with HA-Brca1 plus myc-Wwox and corresponding IP using IgG and HA (Covance) antibodies as bait. (c) Schematic of Wwox domain. (d) Schematic of Brca1 domain and deletion mutants. (e) WB and corresponding HA (SCBT) co-IP of MDA MB-231 cells following transfection with ΔN, ΔM1, ΔM2, ΔM3 and ΔC Brca1 deletion mutants as well as myc-Wwox. Brca1 deletion mutants are detected by polyclonal anti-Brca137 directed against Brca1 amino acids 400–1100, resulting in weaker detection of ΔM1 and ΔM2 mutants. (f) Graph depicting % survival of doxycycline-inducible 231 cells that express Wwox WW1 mutant with (231/WW1 mut-pos) and without (231/WW1 mut-neg) doxycycline after increasing IR doses. (g) Graph depicting % survival of doxycycline-inducible 231 cells that express Wwox SDR mutant with (231/SDR mut-pos) and without (231/SDR mut-neg) doxycycline after increasing IR doses.
To confirm the Brca1–Wwox interaction, we transfected HEK293T cells with HA-Brca1 plus full-length, myc-tagged Wwox plasmid (myc-Wwox) and performed immunoprecipitation using anti-HA and detection with anti-Brca1 (Figure 5b). HA-Brca1 pulled down myc-Wwox (detected by anti-Wwox), whereas immunoglobulin G (IgG) immunoprecipitation did not detectably bind either protein.
Next, to determine which Brca1 domain was participating in the Wwox interaction, we transfected five Brca1 mutants, harboring deletions from the N to C terminus (Figure 5d), into MDA MB-231 cells with myc-Wwox. The lysates were immunoprecipitated with anti-HA to avoid detecting interactions because of the low-level endogenous Brca1 protein expressed in the MB-231 cells. The HA antibody precipitated Wwox in lysates harboring Brca1 deletions at the N, M3 and C regions (lanes 6, 9 and 10; Figure 5e) but not in lysates expressing Brca1 with the M1- and M2-deleted regions (lanes 7 and 8; Figure 5e). These interactions were confirmed in the opposite direction, in HEK293T cells transfected with myc-Wwox plus each of the Brca1 deletion mutants (Supplementary Figure 5). Immunoprecipitation using Myc antibody as bait revealed that all Brca1 deletion mutants except M1 and M2 detectably bound myc-Wwox (lanes 6–10; Supplementary Figure 5), suggesting that sequences within the Brca1 amino-acid 305–1292 region are responsible for the Wwox–Brca1 interaction.
Wwox interaction with Brca1 through the WW domain is responsible for decreased survival of DSBs
To confirm whether interaction with Brca1 through the WW domain is responsible for suppression of HDR and sensitivity to DSBs, the MDA MB-231 cells were stably transfected with doxycycline-inducible full-length Wwox harboring WW1 mutations31 W44F/P47A (231/WW1 mut) or full-length Wwox harboring a mutation (Y293F) at the catalytic SDR site (231/SDR mut) and performed clonogenicity assays following increasing doses of IR. These cells are denoted as 231/WW1 mut-pos and 231/SDR mut-pos when treated with doxycycline for 24 h and expressing the full-length Wwox with WW1 or SDR mutation. Conversely, 231/WW1 mut-neg and 231/SDR mut-neg cells express only very low levels of endogenous Wwox. We inferred that if Wwox interaction with Brca1 was critical for suppression of HDR and sensitivity to DSBs, the cells expressing Wwox WW1 mutant would show no difference in IR survival with or without induction, that is, they would both respond as if Wwox-neg, whereas cells induced to express the Wwox SDR mutant, which maintains a functional WW1 domain, would be IR-sensitive. Supplementary Figure 6b demonstrates the inducibility of Wwox expression in these cell lines. As predicted, Figure 5f shows that 231 cells expressing a non-functional WW1 mutant Wwox protein (231/WW1 mut-pos) exhibit an IR survival curve similar to the same cells uninduced for Wwox (231/WW1 mut-neg). This suggests that without a functional WW1 domain, Wwox does not sensitize cells to radiation. Conversely, upon expression of full-length Wwox harboring an SDR mutant, the cells exhibit significantly decreased survival relative to the same cells uninduced (Figure 5g); that is, the SDR mutant Wwox-expressing cells are sensitive to radiation because they retain a WW1 domain, which interacts with Brca1.
