
Abstract
Mechanisms regulating the transition of mammary epithelial cells (MECs) to mammary stem cells (MaSCs) and to tumor-initiating cells (TICs) have not been entirely elucidated. The p53 family member, p63, is critical for mammary gland development and contains transactivation domain isoforms, which have tumor-suppressive activities, and the ΔN isoforms, which act as oncogenes. In the clinic, p63 is often used as a diagnostic marker, and further analysis of the function of TAp63 in the mammary gland is critical for improved diagnosis and patient care. Loss of TAp63 in mice leads to the formation of aggressive metastatic mammary adenocarcinoma at 9–16 months of age. Here we show that TAp63 is crucial for the transition of mammary cancer cells to TICs. When TAp63 is lost, MECs express embryonic and MaSC signatures and activate the Hippo pathway. These data indicate a crucial role for TAp63 in mammary TICs and provide a mechanism for its role as a tumor- and metastasis-suppressor in breast cancer.
Similar content being viewed by others


Introduction
Mammary stem cells (MaSCs) have key roles in the development of breast cancer, in its progression and in the effectiveness of breast cancer therapy. Upstream genes controlling this process are still poorly understood. One critical transcription factor involved in epithelial stem cell maintenance of the mammary gland and skin is the p53 family member and tumor-suppressor gene, p63.1, 2, 3, 4, 5 p63 is composed of multiple isoforms with overlapping and unique activities. It is important to note that p63 is used as a diagnostic marker in metaplastic breast cancer with no regard to the existence or activities of p63 isoforms,6 and research to date has been focused on the highly expressed isoform, ΔNp63. The p63 isoforms can be placed into two groups: the transactivation domain isoforms, which structurally resemble p53 and act as tumor suppressors, and the ΔN isoforms, which bind to p53, TAp63 and TAp73 and inhibit their function, thus acting as oncogenes.7, 8, 9 In the skin, TAp63 is required to maintain adult dermal stem cells and epidermal progenitor cells, required for wound healing and hair regeneration, in quiescence.5, 10 ΔNp63 also has an important role in the skin. Its expression in the basal compartment of the epidermis is required for epidermal stratification and terminal differentiation in the developing and adult skin.10, 11, 12 Likewise, the expression patterns of the TAp63 and ΔNp63 isoforms of p63 in distinct mammary progenitor and stem cells suggest different roles for these isoforms in mammary gland development and homeostasis.13 Although ΔNp63 is highly expressed in basal cells and is critical for mammary gland development and maturation,4 the roles of TAp63 have not been investigated in vivo using knockout mouse models. Additionally, mechanisms for TAp63 regulation in MaSCs and how this may impinge on mammary tumorigenesis have yet to be elucidated and are critical for further understanding of how p63 can be used as a diagnostic marker for breast cancer and for therapy.
Recent studies have shed light on functions for the p63 isoforms in breast cancer. TAp63 is not expressed or is present at low levels in high-grade mammary adenocarcinoma, and regulation of microRNA biogenesis through transcriptional regulation of Dicer has been implicated in its ability to suppress tumor progression and metastasis.9, 14 Other mechanisms for p63’s role as a suppressor of tumorigenesis and metastasis have also been shown, including integrin recycling and interactions with transforming growth factor-β.9, 15, 16 In mouse models, TAp63-deficient mice are highly prone to metastatic mammary adenocarcinoma.14 Given the functions of TAp63 in stem cells in the skin, its clear role as a mammary tumor and metastasis suppressor5, 11, 14 and the use of p63 as a diagnostic marker in breast cancer, we sought to understand the roles of TAp63 in the regulation of MaSCs and tumor-initiating cells (TICs) using a TAp63 isoform specific knock out mouse model.5
Mechanisms regulating MaSCs and breast cancer stem cells (CSCs) or TICs have not been completely delineated.17 For example, aggressive luminal breast cancer subtypes can acquire basal cell and CSC features during their progression,18 and basal cell breast cancer may originate from luminal cells.19 Recent studies have also revealed that normal breast stem cells and CSCs share some regulatory mechanisms in certain types of breast cancer. For example, coexpression of Sox9 and Slug is sufficient to convert luminal mammary cells into MaSCs capable of mammary gland reconstitution20 and tumor development.21 Additionally, coexpression of Sox9 and Slug were found to be predictive CSC markers and promoted tumor development and metastasis.20, 21 TAZ, a transducer of the Hippo pathway, has been shown to also confer CSC properties onto mammary epithelial cells (MECs) through regulation of genes that regulate cell polarity, such as Scribble (Scrib).22 Although it is clear that developmental genes such as p63 regulate MaSCs and the development of breast cancer, the complexity of genes such as p63 with its many isoforms and duplicitous activities in tumorigenesis make it essential to further dissect their functions in the regulation of MaSCs in cancer. Here we examine the roles of the tumor-suppressor gene, TAp63, in the development of breast cancer through TICs.
Similar to the p63 isoforms, components of the Hippo pathway have been implicated in breast cancer and progression.22, 23, 24, 25 The Hippo pathway functions to regulate cell proliferation, stem cell properties, cell polarity and tumorigenesis.25 Some cross-talk between p63 and the Hippo pathway has been discovered recently. ΔNp63 directly interacts with the Hippo effector YAP and is a mediator of YAP function in the epithelium of lung airways.26 Additionally, LKB1, a transcriptional target of TAp63 in the liver, is mutated in breast cancers27 and serves to maintain cell polarity through upstream regulation of Scrib and TAZ, a transducer of the Hippo pathway.28 Moreover, TAZ has been shown to confer CSC properties of MECs through regulation of Scrib and cell polarity.22 Although connections between cell polarity, the Hippo pathway and the properties of MaSCs have been well established, the upstream regulation of this process by TAp63 was previously not known.
Here we show that the tumor suppressor and transcription factor, TAp63, regulates cell polarity and MaSCs through transcriptional regulation of LKB1 and downstream regulation of Scrib and components of the Hippo pathway. Using genetically engineered mice, human mammary cancer cell lines, orthotopic xenograft mouse models and patient-derived mammary adenocarcinomas, we show that TAp63 serves to regulate the stem cell potential of MEC and TICs. When TAp63 is lost, MECs and mammary cancer cells express embryonic stem cell (ESC) and human normal mammary stem cell (hNMSC) signatures. Loss of TAp63 in MECs and mammary cancer cells also leads to: (1) loss of cell polarity through the regulation of Scrib and TAZ after serial transplantation, (2) an accumulation of early TICs in serially transplanted mammary glands, and (3) an accumulation of cells with increased tumor-initiating capabilities in tumors derived from the MCF7-shTAp63 orthotopic mammary tumor mouse model. These data reveal novel and crucial roles of TAp63 in the regulation of TICs through the Hippo pathway in the mammary gland and in breast cancer and have important implications for the diagnosis and treatment of breast cancer.
