
Key Points
-
Immune sensing of foreign nucleic acids among abundant self nucleic acids is a hallmark of virus detection and antiviral defence.
-
Efficient antiviral defence requires a balanced process of sensing foreign nucleic acids and ignoring self nucleic acids. This balance is accomplished by a multilevel, fail-safe system which combines immune sensing of pathogen-specific nucleic acid structures with specific labelling of self nucleic acids and nuclease-mediated degradation.
-
Cellular localization of nucleic acids, nucleic acid secondary structure, nucleic acid sequence and chemical modification all contribute to selective recognition of foreign nucleic acids.
-
Nucleic acid sensing occurs in immune cells and non-immune cells and results in antiviral responses that include the induction of antiviral effector proteins, the secretion of cytokines alarming neighbouring cells, the secretion of chemokines, which attract immune cells, and the induction of cell death.
-
Vertebrate cells cannot completely avoid the occurrence of endogenous self nucleic acid structures with immunostimulatory properties. Therefore, additional mechanisms involving self-nucleic acid modification and nuclease-mediated degradation are necessary to diminish uncontrolled immune activation.
-
Viruses have established sophisticated mechanisms to exploit and adopt endogenous tolerance mechanisms or to avoid the presentation of characteristic molecular features recognized by nucleic acid sensing receptors.
Abstract
Innate immunity against pathogens relies on an array of immune receptors to detect molecular patterns that are characteristic of the pathogens, including receptors that are specialized in the detection of foreign nucleic acids. In vertebrates, nucleic acid sensing is the dominant antiviral defence pathway. Stimulation of nucleic acid receptors results in antiviral immune responses with the production of type I interferon (IFN), as well as the expression of IFN-stimulated genes, which encode molecules such as cell-autonomous antiviral effector proteins. This Review summarizes the tremendous progress that has been made in understanding how this sophisticated immune sensory system discriminates self from non-self nucleic acids in order to reliably detect pathogenic viruses.
Similar content being viewed by others
Main
The induction of type I interferon (IFN) is a hallmark of immune sensing of nucleic acids by the innate immune system. Type I IFN was identified on the basis of its functional ability to restrict viral replication. Soon after its discovery, specific types of nucleic acids were found to potently induce this key antiviral cytokine and thus mimic the presence of virus1. The identification of the corresponding immune sensing receptors was a prerequisite to any real understanding of how self and non-self nucleic acids can be distinguished.
The basic molecular structure of DNA and RNA is universal throughout biology. Thus, the recognition of foreign nucleic acids among an abundance of self nucleic acids in an organism is a substantial biochemical challenge. There is now a broad consensus within the field that most of the receptors involved in the immune sensing of nucleic acids have been identified. Given the substantial progress over the last few years, it seems timely to review the current literature and to convey an updated concept of nucleic acid sensing. First, we provide a brief overview of the general principles of nucleic acid sensing. Then, the current literature on RNA and DNA sensing is reviewed with a focus on self versus non-self recognition. We conclude with open questions, future perspectives and implications for human disease. For more detailed information about the molecular structure of the receptors, their specific immune functions and therapeutic development, we refer to other in-depth reviews2,3,4,5,6,7,8,9,10.
Principles of nucleic acid sensing
Most non-vertebrates, such as insects, plants and nematodes, rely mainly on RNA interference (RNAi) for antiviral defence. However, in vertebrates, although there are indications that RNAi is involved in the degradation of genomic RNA of some viruses in some cell types (reviewed in Ref. 11), other reports suggest that there is competition rather than cooperation between small interfering RNA (siRNA)-mediated RNAi and type I IFN-dominated antiviral pathways12 (see below). Overall, RNAi does not seem to have an essential function in vertebrate antiviral defence but, if not the sequence, which biochemical and cell biological features of nucleic acids are used for the reliable discrimination of foreign and self?
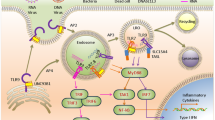
Availability, localization and structure. In vertebrates, the principle of self versus non-self nucleic acid sensing is based on three central criteria (Fig. 1): first, the availability of nucleic acid ligands as determined by their local concentration, by the rate of degradation by endogenous nucleases and by the level of shielding (for example, by nucleocapsid proteins); second, the localization of nucleic acid ligands, such as outside the cell membrane, in the endolysosomal compartment or in the cytosol; and third, the structure of nucleic acid ligands as characterized by sequence motifs, conformation (for example, base-paired versus unpaired) and chemical modification. In many cases, a combination of all three aspects contributes to the reliable recognition of potentially dangerous foreign nucleic acids leading to the induction of appropriate innate immune responses.