Loss of Wwox expression leads to significant increases in radiation-induced nuclear Rpa32 and Rad51 foci
As Brca1 forms complexes comprised of varying interacting proteins and these complexes have distinct functions during DSB recognition and repair, we sought to understand how Wwox–Brca1 might enhance NHEJ and suppress HDR, through immunofluorescent detection of critical DSB repair-associated proteins: γH2AX, 53BP1, RIF1, Brca1, Rad51 and Rpa32 (Figure 6). These proteins form foci at DSBs upon irradiation and are involved in DSB recognition, repair pathway choice and end resection (Figures 7a and b). Briefly, γH2AX detects DSBs and recruits 53BP1/RIF1 complex, which protects DSB ends and promotes NHEJ. However, in the S phase, Brca1 is abundant and complexes with the endonuclease CtIP, displaces 53BP1/RIF1 and promotes end resection through interaction with the MRN complex composed of Mre11, Rad50 and Nbs1. DSB end resection commits cells to HDR or SSA. The resulting single-stranded DNA is coated with replication protein A and subsequent formation of Rad51 nucleofilament initiates strand invasion for HDR.

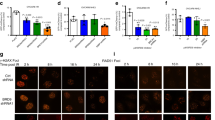
Wwox significantly decreases Rpa32 and Rad51 foci formation after IR. Representative immunofluorescence images of γH2AX, RIF1, 53BP1, Brca1, Rpa32 and Rad51 foci in 231/Wwox cells untreated or treated with doxycycline (24 h) at various time points after 10 Gy IR (left). Wwox expression significantly decreases Rpa32 and Rad51 foci 1 h after IR and increases γH2AX foci 3 h after IR. Graphs (right) represent protein foci averages of three independent experiments. Foci were quantified using the Image J software (Bethesda, MD, USA). Experiments were performed in triplicate and statistical significance was determined using the two-tailed Student’s t-test and error bars depict standard error.

IR induced foci-based hypothetical model illustrating a role for Wwox in DSB repair pathway choice. (a) γH2AX detects DSBs and recruits RIF1/53BP1 complex, which protects DSB ends. In the S phase, Brca1 becomes more abundant, complexes with CtIP, displacing RIF1/ 53BP1 complex. (b) Although bound to CtIP, Brca1 binds MRN complex through Rad50, stimulating nuclease activity of CtIP and MRN (complex of Mre11/Rad50/Nbs1) resecting DNA from the 5′ end. End resection commits cells to DSB repair via HDR or SSA. Following resection, RPA coats and protects 3′ ssDNA and Rad51 nucleofilament invades homologous DNA. (c) A model of putative role of Wwox in DSB repair pathway choice: in Wwox-sufficient cells, Wwox competes with Rad50 for binding to Brca1, impeding Brca1/CtIP complex binding to MRN, impairing resection. Impaired resection hampers HDR and promotes NHEJ in Wwox-sufficient cells.