Results
Mammary glands from TAp63−/− mice are hyperplastic and have increased regenerative potential
We have shown previously that TAp63 is critical for the maintenance of stem cells within the dermis by employing mechanisms that keep these cells in quiescence.5 To determine whether TAp63 may similarly regulate stem cells in the mammary gland, we assayed for the regenerative potential of mammary glands from TAp63−/− mice. In virgin females at 10 weeks of age, no difference was observed in the development and morphology of mammary glands from wild-type (WT) and TAp63−/− mice (Figures 1a and e). However, following serial transplantation, we observed a number of differences between the two genotypes (Figures 1a–t). TAp63−/− mammary glands are capable of repopulating WT donor mammary fat pads for eight generations while WT mammary glands only do so for five generations (Figures 1a–h, q and r). The outgrowth coverage, the area of the fat pad repopulated by the MECs, of transplanted WT mammary glands drops from 100% in transplantation generation 1 (TG1) to 0% in generation 6 (TG6) (Figures 1b–d; Supplementary Table S1). In contrast, the outgrowth coverage of TAp63−/− mammary glands is 25%±32% in TG6, 12%±27% in TG7 and 8%±11% in TG8 (Figures 1h, q and r; Supplementary Table S1). The outgrowth rate, calculated by dividing the number of fat pads with mammary outgrowth coverage by the total number of mammary fat pads inoculated with MECs, of transplanted TAp63−/− mammary glands is significantly higher than WT mammary glands from TG5 to TG8 (Supplementary Figure S1). TAp63−/− mice had 83% mammary outgrowth coverage compared with 58% in WT mice at TG5. Although WT mice had no outgrowth coverage at TG6, TG7 and TG8, TAp63−/− mice had outgrowth coverages of 58, 36 and 62% for each of these transplant generations, respectively (Supplementary Figure S1). Additionally, the morphology of TAp63−/− mammary glands differed from that of WT-transplanted glands (Figures 1i–p, s and t). The TAp63−/− mammary glands lacked lumens, and these areas were filled with cells (Figures 1o, p, s and t). Detailed analyses using immunofluorescence (IF) staining for the basal marker, smooth muscle actin (SMA) and the luminal marker, Na-K-Cl cotransporter (NKCC), revealed that the basal and luminal layers of the mammary gland are highly disorganized in TAp63−/− glands. This disorganization was apparent as early as the first transplantation (TG1), with severe defects noted by TG5 (Figures 1u–b’). In particular, we found that basal cells detached from the basement membrane and localized to the luminal cell compartment (Figures 1a’ and b’). We asked whether the TAp63−/− mammary glands are hyperplastic by immunostaining with an antibody for Ki67 (Figures 1c’–j’). Indeed, we found an increase in Ki67-positive cells within the lumen of the mammary glands of 5-week-old TAp63−/− virgin females (50%±10%) compared with WT females of the same age (5%±3%; Figures 1g’ and k’). We also found increased Ki67-positive cells in TAp63−/− mammary glands after each transplantation (Figures 1h’–k’). The TAp63−/− TG1, TG3 and TG5 glands contained 40%±12%, 40%±2% and 35%±5%, respectively, while the WT-transplanted glands had 3%±5% for each passage (Figure 1k’), indicating an increase in proliferating cells within the mammary gland of TAp63−/− mice even in virgin females.

TAp63−/− mammary glands are hyperplastic and have increased regenerative potential. (a–h) Representative whole-mount staining and (i–p) H&E staining of WT and TAp63−/− mammary glands (MG) at 10 weeks of age and subsequent to 1, 3 or 6 serial transplantation passages in vivo (TG1, TG3, TG6). (q and r) Representative whole-mount staining and (s and t) H&E staining of TAp63−/−-transplanted MG at TG7 and TG8. Insets of pie graphs (a–h and q–r) indicate the transplanted MG outgrowth coverage of fat pads. For panels (a–t), n=6. Representative micrographs of IF staining of WT and TAp63−/− MG at 5 weeks and serially transplanted MG indicated using antibodies for (u–b’) SMA and NKCC, (c’–j’) SMA and Ki67. (k’) Bar graph showing quantification of Ki67-positive cells from panels (c’–j’). Asterisks indicate statistical significance, P<0.05. (l’–o’) Representative micrographs of IF staining of WT and TAp63−/− MG that are involuting and at TG5 using antibodies for Casp 3 and K18. Scale bar represents 5 mm in panels (a–h) and (q–r), 100 μm in (i–p) and (s–t), 50 μm in (u–j’) and (l’–o’). For panels (u–o’), n=5.
Apoptosis is a critical step in lumen formation of the mammary gland and in mammary gland involution after pregnancy and weaning. We therefore asked whether the TAp63−/− mammary glands have defects in apoptosis. We found cleaved caspase 3-positive cells within WT and TAp63−/− involuting mammary glands in addition to positive cells within the disorganized TAp63−/− TG5 mammary glands. The number of cleaved caspase 3-positive cells was comparable to what is detected in WT TG5 mammary glands, indicating that TAp63−/− mammary glands do not have defects in the induction of apoptosis (Figures 1l’–o’). The apoptotic cells in TAp63−/− mammary glands appear to be mislocalized in TG5 glands (Figure 1o’).
Transplanted TAp63−/− mammary glands have defects in cell polarity
Cell polarity are common pathways altered in human breast cancer, which are also associated with cellular hyperproliferation and disorganization of the mammary gland structure.27, 29, 30, 31 To determine whether these pathways are deregulated in the absence of TAp63 leading to the observed solid gland structure and hyperplastic phenotype of TAp63−/−transplanted glands, we performed IF staining for markers of cell polarity at 10 weeks after birth in virgin females and after 1, 3 and 5 rounds of transplantation (TG1, TG3 and TG5). Both WT and TAp63−/− luminal epithelial cells express AQP5, an apical marker of luminal cells, on one side of the cells indicating no defects in apical–basal cell polarity in the absence of TAp63 (Figures 2a–h). With later transplantation passages (TG3 and TG5), the AQP5-positive cells within the TAp63−/− mammary glands are disorganized as indicted by the arrows, AQP5 is still expressed on one side of the cell (Figures 2g and h) in some cells, but most TAp63−/− cells lost apical–basal cell polarity during transplantation (compare Figures 2e–h). In contrast, the AQP5-positive cells within the WT mammary gland line up on the apical side to form the lumen (Figures 2c and d). These data suggest that the TAp63−/− luminal MECs have defects in cell polarity. Scrib and Vangl2 serve to maintain cell polarity.29, 32, 33 To ask whether the TAp63−/− mammary glands have defects in cell polarity, we performed IF staining for Scrib and Vangl2. Although Scrib and Vangl2 were expressed similarly in 10-week old WT and TAp63−/− mammary glands from virgin females, we found that these proteins are mislocalized or decreased in expression in TAp63−/−-transplanted glands (Figures 2i–x). These data indicate that TAp63−/− mammary glands have defects in cell polarity. Scrib has also been shown to control polarization of the Golgi and is required for the establishment of cell polarity.31 We therefore used a marker to label the Golgi (GM130) and found that transplanted TAp63−/− glands as early as passage 1 had mislocalized staining of GM130, further indicating defects in cell polarity and suggesting regulation of Scrib by TAp63 in the mammary gland (Figures 2y–f’).

TAp63−/− mammary glands have cell polarity defects after transplantation. Representative IF micrographs of WT and TAp63−/− mammary glands at 10 weeks of age and subsequent to 1, 3, or 5 serial transplantation passages in vivo (TG1, TG3, TG5) using antibodies for: (a–h) APQ5, (i–p) Scrib, (q–x) Vangl2 and (y–f’) GM130. Arrows indicate positive cells in panels (g–h) and areas further magnified in insets in the upper right corner of panels (z–b’) and (d’–f’). Scale bar represents 50 μm in panels (a–f’). n=5.
TAp63−/− mammary glands acquire an increased number of early TICs through the activation of an epithelial–mesenchymal transition (EMT) program
The TAp63−/− mammary glands have increased regenerative potential as assessed by an increased capacity to transplant these mammary glands into syngeneic mice, suggesting that the TAp63−/− mammary glands have an increased number of TICs. To determine whether TAp63−/− mammary glands contain a greater number of TICs than WT mammary glands, we performed limiting dilution assays. These assays were performed by isolating single MECs from WT and TAp63−/− mouse mammary glands and injecting 250, 500, 1000, and 2500 cells into the cleared fat pads of syngeneic WT mice. We found that TAp63−/− MECs had a greater outgrowth rate and outgrowth coverage than WT MECs (Figures 3a–c and Supplementary Figure S2). We found the outgrowth rate from TAp63−/− MECs, calculated by dividing the number of fat pads with mammary outgrowth by the total number of mammary fat pads inoculated with MECs, to be five times greater than that of MECs derived from WT mice (Figures 3b and c). We also calculated the amount of outgrowth coverage by calculating the area of the fat pad repopulated by the MECs. Using this calculation, we found that TAp63−/− MECs have an outgrowth coverage that is 10 times greater than that of WT MECs (Supplementary Figure S2). By histological analysis of these glands, we found that TAp63−/− MECs formed disorganized mammary glands lacking a luminal space (compare Figures 3d and h) as we had previously noted after serial transplantation (Figure 1). We also found the TAp63−/− mammary glands from the limiting dilution experiment to be highly disorganized by analyzing well-characterized markers of the mammary gland (Figures 3h–k). By IF staining for the luminal marker, keratin 8, and the basal marker, SMA, we found basal cells in TAp63−/− mammary glands to be mislocalized to the luminal compartment (compare Figures 3e and i). Additionally, the TAp63−/− mammary glands had an increase in the number and location of proliferative cells as assessed by Ki67 staining (Figures 3f and j). TAp63−/− mammary glands had 75%±1% Ki67-positive cells compared with 38%±2% in WT mammary glands (Figure 3l). Taken together, these results recapitulate the defects detected after serial transplantation of TAp63−/− mammary glands and suggest that these mammary glands are hyperplastic and may be enriched for early TICs.