The detection of foreign nucleic acids is based on their availability, localization and structure. Nucleases rapidly degrade most self nucleic acids before they can be sensed by nucleic acid receptors, and the localization of nucleic acids determines if the potentially immunoactive nucleic acids are accessible for their detectors. The nucleic acid receptors can be divided into two main categories: immune sensing receptors, which include Toll-like receptor 3 (TLR3), TLR7, TLR8, TLR9, retinoic acid inducible gene I (RIG-I), melanoma differentiation associated gene 5 (MDA5), absent in melanoma 2 (AIM2) and cyclic GMP–AMP synthetase (cGAS); and nucleic acid receptors with direct antiviral activity, including double-stranded RNA (dsRNA)-activated protein kinase R (PKR), IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), 2′-5′-oligoadenylate synthetase 1 (OAS1) and ribonuclease L (RNase L), and adenosine deaminase acting on RNA 1 (ADAR1). Immune sensing receptors can detect structural features such as dsRNA or 5′-triphosphate RNA, which indicate non-self, and indirectly or directly induce transcription factors that upregulate the expression of antiviral effector proteins, chemokines and cytokines, including type I interferon (IFN), to promote an antiviral immune response. In addition, these immune sensing receptors induce the expression of nucleic acid receptors with direct antiviral activity through the induction of IFN-stimulated genes. Those effector proteins primarily do not induce transcription factors but act directly on the target RNA. Of note, in addition to its primary role as a sensor, RIG-I has direct antiviral activity. ssRNA, single-stranded RNA.
Two categories of nucleic acid receptors. The first category of nucleic acid receptors comprises pattern recognition receptors (PRRs) of the Toll-like receptor (TLR) family (specifically, TLR3, TLR7, TLR8 and TLR9), the RIG-I-like receptor (RLR) family of RNA sensors (such as retinoic acid inducible gene I (RIG-I; also known as DDX58) and melanoma differentiation associated gene 5 (MDA5; also known as IFIH1)), and the DNA sensors absent in melanoma 2 (AIM2) and cyclic GMP–AMP synthetase (cGAS). These PRRs directly or indirectly induce transcription factors, including nuclear factor-κB (NF-κB) and IFN-regulatory factor 3 (IRF3), that upregulate the expression of antiviral effector proteins, chemokines and cytokines (Fig. 1). This antiviral response is dominated by type I IFN and IFN-stimulated genes (ISGs).
Although the expression of some PRRs (for example, AIM2, RIG-I and MDA5) are enhanced by type I IFN in a positive feedback loop, the initial expression of most of the second category of receptors requires type I IFN or PRR signalling. Second category receptors comprise nucleic acid receptors with direct antiviral activity — for example, double-stranded RNA (dsRNA)- activated protein kinase R (PKR; also known as eIF2AK2), 2′-5′-oligoadenylate synthetase 1 (OAS1) and adenosine deaminase acting on RNA 1 (ADAR1). Like PRRs, they recognize the biochemical features of foreign nucleic acids. However, unlike PRRs, their major function is not to induce immune responses via transcription factors and cytokines but rather to directly act on viral RNA (for example, by inhibiting translation or by chemical modification or degradation of their target RNA). Recent findings have shown that these categories can overlap: as detailed below, RIG-I can also act as an effector molecule on viral RNA13,14, translational inhibition of PKR was reported to enhance NF-κB signalling15, and RNA modifications or cleavage can inactivate or generate target RNA structures that are recognized by PRRs.