To quantify the accumulation of repair proteins at DSBs, the number of positive foci per nucleus were determined in 231/Wwox-pos and 231/Wwox-neg cells following 10 Gy IR as indicated in Figure 6. We observed a statistically significant increase in γH2AX foci at 3 h after IR in 231/Wwox-neg cells compared with 231/Wwox-pos cells, likely reflecting the more time-consuming repair process associated with HDR vs rapid NHEJ. There were no significant differences in foci of RIF1, 53BP1 or Brca1 proteins at 0, 1, 3 and 6 h after IR, suggesting that Wwox interaction with Brca1 does not interfere with recruitment of Brca1 to DSBs and displacement of the 53BP1/RIF1 complex. However, Rpa32 and Rad51 showed significantly increased foci in 231/Wwox-neg cells compared with 231/Wwox-pos cells 1 h after IR. Increased Rpa32 foci indicate that 231/Wwox-neg cells exhibit enhanced DSB end resection; therefore, we hypothesize that Wwox interaction with Brca1 suppresses DSB end resection in Wwox-sufficient cells, ultimately promoting NHEJ over HDR.
Discussion
We have established a role for Wwox in the regulation of DSB repair, such that Wwox-deficient cells exhibit enhanced HDR and survival of DSB-inducing agents. We also reveal a novel interaction of Wwox protein with Brca1, the genome caretaker that mediates HDR repair of DSBs and hypothesize that Wwox suppresses HDR through its interaction with Brca1, tipping the pathway choice to NHEJ repair. Unlike a recently reported interaction between Wwox and the kinase, ATM,16 the interaction of Wwox with Brca1 appears not to be dependent on DNA damage. Our co-immunoprecipitation experiments confirmed that Wwox may interact with Brca1 near the 981PPLF984 motif as Brca1 mutants lacking amino acids 305–1292 did not bind Wwox. Therefore, we propose that in Wwox-sufficient cells, Wwox binds Brca1, directly or indirectly, along this central region, competing with other Brca1-interacting proteins critical for promoting HDR. One candidate, Rad50, a component of the MRN complex, binds Brca1 at amino acids 341–758.32 This interaction is responsible for stimulating nuclease activity of CtIP and MRN to enable end resection, which commits cells to HDR or SSA. We offer a model (Figure 7c) that proposes Wwox competes with Rad50 for binding to Brca1 and subsequently impairs end resection in Wwox-sufficient cells. In this way, Wwox deficiency, which commonly occurs in cancer cells, lacks an inhibitory regulatory step, resulting in enhanced end resection and HDR repair, ultimately enabling the cells to survive DNA damage-inducing cytotoxic treatments.
With this delineation of a role for Wwox in DNA repair pathway choice, we have established that Wwox is a genome caretaker, apparently paradoxically, through its role in supporting NHEJ repair, an imperfect pathway. If Wwox is involved in genome caretaking how can it be that the absence of Wwox pushes cells toward HDR, the presumptive pathway of choice for best repair, and in this case, the pathway to increased survival cellular survival of severe induced DNA damage? We believe that the enhancement of HDR because of Wwox absence is mutagenic in Wwox-deficient cells based on the cell cycle analysis data (Figure 4), which demonstrates that Wwox expression does not affect cell cycle phase. This implies that although HDR is enhanced, it is not restricted to S or G2 phases of the cell cycle in Wwox-deficient cells. If HDR should occur in the G1 phase, when sister chromatids are unavailable, the homology search may take place across the entire genome, increasing opportunities for mutagenesis because of inappropriate pairing.33 In addition, we have shown that loss of Wwox significantly alters the efficiencies of the remaining DSB repair pathways (summarized in Figure 3f), impairing NHEJ and Alt-NHEJ, but enhancing SSA, a mutagenic repair pathway that typically generates large deletions with no insertions.27 As the DSB repair pathways are complex and interconnected, it is difficult at this point to assign specifically the mutational burden or consequences of Wwox dysregulation to individual altered repair pathways following DNA damage. However, the chromosomal instability and large deletions observed through karyotype and CNV analysis associated with Wwox loss supports the idea that Wwox deficiency is mutagenic. As loss of Wwox has been well documented in association with the progression of numerous human cancers cancers,1, 10 we propose that long-term, Wwox-deficient cells carry a higher mutational burden despite short-term cellular survival of DNA DSBs because of enhanced HDR.