TAp63−/− mammary glands have increased numbers of TICs. (a) Representative whole-mount staining of MG generated from WT and TAp63−/−MECs in limiting dilution assay (LDA). Scale bar represents 50 mm. (b) Pie charts indicating MG outgrowth rate from LDA. Colored pie slices with numbers indicate outgrowth coverage. (c) Table indicating MG outgrowth of LDA using MECs from WT and TAp63−/− mammary glands from panel (a). (d–k) H&E staining (d and h) or IF staining (e–g and i–k) of outgrown MG from LDA using: keratin 8 (K8; green), SMA (red), Ki67 (green), Sox9 (green), and slug (red). Scale bar represents 50 μm in panels (d–k). (l) Bar graph showing quantification of Ki67-positive cells from panels (f) and (j). (m) Bar graph showing quantification of Sox9 and Slug double-positive cells from panels (g) and (k). Asterisk represents statistical significance, P<0.05. (n) Representative western blotting analysis of WT and TAp63−/− mouse MECs using antibodies for slug, Sox9, Sox2, ALDH and αSMA. Actin was used as a loading control. (o–x) IF or IHC staining of mammary adenocarcinomas from TAp63−/− mice (o–s) or MG from 5-week-old WT mice (t–x) using antibodies for Sox9 (green), slug (red), Sox2 (brown), ALDH (brown), vimentin (brown) and E–cadherin (green). DAPI (blue) or hematoxylin (purple) were used as counterstains. Arrow indicates example of double-positive nuclei (yellow). Scale bar represents 50 μm in panels (o–s). All scale bars represent 50 μm. n=6 for all experiments.
Sox9 and Slug double-positive cells have been shown to mark MaSCs, TICs and CSCs.20, 21 Further, Sox9 and Slug activate an EMT program in MECs maintaining them in the stem cell stage and have been implicated as markers of MaSCs, TICs and CSCs based on this activity.20, 21 To ask whether TAp63−/− mammary glands formed from the limiting dilution experiment have increased number of MaSCs, TICs and CSCs, we performed IF staining for Sox9 and Slug (Figures 3g and k). Indeed, we found that TAp63−/− mammary glands from limiting dilution experiments had 22%±8% Sox9 and Slug double-positive cells compared with just 5%±2% in WT mammary glands from limiting dilution assays (Figures 3g, k and m), indicating that these mammary glands are enriched for early TICs. We also performed western blotting analysis using MECs from 10-week-old virgin WT and TAp63−/− female mice and found that both Sox9 and Slug are expressed at much higher levels in TAp63−/− versus WT MECs (Figure 3n). Further, we analyzed additional markers for TICs, Sox2 and ALDH1 and found these to be highly expressed in TAp63−/− MECs, suggesting an enrichment for early TICs in TAp63−/− mammary glands (Figure 3n). Finally, we found the mesenchymal marker, SMA, present at higher levels in MECs extracted from TAp63−/− compared with WT mammary glands (Figure 3n), in agreement with previous data showing the association of EMT with an acquisition of early TIC characteristics.20, 21 We further demonstrated that TAp63−/− mammary adenocarcinomas express the luminal marker NKCC1, are ER positive and lose expression of the polarity protein, Scrib (Supplementary Figures S3a–c). Given the expansion of early TICs in transplanted TAp63−/− mammary glands, we asked whether this population was expanded in TAp63−/− mammary adenocarcinomas. Indeed, we found an enrichment of TICs in mammary adenocarcinomas from TAp63−/− mice by IF using markers for MaSCs (Slug, Sox9, Sox2 and ALDH) (Figures 3o–q and t–v and Supplementary Figures S6a–r and y–a’) and an upregulation of the mesenchymal marker, vimentin (Figures 3r and w and Supplementary Figures S6s–x and b’), with a concomitant downregulation of the epithelial marker, E-cadherin (Figures 3s–x), demonstrating EMT in these tumors and suggesting that cells characterized are TICs. Mammary glands from 5-week-old WT mice were used as controls (Figures 3t–x). Taken together, our data indicate that TAp63 serves to maintain MECs. When TAp63 is lost, MECs undergo EMT and transition to acquire characteristics of TICs.
TAp63−/− MECs are significantly enriched in MaSC and ESC signatures
To determine critical pathways present in TAp63−/− MECs, we performed RNA-seq analysis using RNA extracted from WT and TAp63−/− MECs. Using Gene Set Enrichment Analysis, we found that TAp63−/− MECs are significantly enriched in hNMSC and ESC signatures (q<0.0001)22, 28, 34, 35 (Supplementary Table S2 and Figures 4a–d). Using pathway analysis for genes that are common in TAp63−/− MECs and hNMSC and ESC gene signatures, we found a significant enrichment for pathways involved in tubulin folding, cell adhesion and tight and gap junctions, all pathways influencing cellular polarity and affecting stem cell potential22, 36, 37(Figure 4e).

TAp63−/− MECs are significantly enriched in hNMSC and ESC signatures. Gene set enrichment analyses of upregulated genes in hNMSCs (a) and ESCs (b) reveals positive enrichment of TAp63−/− MECs, while downregulated genes of hNMSCs (c) shows negative enrichment of TAp63−/− MECs. (d) Normalized enrichment scores for hNMSC, ESC, Hippo and Yap pathway signatures in TAp63−/− MECs. (e) Pathway enrichment analysis reveals enrichment of tubulin, tight and gap junction and epithelial-to-mesenchymal transition pathways as analyzed from BP, KEGG and Reactome databases. (f) qRT-PCR validation of targets that are common to TAp63−/− MECs for each of the signatures.
We then performed quantitative real-time PCR (qRT-PCR) in order to validate genes that are significantly enriched in the hNMSC and ESC signatures. As shown in Figure 4e, GAL, PMAIP1 and NFE2L3, which were previously reported to be upregulated in the ESC1 signature,34 were also upregulated in TAp63−/− MECs. Genes enriched in the hNMSC signature were also validated; we found Sox4, TUBB2B and F11R to be upregulated and SPON1, Slc41a2 and P4HB to be downregulated in TAp63−/− MECs (Figure 4f) similar to the expression levels found in hNMSCs.35 These data indicate that TAp63−/− MECs express a transcriptional profile resembling that of both embryonic and normal MaSCs and suggest that MaSCs accumulate in TAp63-deficient mammary glands through transcriptional regulation of these stem cell pathways. The Hippo pathway has also been implicated in conferring stem cell properties in multiple tissues,38, 39 and we found the Hippo pathway to be deregulated in TAp63−/− MECs (Figure 4d). Therefore, we also assessed the expression of genes in the published Hippo- and YAP-conserved signatures.22, 28 We found that several of these mRNAs, including CTGF, NT5E, Slc2a3, BMP4, SLIT2 and SPDR, were upregulated in TAp63−/− MECs, suggesting regulation of the Hippo pathway by TAp63.
TAp63 regulates the Hippo pathway
To understand the mechanism employed by TAp63 to acquire characteristics of TICs and cell polarity, we cultured WT and TAp63−/− mouse MECs in three-dimensional (3D) culture (Figures 5a–h). TAp63−/− 3D cultures formed more solid (85%) than hollow (15%) acinar structures compared with 67% solid and 43% hollow acini in WT cultures (Figure 5i). To determine whether TAp63−/− mammary acini had defects in cell polarity, as we had detected in transplanted TAp63−/− mammary glands, we performed IF staining using the polarity and Golgi marker, GM130. Although WT acini had normal polarity (Figures 5e and f), TAp63−/− acini contained random GM130 staining and were highly disorganized (Figures 5g and h). This result recapitulates the polarity defects noted in the transplanted TAp63−/− mammary glands.