RNA sensing
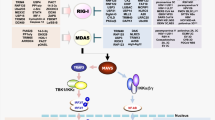
RNA-sensing receptors. The transmembrane TLRs TLR3, TLR7, and TLR8 recognize RNA in the endosome16,17,18,19 and, in the case of TLR3, at the surface of some cell types, such as fibroblasts and tumour cell lines20 (Fig. 2). In the cytosol, the RLRs RIG-I and MDA5, which are DExD/H box RNA helicases with caspase recruitment domains (CARDs)21, signal via mitochondrial antiviral signalling protein (MAVS; also known as CARDIF, IPS1 and VISA) and IRF3–IRF7 to induce type I IFNs following RNA recognition4 (Fig. 2). In addition to its signalling function, RIG-I can act as a direct antiviral effector protein that binds viral genomic RNA and interferes with viral polymerases13,14. A third cytosolic RIG-I-like helicase is LGP2 (also known as DHX58), which lacks a CARD and appears to contribute to the fine tuning of immune responses by inhibiting RIG-I and supporting MDA5 signalling22. Although LGP2 can recognize RNA, it is unclear whether this is relevant for its function23.

Double-stranded RNA (dsRNA) in the endosome is detected by Toll-like receptor 3 (TLR3) and TLR7, whereas single-stranded RNA (ssRNA) is sensed by TLR7 and TLR8. These receptors signal via myeloid differentiation primary response protein 88 (MYD88) and TIR domain-containing adaptor protein inducing IFNβ (TRIF) to induce interferon (IFN)-regulatory factor 3 (IRF3)–IRF7 and type I IFN production and via nuclear factor-κB (NF-κB) to induce pro-interleukin-1β (pro-IL-1β) and the inflammasome component NLRP3. The helicases retinoic acid inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) detect dsRNA in the cytosol and signal through mitochondrial antiviral signalling protein (MAVS) to induce type I IFN production and pro-apoptotic signalling via IRF3–IRF7, and to activate the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome resulting in IL-1β production. The RNA receptor 2′-5′-oligoadenylate synthetase 1 (OAS1) has direct antiviral activity and degrades RNA via the generation of 2′-5′-oligoadenylates that activate ribonuclease L (RNase L). RNase L generates 5′ OH- and 3′ phosphate-containing RNA fragments that can stimulate RIG-I. The crucial RIG-I-stimulating structure has not been determined to date. The RNA receptors IFN-induced protein with tetratricopeptide repeats 1 (IFIT1) and double- stranded RNA (dsRNA)-activated protein kinase R (PKR) inhibit cap-dependent translation. BAX, BCL-2-associated X protein; BCL-2, B cell lymphoma 2; BIRC3, baculoviral IAP repeat-containing protein 3; IFNAR, interferon-α/β receptor; PRKCE, protein kinase Cε; PUMA, p53-upregulated modulator of apoptosis; TRAIL, TNF-related apoptosis-inducing ligand.
In addition to IFN production, MAVS activation promotes apoptosis through several pathways: recruitment and activation of caspase 8 (Ref. 24); IRF3–IRF7-dependent upregulation of TRAIL and the pro-apoptotic proteins p53-upregulated modulator of apoptosis (PUMA; also known as BBC3) and NOXA (also known as PMAIP1); downregulation of the anti-apoptotic genes BCL2, BIRC3 and PRKCE; and direct activation of the pro- apoptotic BCL-2-associated X protein (BAX)25,26,27,28 (Fig. 2). Furthermore, MAVS was implicated in the activation of the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome, thereby contributing to the induction of an interleukin-1β (IL-1β)-dependent pro-inflammatory cytokine response29,30,31,32,33 (Fig. 2).
RNA-sensing receptors with direct antiviral activity. In addition to immune receptors that induce antiviral immune responses, there are several receptors that, upon RNA recognition, directly inhibit viral replication and propagation. Since viral replication crucially relies on the host's translation machinery, most antiviral mechanisms specifically or nonspecifically target the mRNA translation process (Fig. 2).
One such receptor is PKR, which inhibits viral and host cap-dependent translation by phosphorylating eukaryotic translation initiation factor 2A (eIF2A)34. Further antiviral functions of PKR have been postulated but are controversial. IFN-induced protein with tetratricopeptide repeats 1 (IFIT1) is an antiviral effector protein35,36 that binds to the 5′ cap of mRNA lacking a 2′-O-methyl modification at the N1 position and blocks its translation37,38,39. IFIT1 has been proposed to compete with eIF4E (a 5′ cap binding and translation factor)38, which is supported by the observation that IFIT1 interferes with the formation of the 48S translation initiation complex39. To date, a crucial role for IFIT1 in the antiviral immune response was reported for negative sense single-stranded RNA ((−)ssRNA) viruses (vesicular stomatitis virus and influenza virus) and positive sense ssRNA ((+)ssRNA) viruses (West Nile virus and mouse hepatitis virus) but not picornaviruses which do not have a 5′ cap35,36,38.