Our findings have important, broad therapeutic implications as Wwox is among the most commonly deleted genes in a wide variety of common human cancers.1, 8, 9, 34 We propose that determining Wwox expression levels could be an important predictor of response to radiation, cisplatin and possibly other chemotherapeutic agents. Stratifying cancers by Wwox expression could allow physicians to determine whether particular cancers would be good candidates for radiation and perhaps also inform the dosage of DSB-inducing treatments. Most importantly, we show that selectively inhibiting the HDR pathway in Wwox-deficient cells abolishes their radioresistance and resensitizes them to radiation, suggesting the use of HDR inhibitors in conjunction with radiation for treatment of Wwox-deficient tumors.
Materials and methods
Ethics statement and xenograft
Animal experiments were in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ of the National Institutes of Health and the Ohio State University IACUC Protocol 2014A00000017. A total of 10 athymic nude female mice (JAX stock no. 007850) were injected at 8 weeks of age with 1 × 107. Positive control mice (n=2) received unirradiated cells, whereas experimental mice (n=4 for each group) received cells exposed to 5 Gy. There were two groups of experimental mice: Group no. 1 received 231/Wwox-pos cells and Group no. 2 received 231/Wwox-neg cells.
Cell lines
MEFs were isolated from individual 13-day embryos of Wwox+/+ and Wwox−/− mixed background (B6 × 129 SvJ) strain pregnant females and designated MEF WT4, WT7, KO3 and KO5 cell lines. They were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 100 μg/ml gentamicin and established as described.35 Wwox3 (Wwox+/+) and Wwox5 (Wwox−/−) MEFs have been described.3 Breast cancer cell line, MDA MB-231, and Wwox-inducible derivatives were maintained under standard conditions. The MCF10A cell line and Wwox stably silenced clones, shWWOXA and shWWOXB, have been described previously.15 Supplementary Figures 6a and b shows relative Wwox expression of cell lines used throughout the manuscript.
Chemicals and plasmids
MMC (SCBT, Santa Cruz, CA, USA), cisplatin (Sigma-Aldrich, St Louis, MO, USA), ABT-888 (SelleckChem, Houston, TX, USA) and bleomycin (SCBT) were purchased in dimethyl sulfoxide or prepared as stocks in dimethyl sulfoxide. The Brca1 deletion mutants36 are HA tagged and correspond to deletions within the Brca1 protein as illustrated in Figure 6d. The Brca1 ΔM3 mutant (amino acids 1290–1530) was generated from full-length HA-Brca1 plasmid37 using methods described in Gibson Assembly Cloning Kit (New England BioLabs, Ipswich, MA, USA) and primer sequences: BRCA1-305-For, 5′-CAGAATGAATGTAGAAAAGGCTG-3′ and BRCA1-305-1290-del-Rev, 5′-GTTGCTCCTCCACATCAACAACACATTTTGTTTCCTCACTAAG-3′; BRCA1-305-1290-del-For, 5′-CTTAGTGAGGAAACAAAATGTGTTGTTGATGTGGAGGAGCAAC-3′ and pcDNA-Xho-rev, 5′-TAGGGCCCTCTAGATGCATGC-3′. XhoI and EcoRI restriction digestion and sequencing validated the correct insertion and sequence. The full-length myc-Wwox plasmid13 and deletion mutants15 were described previously.
Transient transfections
The MDA MB-231 cell line with doxycycline-inducible Wwox lentiviral-based system was described previously15 and is referred to as 231/Wwox. For transient knockdowns, mouse MEFs WT4, WT7, KO3 and KO5 were transfected with small interfering RNAs targeting Rad51 or a nonspecific control small interfering RNA using standard Dharmacon protocol (Dharmacon, Lafayette, CO, USA).