TAp63 regulates mammary gland cell polarity through the Hippo pathway. (a–d) Bright-field micrographs of primary mouse MECs grown in 3D cultures. Acinar structures from WT MECs grown in 3D culture (a and b) and solid structures from TAp63−/− MECs grown in 3D culture (c and d). (e–h) Representative confocal micrographs of WT and TAp63−/− 3D cultures immunostained with K18 (green) and GM130 (red). DAPI (blue) was used as a counterstain. (i) Bar graph showing the relative formation rate of hollow and solid acinar structures from a total of 268 WT and 207 of TAp63−/− 3D structures. Asterisks indicate statistical significance, P<0.05. (j) Representative western blotting (WB) of MECs of the indicated genotypes using antibodies against Scrib, YAP, TAZ and BMP4. Actin was used as a loading control. (k–l) Semiquantitative RT-PCR for TAp63 (k) and ΔNp63 (l) in MCF10A-shRNA specific for TAp63 and a non-specific control shCON. (m) Representative WB of MCF10A-shCON or -shTAp63 using antibodies for TAp63 and ΔNp63. Actin was used as a loading control. (n) Bar graph showing relative acinar formation of MCF10A-shCON or -shTAp63 cells. Six hundred structures were counted for each genotype. Asterisks show statistical significance, P<0.05. (o) Representative WB of MCF10A-shCON or -shTAp63 using antibodies for Scrib, YAP, TAZ and BMP4. (p and q) Representative confocal micrographs of MCF10A-shCON cells (p) or -shTAp63 (q) and immunostained with TAZ (green). DAPI (blue) was used as a counterstain. (r and s) IHC staining of MCF10A-shCON or -shTAp63 cells using an antibody for BMP4 (brown). Hematoxylin (purple) was used as a counterstain. (t and u) Representative bright-field micrographs of MCF10A-shCON or -shTAp63 in 3D culture. (v and w) Representative confocal micrographs of MCF10A-shCON (v) or -shTAp63 (w) and immunostained GM130 (red). DAPI (blue) was used as a counterstain. All scale bar represents 50 μm. n=3 for all experiments. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
To further understand whether polarity is regulated through Scrib, as our previous data suggested (Figures 2i–x), we performed western blotting analysis and found that Scrib is present at low levels in TAp63−/− MECs (Figure 5j). Because Scrib is upstream of the Hippo pathway, we asked whether YAP and TAZ are increased and whether their downstream target, bone morphogenetic protein 4 (BMP4), of this pathway is activated. We found that TAZ but not YAP levels are increased in TAp63−/− MECs and that their downstream target, BMP4, is also increased, suggesting that the Hippo pathway is activated in MECs deficient for TAp63 (Figure 5j).
The Hippo pathway is activated after TAp63 knockdown in MCF10A cells
To further determine whether the Hippo pathway is activated by TAp63, we knocked down TAp63 in human MECs, MCF10A cells (Figures 5k–m and Supplementary Figure S4). This resulted in downregulation of TAp63 mRNA and protein and no change in the expression of ΔNp63 (Figures 5k–m). As seen in the TAp63−/− MECs, MCF10A cells expressing the short hairpin RNA (shRNA) for TAp63 had fewer hollow acini (Figure 5n) and a decrease in Scrib expression with downstream activation of TAZ and BMP4 (Figure 5o). We further demonstrated using confocal imaging that TAZ localized to the nucleus of MCF10A cells deficient for TAp63 (Figures 5p and q) consistent with the activation of TAZ. Finally, the downstream target of TAZ, BMP4 was upregulated in MCF10A cells deficient for TAp63 (Figures 5r and s). 3D cultures of MCF10A cells deficient for TAp63 exhibited a loss of cell polarity as assessed by GM130 IF staining (Figures 5t–w). These data further suggest that TAp63 regulates cell polarity through the Hippo pathway.
TAp63 activates the Hippo pathway through transcriptional regulation of LKB1
LKB1 is upstream of the MARK kinase family members (MARK1, MARK2 and MARK4), which controls Scrib expression and localization to regulate the Hippo pathway.28 We have shown previously that Lkb1 is transcriptionally activated by TAp63 to regulate glucose metabolism.40 To determine whether Scribble or Lkb1 are transcriptional targets of TAp63 in MECs, we assessed their mRNA levels in MCF10A and MCF10A cells expressing an shRNA for TAp63 (Figure 6a). We found the levels of Scrib mRNA to be unchanged in cell lines deficient for TAp63, but LKB1 mRNA was present at significantly lower levels in the absence of TAp63, suggesting that TAp63 may be transcriptionally regulating LKB1 in MECs (Figure 6a). We therefore performed a chromatin immunoprecipitation analysis using an antibody for TAp63 and the LKB1 promoter in mouse and human MECs deficient for TAp63. We identified two sites within the LKB1 promoter and within intron 1 where TAp63 binds (Supplementary Table S3), indicating that TAp63 transcriptionally activates LKB1 in MECs as it did in other tissues (Figures 6b and c).40

TAp63 regulates cell polarity and the Hippo pathway through transcriptional regulation of LKB1. (a) qRT-PCR for Scrib and LKB1 in MCF10A-shTAp63 or -shCON cells. Asterisks show statistical significance, P<0.05. (b and c) qRT-PCR of chromatin immunoprecipitation assay indicating the percentage of TAp63 bound to the LKB1 promoter in WT and TAp63−/− mouse MECs (b) and MCF10A-shCON or -shTAp63 cells (c). NSBS indicates non-specific binding site. Asterisks show statistical significance, P<0.05. (d) Representative western blotting analysis of MCF10A-shCON or -shTAp63 cells and transduced with Ad-GFP or Ad-LKB1 using antibodies for LKB1, Scrib, YAP, TAZ, BMP4, CTGF, Sox9 and Slug. Actin was used as a loading control. (e–l) Bright-field micrographs (e, g, l, k) and fluorescent micrographs (f, h, j, l) of MCF10A-shCON or -shTAp63 cells and transduced with Ad-GFP or Ad-LKB1 using antibodies for GM130 (red). DAPI (blue) was used as a counterstain. (m) Bar graph showing relative acinar formation in 3D cultures of MCF10A-shCON and -shTAp63 cells and MCF10A-shCON and -shTAp63 transduced with Ad-GFP or Ad-Lkb1. Six hundred structures were counted for each condition. Asterisks show statistical significance, P<0.05. (n–y) Frame-by-frame in vivo tracking of cell movement within 24 h after wounding. (z) Bar graph showing the average distance travelled by 20 cells within 24 h after wounding. Asterisks show statistical significance, P<0.05. All scale bars represent 50 μm. n=3 for all experiments.
Re-expression of LKB1 rescues cell polarity and stem cell defects of TAp63-deficient MECs
We performed immunoblotting on MCF10A and MCF10A cells expressing shTAp63 and found that MCF10A cells deficient for TAp63 expressed low levels of LKB1 and Scrib and increased levels of TAZ, indicating activation of the Hippo pathway subsequent to deletion of TAp63 (Figure 6d). Re-expression of LKB-1 in TAp63-deficient MCF10A cells inactivated the Hippo pathway as indicated by downregulation of BMP4 and connective tissue growth factor (CTGF) (Figure 6d and Supplementary Figure S4), indicating that TAp63 activates the Hippo pathway through transcriptional activation of LKB1. TAZ has been found to confer CSC-related traits onto MECs,22 indicating that loss of TAp63 may likewise confer these traits through activation of TAZ. We found that re-expression of LKB-1 in TAp63-deficient MCF10A resulted in a downregulation of markers associated with CSC, TAZ, Slug and Sox9 (Figure 6d). To determine whether transcriptional regulation of LKB1 by TAp63 is causal in the cell polarity defects in TAp63-deficient MCF10A cells, we performed assays in 3D and 2D tissue culture to assess cell polarity before and after the forced expression of LKB1 in TAp63-deficient MCF10A cells (Figures 6e–z). In 3D cultures, TAp63-deficient MCF10A cells have fewer acinar structures and the polarity marker, GM130, is expressed randomly (Figures 6e–l). Re-expression of LKB1 in TAp63-deficient MCF10A cells rescued the acinar structures (Figure 6m) and polarity defect (compare Figures 6j and l). We also performed wound-healing assays in 2D cultures to assess polarity using live imaging (Figures 6n–z and Supplementary Figures S5a and b). We found that TAp63-deficient MCF10A cells exhibited a delay in wound closure owing to loss of cell polarity (Figures 6e–z and Supplementary Figure S5). Re-expression of LKB1 in TAp63-deficient MCF10A cells reinstall cell polarity (Figures 6e–m) and appropriate wound closure (Figures 6n–z and Supplementary Figures S5c and d), indicating that activation of LKB1 and the downstream regulation of the Hippo pathway by TAp63 are critical for maintenance of cell polarity in MECs. Taken together, our data indicate that TAp63 maintains TICs through the maintenance of cell polarity by the transcriptional regulation of LKB1 and the downstream regulation of the Hippo pathway and suggest this as a possible mechanism for its action as a tumor and metastasis suppressor.