RNA recognition by OAS1 provides another cell autonomous antiviral mechanism. Upon binding to double-stranded RNA (dsRNA), OAS1 synthesizes 2′-5′ oligomers of adenosine (2′-5′-oligoadenylate) as a second messenger40,41. 2′-5′-oligoadenylate in turn activate ribonuclease L (RNase L)42, which degrades viral and cellular RNA molecules (Fig. 2). The antiviral molecule ADAR1 catalyses the C6 deamination of adenosine in base-paired RNA, which results in A-to-I conversions. A-to-I conversions change the coding of RNA, as inosine (I) is read as guanosine (G) instead of adenosine (A) by ribosomes, leading to the translation of altered, potentially non-functional, proteins43. Furthermore, conversion of A-U to I-U further destabilizes potential secondary structures important for viral RNA regulation43. However, as detailed below, the destabilization of secondary structures can also impair recognition by PRRs, thereby supporting immune escape of the virus. In conclusion, the role of ADAR1 in the context of viral infections remains unclear and might depend on the type of the virus44. Notably, the expression and functions of PKR, IFIT1, OAS1 and ADAR1 are all downstream of the type I IFN-inducing nucleic acid immune receptors (such as RLRs).
RNA recognition motifs
In the following section, we discuss structural motifs of RNA that are recognized by sensing receptors, such as secondary structure, RNA modification and RNA sequence (Fig. 3). Interestingly, the same structural feature or modification is often recognized by evolutionarily unrelated receptors, pointing to a convergent evolution of PRRs.

Double-stranded RNA (dsRNA), which is the prototypic non-self nucleic acid stimulus, is detected by three signalling receptors: Toll-like receptor 3 (TLR3), which is located on the cell membrane and in the endosomal membrane, and retinoic acid inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5), which are located in the cytosol. Long forms of dsRNA are recognized independently of the structure at the ends (TLR3 recognizes dsRNA >35 bp, and MDA5 recognises dsRNA >300 bp). A short stretch of dsRNA (>19 bp) is sufficient for recognition by RIG-I if a triphosphate or a diphosphate is present at the 5′ end, and if the end is blunt with no overhangs. A 2′-O-methyl group at the first nucleotide (N1) of the 5′ end is part of the cap 1 structure of self RNA that labels it as self, and thus prevents recognition by RIG-I.
RNA secondary structures recognized as non-self RNA. Long dsRNA in the cytosol is a hallmark of DNA and RNA virus replication and is absent from an uninfected host cell45. Polyinosinic–polycytidylic acid (poly(I:C)), which mimics dsRNA, binds to and triggers the activation of the RNA sensors PKR, OAS1, TLR3, MDA5 and RIG-I, and thus poly(I:C) or its derivatives have been used as a tool to identify and characterize dsRNA recognition receptors and ligand requirements16,21,46,47,48,49. TLR3-mediated NF-κB activation requires dsRNA of 35–39 bp in length50 (Table 1). Of note, mouse TLR3 responds to shorter dsRNA, including siRNAs that are not recognized by human TLR3 (Refs 51,52). This observation calls into question the use of mouse models for testing siRNA-based therapeutic approaches. In addition to long dsRNA, ssRNA segments harbouring stem structures with bulge loops have been proposed as possible ligand structures that are recognized by TLR3 (Ref. 53).
The cytosolic helicase MDA5 is potently activated by very long dsRNA (>300 bp) — for example, poly(I:C) — but the minimal length requirement of a biologically relevant dsRNA remains unclear46,47,48. The current model for the recognition of continuous dsRNA by MDA5 is that MDA5 uses the long dsRNA as a signalling platform to cooperatively assemble a filament of MDA5 molecules in a head-to-tail arrangement with exposed CARDs along the long dsRNA; the exposed CARDs in turn recruit and activate the downstream adaptor MAVS54. It has recently been shown that the RNA-modifying activity of ADAR1 (that is, the conversion of A to I) is required to prevent endogenous dsRNA from activating MDA5 (Refs 55,56); given that there is no evidence for endogenous dsRNA of that length, these data suggest that MDA5 senses ligand motifs in addition to very long dsRNA. Several studies demonstrate that MDA5 activation concurs with the formation of dsRNA in the course of viral replication23. Recent data suggest that MDA5 contributes to the detection of (−)ssRNA viruses despite the absence of long dsRNA45. MDA5 was found to bind to and be activated by the mRNA of paramyxovirus (a (−)ssRNA virus)57,58, with paramyxovirus V protein binding to and inhibiting MDA5 but not RIG-I59. Furthermore, an in vivo short hairpin RNA screen identified MDA5 as one of the key antiviral proteins involved in the antiviral defence response against influenza A virus60. These data indicate that (−)ssRNA viruses, which do not produce dsRNA, are sensed by MDA5 and can actively interfere with MDA5 function. It is therefore tempting to speculate that, in addition to long dsRNA, ssRNA with short base-paired secondary structures is recognized by MDA5.