Clonogenicity
Clonogenicity assays were performed as described.35 Cells were harvested immediately after exposure to IR and 4 h following exposure to MMC, 24 h after ABT-888 and 2 h after bleomycin treatment. Survival (%) was calculated based on plating efficiencies of cells with no exposure to treatment.
DSB repair assays
To assess HDR, two cell lines with DR-GFP-stable integrations were used: HeLa cells,22 which are Wwox-deficient, and U87 cells,23 which are Wwox-sufficient. The NHEJ experiment was performed in Wwox-sufficient HEK293 cells with stably integrated NHEJ substrate, HW1. HDR, NHEJ, Alt-NHEJ and SSA assays were performed as described previously.22, 26 Wwox-deficient HeLa cells were transfected with 300 ng of myc-Wwox plasmid, empty myc vector or myc-Wwox+siWwox where the myc-Wwox plasmid is resistant to silencing by the small interfering RNAs. Wwox-sufficient cell lines, U87, HEK293 and H1299 cells, were transfected with siWwox or scrambled control small interfering RNAs (Dharmacon).
Immunoblot and co-immunoprecipitation assays
For immunoblot analysis, cells were lysed with RIPA buffer (TFS, Waltham, MS, USA) supplemented with Halt Protease Cocktail Inhibitors (TFS) or when detecting Brca1, and lysed with low salt NP40 buffer as described.26 When transfecting plasmids, ~75–85% confluent cells were overlayed with a mixture containing 2 μg of plasmid DNA, 12 μl Lipofectamine 2000 (Invitrogen, Grand Island, NY, USA) in 200 μl Opti-Mem and the medium was changed 4–6 h later. Cells were harvested for co-immunoprecipitation 24 h later. Approximately 20 μg of antisera was used in binding of protein baits for co-immunoprecipitation: Brca1,37 Wwox,38 myc,39 HA40 or HA (Covance, Princeton, NJ, USA) and IgG (SCBT; no. 2025) (Supplementary Table 1). GST pulldown with Wwox-wild-type GST-fusion protein and Wwox derivatives was performed as described previously.31 The GST-WW domain fragments harbored mutations at W44F+P47A (WW1) and Y85A+P88A (WW2).
Human brain cancer analysis (REMBRANDT)
Brain cancer expression data and patient clinical data were accessed through Georgetown Database of Cancer (https://gdoc.georgetown.edu/gdoc/). Two patient groups were created: group 1, detailed in Supplementary Table 2 and depicted in Figure 2c, was made up of all brain cancer patients who had both clinical data and expression data, whereas group 2, detailed in Supplementary Table 3 and depicted in Figure 2d, was composed of patients from group 1 who were treated with radiation. Kaplan–Meier plots were generated through the KM Clinical Plot application, a fold change of 2 was used to designate patients as downregulated Wwox expression.
References
Aldaz CM, Ferguson BW, Abba MC . WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta 2014; 1: 188–200.
Bednarek AJ, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM . WWOX, a novel WW domain-containing proteins mapping to human chromosome 16q23.3–16q24.1, a region frequently affected in breast cancer. Cancer Res 2000; 60: 2140–2145.
Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y et al. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci USA 2007; 10: 3949–3954.
Abdeen SK, Salah Z, Maly B, Smith Y, Tufail R, Abu-Odeh M et al. Wwox inactivation enhances mammary tumorigenesis. Oncogene 2011; 36: 3900–3906.
Aqeilan RI, Donati V, Palamarchuk A, Trapasso F, Kaou M, Pekarsky Y et al. WW domain-containing proteins, WWOX and YAP, compete for interaction with ErbB-4 and modulate its transcriptional function. Cancer Res 2005; 15: 6764–6772.
Iliopoulos D, Fabbri M, Druck T, Qin HR, Han SY, Huebner K . Inhibition of breast cancer cell growth in vitro and in vivo: effect of restoration of Wwox expression. Clin Cancer Res 2007; 13: 268–274.