TAp63 loss unleashes the tumor-initiating potential of human mammary cancer cells
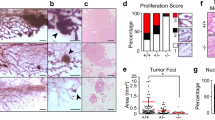
We have shown previously that loss of TAp63 results in metastatic mammary adenocarcinoma in mouse models (Figures 3o–s).14 We have further shown that TAp63-deficient tumors are enriched in early TICs (Figures 3n–q). Additionally, we have found that aggressive human mammary adenocarcinomas lose the expression of TAp63.14 To further demonstrate that TAp63 is a tumor suppressor in human mammary adenocarcinoma and regulate the tumor-initiating potential of mammary cancer cells, we knocked down TAp63 in the non-tumorigenic MEC lines, MCF10A, and investigated the tumorigenic potential using an orthotopic xenograft mouse model. Twenty-one percent of MCF10A-shTAp63 glands formed tumors (5 out of a total of the 24 injected glands) while no tumors formed in mice injected with MCF10A-shCON (0 out of a total of the 18 injected glands; Figures 7a, b, e and f). These data indicate the tumor-initiating potential of loss of TAp63 given the non-tumorigenic nature of MCF10A cells. Further immunohistochemical (IHC) staining revealed that MCF10A-shTAp63 tumors contained a higher level of Sox2-positive cells (Figures 7c and g), indicating an enrichment in TICs in these tumors. To future explore the mechanism of TAp63 in TICs, MCF7 mammary adenocarcinoma cells were used. MCF7 cells formed colonies in soft agar, MCF7 cells lacking TAp63 formed almost two times more colonies in soft agar (Figures 7i–k). We also assayed the tumorigenic potential of MCF7 cells deficient for TAp63 using an orthotopic xenograft mouse model. Tumors arising from MCF7-shTAp63 cells were three times bigger in volume then MCF7-shCON cells (Figures 7l and m), indicating that loss of TAp63 provides a growth advantage of MCF7 mammary cancer cells. To determine whether TAp63 is regulating the stem cell potential in tumors derived from the MCF7 orthotopic mammary tumor model, we assessed the expression of gene signatures in the ESC, hnMSC and Hippo pathways as we showed in the MECs in Figure 4f. We found that MCF7-shTAp63 tumors were enriched for these signatures as we had shown in TAp63−/− MECs (Figure 7n). We also assessed the expression of the TAp63 target, LKB1, in MCF7-shTAp63 as well as MCF10A-shTAp63 tumors and found that the expression was decreased in these tumors compared with those derived from MCF10A-shCON or MCF7-shCON cells (Figures 7d, h, o and r). Concomitantly, we found that the downstream Hippo target BMP4 was upregulated in MCF7-shTAp63 tumors (Figures 7p and s). Finally, in agreement with our previous data, the CSC marker, Sox2, was highly upregulated in MCF7-shTAp63 tumors compared with MCF7-shCON tumors (Figures 7q and t). These data emphasize the crucial role of TAp63 in controlling the tumor-initiating potential of mammary cancer cells through regulation of LKB1 and the Hippo pathway.

Enrichment of TICs in MCF10A and MCF7 tumors lacking TAp63. (a and e) Representative micrographs of orthotopic xenograft mouse tumors from mice injected with MCF10A-shCON and -shTAp63. Numbers under micrographs indicate the number of glands with tumor over the total injected glands. Black arrow indicates tumor. (b and f) Representative H&E and immunostaining of mammary tumors derived from MCF10A-shCON and -shTAp63 orthotopic xenograft mice using antibodies for Sox2 (brown) (c and g) and Lkb1 (brown) (d and h). Hematoxylin (purple) was used as a counterstain. Black squares indicate areas further magnified in insets in the upper right corner of each panel. All scale bars represent 50 μm. For panels (a–h), n=6. (i) Representative western blotting analysis of MCF7-shCON or -shTAp63 and using antibodies for TAp63 and ΔNp63. Actin was used as a loading control. (j) Representative micrographs of MCF7-shCON and -shTAp63 cells cultured in soft agar. (k) Bar graph showing the quantification of colonies in soft agar generated from the experiment in panel (j). Asterisks show statistical significance, P<0.05. For panels (i–k), n=6. (l) Representative photographs of orthotopic xenograft mouse tumors from mice injected with MCF7-shCON and -shTAp63. (m) Average volume of xenograft mouse tumors from mice injected with MCF7-shCON and -shTAp63 cells. (n) qRT-PCR for the indicated mRNAs from mammary tumors derived from MCF7-shTAp63 versus -shCON orthotopic xenograft mice. Data are mean±s.d., n=3, *versus MCF7-shCON cells, P<0.01, two-tailed t-test. (o–t) Representative IHC staining of mammary tumors derived from MCF7-shCON and -shTAp63 orthotopic xenograft mice. For panels (l, m and o–t), n=6.
TAp63, components of cell polarity and the Hippo pathway are deregulated in human mammary adenocarcinoma
Our data indicate that TAp63 transcriptionally activates LKB1, which in turn regulates Scrib and the Hippo pathway through TAZ. To ask whether this pathway is deregulated in human mammary cancer through loss of TAp63, we tested 48 human breast samples, including normal, ductal carcinoma in situ and infiltrative mammary adenocarcinoma. We found that TAp63 is downregulated along with LKB1 and Scrib in ductal carcinoma in situ and infiltrative mammary adenocarcinomas (Figures 8a–f and Supplementary Table S4).14 We also found that TAZ is overexpressed in these samples along with its downstream target BMP4 (Figures 8g–l and Supplementary Table S4), indicating activation of the Hippo pathway in tumors deficient for TAp63. We next asked whether activation of the Hippo pathway through TAZ led to an increase in breast CSCs. Indeed, Sox2 and ALDH are expressed in these samples (Figures 8m–r and Supplementary Table S4). Taken together, these data further indicate that the Hippo pathway is activated through the TAp63 pathway and that this leads to an increase in TICs and CSCs through deregulation of cell polarity in the mammary gland.

Components of cell polarity are deregulated in TAp63-deficient breast cancers. (a–r) Representative fluorescence or bright-field micrographs of normal human mammary tissue (normal), non-invasive mammary tumors (ductal carcinoma in situ) and invasive human mammary adenocarcinomas (infiltrated) using antibodies for LKB1 (brown) (a–c), Scrib (red) (d–f), TAZ (green) (g–i), BMP4 (brown) (j–l), Sox2 (brown) (m–o) and ALDH (brown) (p–r). DAPI (blue) or hematoxylin (purple) were used as counterstains. Arrows indicate areas further magnified in insets in the upper right corner of each panel. All scale bars represent 50 μm. Normal tissue n=6, in situ tissue n=20, infiltrated tissue n=12.
Discussion
p63 is a crucial regulator of epithelial stem cells and is frequently used as a diagnostic marker for breast cancer.2, 5, 6, 10 The N-terminal isoforms of p63 maintain distinct stem cell compartments in the skin.5, 11 TAp63, containing an acidic transactivation domain, serves to maintain dermal stem cells critical for wound healing and hair regeneration known as skin-derived precursors in quiescence.5, 41 ΔNp63, the p63 isoforms lacking the N-terminal acidic transactivation domain, is highly expressed in basal cells within the epidermis and is needed for terminal differentiation of the stratified epidermis.11, 12 Although p63’s function in progenitor and stem cells in the skin has been well studied, the mechanisms employed by p63 and its isoforms to regulate stem cell proliferation remain poorly understood. Moreover, the roles of p63 isoforms in mammary gland development and the regulation of progenitor and stem cells in the mammary gland remain poorly defined. Although the roles of ΔNp63 in the mammary gland have been thoroughly examined3, 4 and ΔNp63 is necessary to sustain the self-renewal of mammary CSC,42 TAp63 has been ignored owing to its low level of expression in spite of its crucial role as a suppressor of tumorigenesis and metastasis.9, 14, 15, 16, 43 Here we used a TAp63-deficient mouse model to probe the function of TAp63 in the mammary gland and found that TAp63 regulates mammary TICs through the Hippo pathway. We found an accumulation of early TICs in transplanted TAp63-deficient mammary glands, in two orthotopic xenograft mammary cancer mouse models and in TAp63−/− mammary adenocarcinomas from mice and humans. We found that EMT occurs in the absence of TAp63 leading to the acquisition of stem cell properties in MECs and cancer cells. These data provide a novel mechanism for TAp63’s ability to suppress mammary adenocarcinoma through regulation of stem cells in the mammary gland.