'Short' fragments of poly(I:C) (<300 bp) and long random dsRNA molecules (>200 bp) have both been reported to activate RIG-I in a manner independent of 5′-modifications48,61. However, these findings are controversial as the recognition by RIG-I of long genomic dsRNA of reovirus was found to depend on the presence of 5′-diphosphates49 (see below).
An examination of poly(I:C) molecules of different molecular size revealed that the minimum length of dsRNA for the activation of PKR and OAS1 was above 30 bp62. For PKR, this was confirmed with dsRNA fragments of heterogeneous sequence and defined length that were derived from phage RNA polymerases in vitro63. Owing to A-to-I conversions, ADAR1 activity can also impair dsRNA recognition by PKR and OAS1 (Refs 55,56), indicating a broad immunomodulatory role of ADAR1 as an inhibitor of the dsRNA sensors MDA5, PKR and OAS1 and explaining its role in preventing autoinflammatory diseases (reviewed in Ref. 2).
Altogether, dsRNA, which is a hallmark of viral replication, and long RNA with base-paired structures appear to be strong recognition motifs for several evolutionarily unrelated PRRs.
Sequence-dependent recognition of foreign RNA. Some RNA-sensing receptors have sequence preferences (Table 1). TLR7 and TLR8 are preferentially activated by poly-uridine (polyU) and by guanosine and uridine rich (GU-rich) sequences18,19,64. Since polyU or GU-rich sequences are not more frequent in microbial RNA compared to vertebrate RNA, it is currently unclear how the preference for these motifs may help distinguish self from non-self RNA. Furthermore, although TLR8 exclusively detects ssRNA, TLR7 primarily detects dsRNA but can also accommodate certain ssRNA oligonucleotides65. However, the biological rationale behind these sequence preferences is still unknown. Of note, in a recent study a binding site for a single ssRNA U nucleotide and another binding site for short RNA degradation products were found in a TLR8–ligand co-crystal, suggesting that TLR8 is activated by degradation products of ssRNA66. Similarly, TLR7 was proposed to sense G derivatives of ssRNA67.
Studies have shown that RIG-I and MDA5 have a preference for AU-rich sequences in long viral transcripts58,68. ADAR1 has binding domains for A-form dsRNA and for Z-form dsRNA, which guide ADAR1 to mediate highly site-selective adenosine deamination of only a few, specific adenosine residues, although non-selective editing of multiple sites in endogenous and viral RNAs has been described43. However, non-selective editing of multiple sites also has the potential to destroy secondary structures, which are potential recognition sites for MDA5, PKR and OAS1.
Another sequence-dependent antiviral mechanism is triggered by high frequencies of CpG and UpA (but not GpC or ApU) dinucleotides in coding RNA, and these dinucleotide motifs are indeed avoided in the RNA sequences of many RNA viruses (for example, picornaviruses). So far, the mechanism behind the antiviral effects of CpG and UpA motifs is unclear; however, it does not involve ADAR1, RIG-I, MDA5 and PKR or translation codon usage effects69,70.
Although many PRRs recognize RNA in a sequence dependent manner, the underlying biological significance is unknown.