Letessier A, Millot GA, Koundrioukoff S, Lachages A, Vogt N, Hansen RS et al. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011; 470: 120–123.
Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM et al. Signatures of mutation and selection in the cancer genome. Nature 2010; 463: 893–898.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 18: 899–905.
Salah Z, Aqeilan R, Huebner K . WWOX gene and gene product: tumor suppression through specific protein interactions. Fut Oncol 2010; 2: 249–259.
Aqeilan RI, Hagan JP, de Bruin A, Rawahneh M, Salah Z, Gaudio E et al. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinol 2009; 3: 1530–1535.
Choo A, O'Keefe LV, Lee CS, Gregory SL, Shaukat Z, Colella A et al. Tumor suppressor WWOX moderates the mitochondrial respiratory complex. Genes Chrom Cancer 2015; 54: 745–761.
Aqeilan RI, Pekarsky Y, Herrero JJ, Palamarchuk A, Letofsky J, Druck T et al. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci USA 2004; 101: 4401–4406.
Aqeilan RI, Palamarchuk A, Weigel RJ, Herrero JJ, Pekarsky Y, Croce CM . Physical and functional interactions between the Wwox tumor suppressor protein and the Ap-2γ transcription factor. Cancer Res 2004; 64: 8256–8261.
Ferguson BW, Gao X, Zelazowski MJ, Lee J, Jeter CR, Abba MC et al. The cancer gene WWOX behaves as an inhibitor of SMAD3 transcriptional activity via direct binding. BMC Cancer 2013; 13: 593.
Abu-Odeh M, Salah Z, Herbel C, Hofmann TG, Aqeilan RI . WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc Natl Acad Sci USA 2014; 111: E4716–E4725.
Ludes-Meyers JH, Kil H, Parker-Thornburg J, Kusewitt DF, Bedford MT, Aldaz CM . Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS One 2009; 4: e7775.
Hartmann C, Kluwe L, Lucke M, Westphal M . The rate of homozygous CDKN2A/p16 deletions in glioma cell lines and in primary tumors. Int J Oncol 1999; 5: 975–982.
Scarpace L, Flanders AE, Jain R, Mikkelsen T, Andrews DW . Data from REMBRANDT. Cancer Imag Arch 2015; doi.og/10.7937/K9/TCIA.2015.588OZUZB.
Driscoll M, Jeggo PA . The role of double-strand break repair-insights from human genetics. Nat Rev Genet 2006; 7: 45–54.
Pierce AJ, Johnson RD, Thompson LH, Jasin M . XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13: 2633–2638.
Ransburgh DJ, Chiba N, Ishioka C, Toland AE, Parvin JD . Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res 2010; 70: 988–995.
Golding SE, Rosenberg E, Khalil A, McEwen A, Holmes M, Neill S et al. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J Biol Chem 2004; 279: 15402–15410.
Lim YC, Roberts TL, Day BW, Stringer BW, Kozlov S, Fazry S et al. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol Oncol 2014; 8: 1603–1615.
Li Y, Wang X, Pan Y, Lee D, Chowdhury D, Kimmelman AC . Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS One 2012; 7: e39588.
Hu Y, Wang C, Huang K, Xia F, Parvn JD, Mondal N . Regulation of 53BP1 protein stability by RNF8 and RNF168 is important for efficient DNA double-strand break repair. PLoS One 2014; 9: 1–13.
Ceccaldi R, Rondinelli B, D’Andrea AD . Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 2016; 26: 52–64.
Bennardo N, Cheng A, Huang N, Stark JM . Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 2008; 6: e1000110.
Towler WI, Zhang J, Ransburgh DJ, Toland AE, Ishioka C, Chiba N et al. Analysis of BRCA1 variants in double-strand break repair by homologous recombination and single-strand annealing. Hum Mutat 2013; 3: 439–445.
Zimmerman M, de Lange T . 53BP1: pro-choice in DNA repair. Trends Cell Biol 2014; 24: 108–117.