The mammary gland is composed of basal and luminal cells. The various types of breast cancer are thought to arise from stem cells within the basal and luminal layers of the mammary gland. Luminal progenitor cells are thought to give rise to the basal-like subtype of breast cancer and MaSCs give rise to the claudin-low subtype of breast cancer.44, 45, 46 The mechanisms employed to maintain these stem cells populations in the mammary gland and their deregulation in cancer are not well understood. p63 is known to be crucial for the development and differentiation of stratified epithelium.1, 2, 11, 12 Mice deficient for all isoforms of p63 lack stratified epidermis and mammary glands.2 Mice lacking TAp63 have fragile skin, develop blisters, wounds that never heal and alopecia.5 The epidermal phenotypes of TAp63−/− are due to the transcriptional regulation of p57Kip2 by TAp63 to maintain skin-derived precursors, a stem cell found to be critical for wound healing and hair regeneration,5, 41 in quiescence. When TAp63 is lost, skin-derived precursor cells proliferate inappropriately and are prematurely depleted resulting in the phenotypes seen.5 In contrast, ΔNp63 was found to be crucial for terminal differentiation of basal keratinocytes and is necessary for the maintenance of basal cells in the epidermis.11, 12 Given that the p63 isoforms have crucial roles to maintain these epidermal and dermal adult stem in the skin, we hypothesized that TAp63 may be having similar roles in the mammary gland owing to its importance as a suppressor of tumorigenesis and metastasis.9, 14, 15, 16, 43 The ΔNp63 is highly expressed in mammary epithelial basal cells and its role in maintaining MaSCs has recently been well described using a ΔNp63-deficient mouse model through regulation of Wnt signaling3; however, in these studies, the role of TAp63 could not be examined and was exclusively focused on the use of ΔNp63-deficient mouse models. Additionally, it is important to note that p63 is used as a diagnostic marker in breast cancer with no regard for the existence of distinct isoforms and their functions potentially leading to improper diagnosis and treatment of breast cancer patients. Here we used the tumor-prone TAp63−/− mouse model to specifically dissect the role of the TAp63 isoforms in the mammary gland. Consistent with previous reports,3 we found that lack of TAp63 does not dramatically affect the developing mammary gland, as mammary glands from virgin TAp63−/− females have a higher proliferative index than WT mammary glands but are otherwise indistinguishable from those of WT females. However, we unveiled striking phenotypes subsequent to serial mammary transplantation, suggesting that TAp63 has similar roles in the mammary gland as it does in the skin, that is, TAp63 is activated in response to stress and is critical to maintain stem and progenitor cells in quiescence. Indeed, loss of TAp63 led to an increase in TICs through the regulation of cell polarity and EMT. Moreover, mouse and mammary adenocarcinomas deficient for TAp63 and an orthotopic xenograft mouse model of mammary adenocarcinoma had an increased number of TICs, indicating that TAp63 serves to regulate stem cell properties and its loss leads to an expansion of TICs.
We previously reported that 8% of TAp63−/− mice spontaneously develop mammary adenocarcinoma late in life (9–16 months of age).14 In this study, TAp63−/− MECs were serially transplanted nine times in 10-week-old WT recipients. We did not observe any frank mammary gland adenocarcinomas but did detect in situ ductal carcinoma in the transplantation model. The difference in the models is most likely due to age and tumor latency. In fact, we did see mammary adenocarcinoma formation in MCF10-shTAp63 cells injected to SCID mice after 8 months (Figure 7e).
The Hippo pathway known to regulate organ size in Drosophila has been found to have important roles in the regulation of cell proliferation, cell polarity, stem cell maintenance and cancer in mammals.22, 24, 25, 38 The upstream regulators of the Hippo pathway are an area of intense investigation and tumor suppressors that may regulate this pathway are still being unveiled. We found that the tumor-suppressor TAp63 regulates the Hippo pathway effector TAZ through transcriptional regulation of LKB1, which has previously been shown to have important roles in stem cell regulation and metastasis.22, 38, 47 Our laboratory had already made two key discoveries: (1) TAp63 transcriptionally activates Lkb1 in liver, muscle and fat tissues to regulate glucose metabolism,40 and (2) loss of TAp63 results in the development of metastatic mammary adenocarcinoma.14 What was unknown were the mechanisms employed by TAp63 that lead to the development of mammary adenocarcinomas. Here we show that TAp63 also transcriptionally regulates LKB1 in MECs and mammary cancer cell lines. Loss of regulation of LKB1 in TAp63-deficient MECs resulted in loss of Scrib expression and activation of the Hippo pathway through TAZ and a subsequent loss of cell polarity and accumulation of TICs. Importantly, re-expression of LKB1 in MECs lacking TAp63 resulted in rescue of the aforementioned phenotypes. Cell polarity was restored and the number of MaSCs was reduced to WT levels. Taken together, we have unveiled mechanisms employed by the tumor- and metastasis-suppressor TAp63 to regulate stem cell properties in MECs, leading to the formation of TICs through regulation of the Hippo pathway. These observations are important for the appropriate use of p63 as a diagnostic marker and the potential treatments chosen for these patients.
Materials and methods
Animal studies and mammary gland transplantation
TAp63−/− and WT female mice on an enriched C57BL/6 background (95%) were used in this study. For mammary gland transplantation, the endogenous mammary epithelium was surgically removed from the four inguinal glands of WT females at 3 weeks of age to provide a cleared mammary fat pad.48 The donor mice were prepared as follows: fragments of 10-week-old WT or TAp63−/− mammary glands were inserted into the cleared mammary fat pads. WT was inserted on the right and TAp63−/− on the left. Ten weeks after mammary gland transplantation, fat pads were excised for histological and IHC analysis. All in vivo mouse procedures were approved by the IACUC at the University of Texas MD Anderson Cancer Center.
Mammary gland limiting dilution assay
Primary mammary gland epithelial cells were isolated from 10-week-old WT and TAp63−/− female mice and injected into cleared mammary fat pads of WT female mice. Transplantation was performed using 2500, 1000, 500 and 250 of MECs of each genotype resuspended in 50% Matrigel and 50% phosphate-buffered saline (PBS).48 WT cells were injected on the right and TAp63−/− cells were injected on the left. The outgrowths of mammary glands were analyzed at 10 weeks posttransplantation by histology and IHC.
Whole-mount mammary glands analyses
Mammary glands were removed from female mice, spread flat and fixed in 4% paraformaldehyde for 2 h at 4 °C. The glands were washed in 70% ethanol for 15 min followed by a brief rinse in PBS. The tissues were then stained in carmine alum solution (1 g of carmine and 2.5 g of aluminum potassium sulfate/500 ml of water) at room temperature overnight. The glands were flattened between glass slides to capture whole-mount images (× 10 magnification) using a Zeiss Stemi 200-CS microscope (Jena, Germany).
Orthotopic xenograft mouse model
Female SCID mice (6 weeks old) were randomized into four groups: MCF10A-shCON (n=9), MCF10A-shTAp63 (n=12), MCF7-shCON (n=12), and MCF7-shTAp63 (n=12). All mice for MCF7 cells were treated by subcutaneous implantation of 17β-estradiol (0.72 mg 17β-estradiol, 90-day release, Innovative Research of America, Sarasota, FL, USA). MCF10A or MCF7 expressing a scrambled shRNA (shCON) or shTAp63 (1 × 106 cells for MCF10A and 100 000 cells for MCF7 in matrigel) were implanted orthotopically into the mammary fat pads of the mice. Tumor xenografts were collected at 8 weeks after injection for MCF7 tumors and 8 months after injection for MCF10A tumors, photographed and analyzed using IHC. Total RNA was isolated from tumor xenografts for RT-PCR analysis.
Cell lines and culture conditions
MCF10A cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (1:1) media containing 5% horse serum, 10 μg/ml insulin, 20 ng/nl epidermal growth factor, 500 ng/ml hydrocortisone and primocin 100 μg/ml. Mouse epithelial cell lines were developed from mammary glands of WT and TAp63−/− mice at 10 weeks of age. Freshly dissected mammary glands were enzymatic dissociated overnight in DMEM/F12 medium containing collagenase (1 mg/ml), hyaluronidase (100 U/ml) and antibiotic–antimycotic solution (10 000 units/ml of penicillin, 10 000 μg/ml of streptomycin and 25 μg/ml of fungizone antimycotic). Cell pellets were washed two times with PBS+5% fetal bovine serum and were digested by 0.25% Trypsin–2.21 mM EDTA to get single cells. Trypsin was inactivated with PBS+5% fetal bovine serum. Cells were filtered through a 40 μm strainer to yield single-cell suspension. Cells were cultured in DMEM/F12 (1:1) media containing the same components used for MCF10A cells. All cultured cells were mycoplasma negative.