Recognition of 5′-modified RNA. RIG-I is activated by blunt end 5′-triphosphate dsRNA that lacks overhangs71,72,73,74 (Table 1). Experimental approaches using synthetic or highly purified enzymatically generated 5′-triphosphate dsRNA revealed a minimum length of 18–19 bp of dsRNA for RIG-I recognition73,75. Structural studies confirmed the molecular requirement of a base-paired RNA region with a 5′-triphosphate end for RIG-I recognition76,77,78,79,80. By contrast, one group reported that 5′-triphosphate RNA that forms a hairpin structure can activate RIG-I even by a shorter 10 bp stretch81. However, it has to be considered that those hairpin RNAs could also form a 20mer heteroduplex dsRNA from two hairpins and in this way meet the minimum length of 18–19 bp (Table 1).
Nascent 5′-triphosphate RNA in the cytosol is indicative of RNA that has formed outside the nucleus and thus denotes viral infection72 or intracellular bacteria82,83,84. Enzymatic RNA biosynthesis is based on the 3′ to 5′ linkage of nucleotide triphosphates and consequently leaves a free triphosphate at the 5′ end in nascent RNA. Endogenous self RNA is generated in the nucleus and further processed (such as backbone or base modifications, cleavage and 5′ capping), resulting in the removal or modification of accessible 5′-triphosphate ends before the RNA is translocation to the cytosol. By contrast, the perfect replication of an RNA strand is a hallmark of many viruses and is always linked to the formation of a blunt end dsRNA structure containing 5′-triphosphate ends, which is recognized by cytosol RNA sensors. Furthermore, some (-)ssRNA viruses (for example, influenza virus) that minimize the occurrence of dsRNA by binding their ssRNA to nucleocapsid proteins possess genomes that form panhandle structures with a blunt end, due to partially complementary terminal sequences23. These partially complementary terminal sequences are a result of two ssRNA replication origins at the ends of the viral genomic RNA that are conserved, as both are recognized by the same viral RNA polymerase. Panhandle structures are recognized by RIG-I, a cytosol RNA sensor.
Certain (-)ssRNA viruses (for example, Sendai virus and vesicular stomatitis virus) separate nascent RNA strands with nucleocapsid proteins45 and are thus primarily devoid of dsRNA intermediates and panhandle structures. However, they still can be recognized by RIG-I via perfectly matched base-paired abortive RNA structures that are generated during erroneous replication (known as 'snap back' defective interfering RNA genomes)85.
In addition to 5′-triphosphate ends, RIG-I recognizes 5′-diphosphate ends of dsRNA, albeit with lower affinity than 5′-triphosphate. 5′-diphosphate ends are a characteristic molecular structure formed by certain viruses (such as reovirus)49. Interestingly, the enzymatic synthesis of poly(I:C) can leave free 5′-diphosphate ends and therefore contribute to the recognition of poly(I:C) by RIG-I49. Thus, poly(I:C) should not be regarded as a simple synthetic mimic of dsRNA, as other forms of dsRNA do not generally bear 5′-diphosphate moieties. The detection of di- or tri-phosphorylated 5′-ends of RNA allows RIG-I to reliably identify much shorter dsRNA molecules compared with TLR3 and MDA5 (Refs 72,73).
Besides RIG-I, three other nucleic acid receptors — IFIT1 and IFIT5 (Refs 35,36,37,39) and PKR86 — have been shown to recognize 5′-triphosphorylated RNA, but, in these cases, the RNA was single-stranded. IFIT1 and IFIT5 were found to recognize either 5′-triphosphate ssRNA or a triphosphorylated ssRNA overhang of at least 5 nt in length in the case of IFIT1, or 3 nt in length in the case of IFIT5 (Ref. 37). Conversely, base pairing within the first 5 or 3 nt abrogates recognition37. The presence of a 5′ 7-methylguanosine cap (m7G cap) enhances IFIT1 binding, confirming that IFIT1 targets mRNA for translational inhibition and does not target viral genomic RNA38,39. Activation of PKR by 5′-triphosphorylated 'single-stranded' RNA with secondary structures starts at a length of about 47 nt, and an m7G cap abrogates PKR binding86. In addition to 5′-triphosphate-dependent binding to ssRNA, PKR also binds long dsRNA independently of 5′-triphosphate (discussed above).
In conclusion, many evolutionarily unrelated PRRs target polyphosphorylated RNA, albeit in the context of different RNA structures.