Ludes-Meyers JH, Kil H, Bednarek AK, Drake J, Bedford MT, Aldaz CM . WWOX binds the specific proline-rich ligand PPXY: identification of candidate interacting proteins. Oncogene 2004; 29: 5049–5055.
Christou CM, Kyriacou K . BRCA1 and its network of interacting proteins. Biology 2013; 2: 40–63.
Rodgers K, McVey M . Error-prone repair of DNA double-strand breaks. J Cell Physiol 2015; 231: 15–24.
Schrock MS, Huebner K . WWOX: a fragile tumor suppressor. Exp Biol Med (Maywood) 2015; 240: 296–304.
Miuma S, Saldivar JC, Karras JR, Waters CE, Paisie CA, Wang Y et al. Fhit deficiency-induced global genome instability promotes mutation and clonal expansion. PLoS One 2013; 8: e80730.
You F, Chiba N, Ishioka C, Parvin JD . Expression of an amino-terminal BRCA1 deletion mutant causes a dominant growth inhibition in MCF10A cells. Oncogene 2004; 23: 5792–5798.
Sankaran S, Starita LM, Simons AM, Parvin JD . Identification of domains of BRCA1 critical for the ubiquitin-dependent inhibition of centrosome function. Cancer Res 2006; 8: 4100–4107.
Guler G, Uner A, Guler N, Han S, Iliopoulos BS, Hauck WW et al. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer 2004; 100: 1605–1614.
Veronese A, Visone R, Consiglio J, Acunzo M, Lupini L, Kim T et al. Mutated beta-catenin evades a microRNA-dependent regulatory loop. Proc Natl Acad Sci USA 2011; 12: 4840–4845.
Huang J, Yan J, Zhang J, Zhu Z, Wang Y, Shi T et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun 2012; 3: 911.
Saldivar JC, Miuma S, Bene J, Hosseini SA, Shibata H, Sun J et al. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet 2012; 8: e1003077.
Acknowledgements
This work was supported by Pelotonia Graduate and Post-Doctoral Fellowships (to MS, BB), 9T32OD010429 (to MS), CA154200 (to KH), R01CA120516 (to KH), NIH/NCI R01 CA102444 (to CMA), CA198228 (to JP), a fellowship from the Scientific and Technological Research Council of Turkey (TUBITAK to BB) and a graduate research scholarship from the Egyptian Cultural and Educational Bureau (to HH). We thank the OSU CMIF shared Facility for confocal microscopy and the Genome Quebec company for assistance with CNA experiments. We also thank Matthew Guggenbiller (OSUMC) for help with the analysis of the brain cancer cohort and Megan Lowery and Richard D Wood (The University of Texas MD Anderson Cancer Center, Smithville, TX, USA) for providing the data in Figure 1d and Jenna Karras and T Banerjee for helpful discussions. We are especially appreciative of the additional funding by the breast cancer support foundations, the Stefanie Spielman Fund of the OSUCCC and the Anne M Wolfe Foundation (Anne’s Army).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Schrock, M., Batar, B., Lee, J. et al. Wwox–Brca1 interaction: role in DNA repair pathway choice. Oncogene 36, 2215–2227 (2017). https://doi.org/10.1038/onc.2016.389
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.389
This article is cited by
-
Unveiling the relationship between WWOX and BRCA1 in mammary tumorigenicity and in DNA repair pathway selection
Cell Death Discovery (2024)
-
WWOX binds MERIT40 and modulates its function in homologous recombination, implications in breast cancer
Cancer Gene Therapy (2023)
-
Loss of fragile WWOX gene leads to senescence escape and genome instability
Cellular and Molecular Life Sciences (2023)
-
Characterization of WWOX expression and function in canine mast cell tumors and malignant mast cell lines
BMC Veterinary Research (2020)
-
Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide
Nature Communications (2019)