In vitro lentivirus and adenovirus infections
shTAp63 and control lentivirus vectors were a gift from Leif Ellisen.4 MCF10A cells were infected with virus-containing media supplemented with 2 μg/ml polybrene for 24 h and were selected by puromycin for 3 days as described previously.14 Suspension of MCF10-shCON and MCF10A-shTAp63 cells were infected with human adenoviruses (Ad-LKB1) (Vector BioLabs, Malvern, PA, USA) or Ad-GFP (Vector Development Lab, Houston, TX, USA) for 1 h at a multiplicity of infection of 50. The efficiency of infection was quantified by assessing green fluorescent protein (GFP)-positive cells.
Soft agar assay
MCF7 cells expressing a scrambled shRNA (shCON) or an shRNA for TAp63 (shTAp63) (50 000 cells) were seeded on the top layer in 0.3% agar in DMEM media in six-well plates. The base layer was composed of 1.5 ml of 0.6% agar in DMEM. Plates were allowed to solidify, then 1.5 ml DMEM was added to each well. Plates were incubated at 37 °C, and the media was changed every 3 days. Colonies were counted and photomicrographs were taken at day 21 using a Celigo instrument (Nexcelom, Lawrence, MA, USA).
Western blotting analysis
Western blotting analysis was performed as described previously.5 Primary antibodies used were as follows: slug (Santa Cruz, 1:1000; Santa Cruz, CA, USA), sox9 (Millipore, 1:2000; Darmstadt, Germany), ALDH1 (BD Bioscience, 1:500; San Jose, CA, USA), αSMA (Sigma, 1:1000; St Louis, MO, USA), LKB1 (Cell Signaling, 1:500; Beverly, MA, USA), Scrib (Santa Cruz, 1:1000), YAP/TAZ (Cell Signaling, 1:500), CTGF (Santa Cruz, 1:500), TAp63 (BioLegend, 1:500; San Diego, CA, USA), ΔNp63 (BioLegend, 1:500), BMP4 (Abcam, 1:20000; Cambridge, MA, USA), E-cadherin (Cell Signaling, 1:2000), and sox2 (Abcam, 1:1000). Corresponding secondary antibodies conjugated to horseradish peroxidase (Amersham Biosciences, 1:5000; Marlborough, MA, USA) were used. Detection was performed using the ECL Plus Kit (Amersham). Actin was used as a loading control.
Immunofluorescence and immunohistochemistry
For IF, paraffin-embedded sections were stained as described previously40 using the following primary antibodies: TAZ (Cell Signaling, 1:1000), K18 (Sigma, 1:200), αSMA (Sigma, 1:250), Ki67 (Abcam, 1:1000), caspase 3 (Cell Signaling, 1:50), AQP (Calbiochem, 1:100; Darmstadt, Germany), Scrib (Santa Cruz, 1:100), Vangl2 (Santa Cruz, 1:50), GM130 (BD Bioscience, 1:150), Sox9 (Millipore, 1:100), NKCC and Slug (Santa Cruz, 1:100). Alexa 488 and Alex 568-conjugated secondary antibodies (ThermoFisher Scientific, Molecular Probes, Waltham, MA, USA) were used at 1:1000. DAPI (6-diamidino-2-phenylindole) was used to counterstain nuclei. Images were acquired using Zeiss Axiovert 40CFL microscope. For IHC, the following primary antibodies were used: vimentin (BD Bioscience, 1:200), LKB1 (Cell Signaling, 1:100), BMP4 (Abcam, 1:100), sox2 (Abcam, 1:100), and ALDH (BD Bioscience, 1:50). For detection, the ImmPRESS REAGENT KIT (Vector Laboratories, MP-7500) was used followed by the DAB kit (Vector Laboratories, SK-4100) and counterstained with hematoxylin (Vector Laboratories, H-3041).
Chromatin immunoprecipitation
Mouse epithelial cells of the indicated genotypes were cultured as previously described.5 Similarly, human MCF10A cells treated with a scrambled non-targeting (NT) shRNA or shTAp63 were grown to near confluence. TAp63 chromatin immunoprecipitation analysis was performed using anti-TAp63 antibody (D20, SantaCruz, 2 μg/mg protein) or immunoglobulin G as described previously.40 Mouse TAp63-binding sites on LKB1 promoter were validated with the primers as described previously.40 Putative human TAp63-binding sites were found by scanning 5000 bp upstream of the 5′ untranslated region and intron 1 of the LKB1 gene utilizing the Genomatix software prediction tool (Ann Arbor, MI, USA). qRT-PCR was performed using primers in the human LKB1 promoter: (1) Site 1 (−3169)—forward 5′-CCTCCCAAAGTGCTGGGATTA-3′ and (−3068)—reverse 5′-CTGGGATTACAGGCATGAGG-3′, (2) Intron 1: Site 2 (+6548)—forward 5′-ATGTTGTCCAGGCTGGTCTC-3′ and (+6635)—reverse 5′-GTGAACGTGGCGAAGCAGT-3′, and (3) non-specific: (−221)—forward 5′-ATGGCAGGTTCAACCAACG-3′ and (−122)—reverse 5′-GCCGCCATCTTGTTTACCT-3′.
Fluorescence-activated cell sorting (FACS) and flow cytometry
MCF10A-shCON and -shTAp63 cells were labeled with CD24-PE (Biolegend, 1:00) and CD44-Alexa 488 (Biolegend, 1:00) at a concentration of 10 million cells/ml for 20 min and were subjected to FACS analysis and sorting on a triple laser MoFlo FACs machine (Cytomation, Fort Collins, CO, USA). Data analysis was performed using the FlowJo software (Ashland, OR, USA).
Mammary epithelial 3D cell culture
Mammary epithelial 3D cell culture (3D culture) was performed as described previously in growth factor-reduced Matrigel (BD Biosciences).49 For MCF10A cell-infected derivative cell lines, 5000 cells were seeded in each well of an eight-well chamber slide. WT and TAp63−/− primary mouse MECs were isolated from mammary organoids.50, 51 Briefly, mammary glands were removed from 8- to 12-week-old female mice and minced 40–50 times with a scalpel. The epithelial compartment was separated from the stromal tissues using collagenase in PBS (2 mg/ml) digestion. Differential centrifugation was used to separate epithelial organoids from single cells. Pellets containing the mammary organoids were digested using 0.05% Trypsin–2.21 mM EDTA to obtain single cells. In all, 10 000 cells were seeded per well of an eight-well chamber slide.
In vitro wound healing assay
Confluent MCF10A cell monolayers were treated with 4 μg/ml mitomycin C for 2 h to stop cell proliferation. The monolayer of cells was then ‘wounded’ by scratching the surface with a P200 micropipette tip. Six hours following the wound, cells were fixed in 4% paraformaldehyde for immunofluorescent staining. Images were captured using an inverted Zeiss Axiovert 40CFL microscope. For time-lapse imaging, the cells were labeled with nuclear stain (Hoechst, no. 33342, 1 μg/ml) for 20 min. Time-lapse imaging was performed for 24 h on an inverted Nikon Ti microscope (Melville, NY, USA) equipped with phase-contrast optics and a Hamamatsu Flash 4.0 camera (Hamamatsu, Japan). Nikon elements software was used for acquisition and analysis of the time-lapse data.
RNA-Seq and bioinformatics analysis
RNA-sequencing of WT and TAp63−/− mouse epithelial cells was performed as described previously.8, 11 ESC, hNMSC and Hippo and Yap-conserved pathway signatures from previously published studies (Supplementary Table S2)22, 28, 34, 35 were compared with TAp63−/− and WT MEC signatures using Gene Set Enrichment Analysis from the Broad Institute (Cambridge, MA, USA). Gene Set Enrichment Analysis settings were 1000 permutations, classic scoring scheme and maximum probe mode. A positive and negative normalized enrichment score indicate same and opposite direction of signature, respectively. In pathway analysis, we utilized BP, KEGG and Reactome sources in order to identify pathways enriched in our signatures. P-value was set at <0.05 for significance.
Statistical analysis
All data are represented as mean±s.e.m. One-way analysis of variance or Student’s t-test was used for statistical comparison between two groups. A P-value of 0.05 was considered significant. Sample sizes were six for all in vivo mouse experiments and at least in triplicate for all cell culture assays.
References
Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A . p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999; 398: 708–713.
Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398: 714–718.
Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol 2014; 16: 1004–1015.
Forster N, Saladi SV, van Bragt M, Sfondouris ME, Jones FE, Li Z et al. Basal cell signaling by p63 controls luminal progenitor function and lactation via NRG1. Dev Cell 2014; 28: 147–160.
Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 2009; 5: 64–75.
Koker MM, Kleer CG . p63 expression in breast cancer: a highly sensitive and specific marker of metaplastic carcinoma. Am J Surg Pathol 2004; 28: 1506–1512.
Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell 2006; 24: 593–602.
Venkatanarayan A, Raulji P, Norton W, Chakravarti D, Coarfa C, Su X et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015; 517: 626–630.
Su X, Chakravarti D, Flores ER . p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer 2013; 13: 136–143.
Koster MI, Roop DR . The role of p63 in development and differentiation of the epidermis. J Dermatol Sci 2004; 34: 3–9.
Chakravarti D, Su X, Cho MS, Bui NH, Coarfa C, Venkatanarayan A et al. Induced multipotency in adult keratinocytes through down-regulation of DeltaNp63 or DGCR8. Proc Natl Acad Sci USA 2014; 111: E572–E581.
Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 2012; 139: 772–782.
Li N, Singh S, Cherukuri P, Li H, Yuan Z, Ellisen LW et al. Reciprocal intraepithelial interactions between TP63 and hedgehog signaling regulate quiescence and activation of progenitor elaboration by mammary stem cells. Stem Cells 2008; 26: 1253–1264.
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010; 467: 986–990.
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009; 137: 87–98.
Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009; 139: 1327–1341.
Ricardo S, Vieira AF, Gerhard R, Leitao D, Pinto R, Cameselle-Teijeiro JF et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol 2011; 64: 937–946.
Tsang JY, Huang YH, Luo MH, Ni YB, Chan SK, Lui PC et al. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res Treat 2012; 136: 407–417.
Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010; 7: 403–417.
Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012; 148: 1015–1028.
Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015; 525: 256–260.
Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147: 759–772.
Skibinski A, Breindel JL, Prat A, Galvan P, Smith E, Rolfs A et al. The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep 2014; 6: 1059–1072.
Shi P, Feng J, Chen C . Hippo pathway in mammary gland development and breast cancer. Acta Biochim Biophys Sin (Shanghai) 2015; 47: 53–59.
Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA et al. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev 2014; 28: 432–437.
Zhao R, Fallon TR, Saladi SV, Pardo-Saganta A, Villoria J, Mou H et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev Cell 2014; 30: 151–165.
Li J, Liu J, Yang J, Li P, Mao X, Li W et al. Loss of LKB1 disrupts breast epithelial cell polarity and promotes breast cancer metastasis and invasion. J Exp Clin Cancer Res 2014; 33: 70.
Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL et al. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol 2014; 16: 108–117.
Anastas JN, Biechele TL, Robitaille M, Muster J, Allison KH, Angers S et al. A protein complex of SCRIB, NOS1AP and VANGL1 regulates cell polarity and migration, and is associated with breast cancer progression. Oncogene 2012; 31: 3696–3708.
Vaira V, Faversani A, Dohi T, Maggioni M, Nosotti M, Tosi D et al. Aberrant overexpression of the cell polarity module scribble in human cancer. Am J Pathol 2011; 178: 2478–2483.
Dow LE, Kauffman JS, Caddy J, Zarbalis K, Peterson AS, Jane SM et al. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene 2007; 26: 2272–2282.
Yates LL, Schnatwinkel C, Hazelwood L, Chessum L, Paudyal A, Hilton H et al. Scribble is required for normal epithelial cell-cell contacts and lumen morphogenesis in the mammalian lung. Dev Biol 2013; 373: 267–280.
Yamben IF, Rachel RA, Shatadal S, Copeland NG, Jenkins NA, Warming S et al. Scrib is required for epithelial cell identity and prevents epithelial to mesenchymal transition in the mouse. Dev Biol 2013; 384: 41–52.
Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008; 40: 499–507.
Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010; 140: 62–73.
Amin N, Vincan E . The Wnt signaling pathways and cell adhesion. Front Biosci (Landmark Ed) 2012; 17: 784–804.
Nusse R . Wnt signaling and stem cell control. Cell Res 2008; 18: 523–527.
Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2014; 34: 681–690.
Hiemer SE, Varelas X . Stem cell regulation by the Hippo pathway. Biochim Biophys Acta 2013; 1830: 2323–2334.
Su X, Gi YJ, Chakravarti D, Chan IL, Zhang A, Xia X et al. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab 2012; 16: 511–525.
Biernaskie J, Paris M, Morozova O, Fagan BM, Marra M, Pevny L et al. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell 2009; 5: 610–623.
Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci USA 2015; 112: 3499–3504.
Muller PA, Trinidad AG, Caswell PT, Norman JC, Vousden KH . Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J Biol Chem 2014; 289: 122–132.
Prat A, Perou CM . Deconstructing the molecular portraits of breast cancer. Mol Oncol 2011; 5: 5–23.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121: 2750–2767.
Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 2007; 8: R76.
Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 2010; 468: 659–663.
Smith GH . Experimental mammary epithelial morphogenesis in an in vivo model: evidence for distinct cellular progenitors of the ductal and lobular phenotype. Breast Cancer Res Treat 1996; 39: 21–31.
Debnath J, Muthuswamy SK, Brugge JS . Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30: 256–268.
Nguyen-Ngoc KV, Shamir ER, Huebner RJ, Beck JN, Cheung KJ, Ewald AJ . 3D culture assays of murine mammary branching morphogenesis and epithelial invasion. Methods Mol Biol 2015; 1189: 135–162.
Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z . Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell 2008; 14: 570–581.
Acknowledgements
We thank the work of Henry P Adams, Microscopy Core of the Genetics Department. We also thank Leif Ellisen for the kind gift of the TAp63 shRNA. This study was funded by R35CA197452, R01CA160394 and R01CA134796 from the National Cancer Institute (NCI) and RP120124 and RP150094 from the Cancer Prevention & Research Institute of Texas (CPRIT) and funding from the Hildegardo E and Olga M. Flores Foundation to ERF, NCI-Cancer Center Core Grant (CA-16672) (University of Texas MD Anderson Cancer Center) and R01CA16303 to JMR. ERF is an NCI Outstanding Investigator, a Moffitt Distinguished Scholar, a scholar of the Leukemia and Lymphoma Society, the Rita Allen Foundation and the V Foundation for Cancer Research. MN is a CPRIT-TRIUMPH Scholar and was supported by a Research Training Award from the Cancer Prevention and Research Institute of Texas (RP140106). HAA is an Odyssey Fellow at MD Anderson Cancer Center. FB was supported in part by an American Cancer Society grant (IRG-09-062-01), awarded to the University of Kansas Medical Center.
Author contributions
ERF, FB and XS initiated the project. XS, MN, AV, NHB, YYG, FK, HAA, CC and PHG performed the experiments and analyzed the data. DM, JMR, FB, XS and ERF discussed the results and commented on the manuscript at each stage. ERF and XS wrote the paper with input from other authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Su, X., Napoli, M., Abbas, H. et al. TAp63 suppresses mammary tumorigenesis through regulation of the Hippo pathway. Oncogene 36, 2377–2393 (2017). https://doi.org/10.1038/onc.2016.388
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.388
This article is cited by
-
Hypoxia-activated XBP1s recruits HDAC2-EZH2 to engage epigenetic suppression of ΔNp63α expression and promote breast cancer metastasis independent of HIF1α
Cell Death & Differentiation (2024)
-
Identification of multiple organ metastasis-associated hub mRNA/miRNA signatures in non-small cell lung cancer
Cell Death & Disease (2023)
-
p63: a crucial player in epithelial stemness regulation
Oncogene (2023)
-
p63 suppresses the ability of pregnancy-identified mammary epithelial cells (PIMECs) to drive HER2-positive breast cancer
Cell Death & Disease (2021)
-
ZNF750 represses breast cancer invasion via epigenetic control of prometastatic genes
Oncogene (2020)