Labels of self and viral escape mechanisms. In contrast to bacterial RNA, 2′-O-methylation is a common modification of eukaryotic RNA, whereby a methyl group is added to the 2′ hydroxyl group of the ribose87. As a eukaryotic marker of self, it prevents the recognition of endogenous RNA by TLR7 and TLR8, thus adding to the repertoire of discriminatory factors of self versus non-self64,88,89 (Fig. 3; Table 2). Notably, 2′-O-methylation at the N1 position of mRNA is an essential component of the universal cap 1 structure of the mRNA of higher eukaryotes. It is now evident that N1 methylation in cap 1 has a key function in nucleic acid sensing as a marker of self, as it abolishes the recognition of self RNA by RIG-I90,91 and by IFIT1 (Ref. 38) (Table 2).
Certain viruses have evolved mechanisms to mimic this self label of RNA by encoding 2′-O-methyltransferases, which mediate 2′-O-methylation of their RNA to escape detection by RIG-I, IFIT1, TLR7 and MDA5 (Refs 35,90,92). Important examples of viruses with their own N1 2′-O-methyltransferase and that replicate in the cytosol include the (+)ssRNA flaviviruses and coronaviruses, the (−)ssRNA paramyxoviruses and rhabdoviruses, the dsRNA reoviruses and the dsDNA poxviruses and vaccinia virus93. Furthermore, the (−)ssRNA orthomyxoviruses, bunyaviruses and arenaviruses cut the 5′-terminal 10–14 nt of host mRNA and use those capped oligonucleotides as a primer for mRNA generation (termed the cap-snatching mechanism)93.
Concealment and modification of 5′-terminal RNA is an important viral strategy to avoid immune recognition. RNAs of pathogenic alphaviruses that lack N1 2′-O-methylation have 5′-terminal structural motifs that interfere with IFIT1 binding and function94 (Table 2). Members of the Picornaviridae family prime RNA transcription using a non-canonical cap-like structure termed VPg that is covalently linked to a uridine dimer (VPg–pUpU), thereby avoiding the presence of a 5′-triphosphate and escaping recognition by both RIG-I and IFIT1 (Ref. 93). Furthermore, Crimean–Congo haemorrhagic fever virus, Hantaan virus and Borna disease virus have been reported to prevent RIG-I-mediated detection of their genomes by an RNA prime-and-realign mechanism that shifts the panhandle structure to generate an 5′ overhang and by cleavage of the 5′ terminal base of their genomic RNA leaving monophosphorylated 5′ ends95. Arenaviruses use a prime and realign mechanism to generate 5′-triphosphate overhangs of 1 nt in their panhandle structures, which are not recognized by RIG-I75 and, in principle, do not allow binding by IFIT1.
Other RNA modifications present within viral transfer RNA (tRNA) and ribosomal RNA (rRNA) were also reported to inhibit immune sensing. Incorporation of pseudouridine (Ψ), 5-methylcytidine (m5C), 2-thio-uridine (s2U) or N6-methyladenosine (m6A) was found to potently inhibit TLR7 and TLR8 and to reduce TLR3 activity, which is the least sensitive receptor to RNA modifications88. Incorporation of Ψ, 5-methyluridine (m5U) or s2U was also found to abolish RIG-I activation72. As mentioned above, ADAR1 prevents recognition of dsRNA by A-to-I conversion in base-paired structures43,55,56, which results in the destabilization of the double-strands due to disrupted A–U base pairs. Other approaches by RNA viruses, such as (−)ssRNA viruses, to escape immune recognition by sensing receptors involves the use of nucleocapsid protein to avoid the formation of dsRNA45 or to introduce mismatches in their genomic panhandles to minimize recognition by RIG-I96 (Table 2).
Overall, viruses have evolved sophisticated mechanisms to mimic endogenous RNA modifications, especially cap 1 structures, and to generate unique viral structures that prevent 5′ end recognition by PRRs.
Recognition of potentially pathogenic, endogenous RNAs. Some endogenous RNAs that are potentially 'pathogenic', such as RNAs from endogenous retroviruses (ERVs), appear to be continuously recognized by TLRs, thereby limiting their replication97. It has been reported that the loss of TLR7 function causes retroviral viraemia and additional loss of TLR3 and TLR9 causes acute T cell lymphoblastic leukaemia in mice97. This indicates that endogenous RNAs transcribed from RNA polymerase II promoters are not generally excluded from TLR-mediated recognition. Furthermore, it has been demonstrated that OAS1-activated RNase L98 and IRE1α99, which is the detector RNase for the unfolded protein response (UPR), cleave endogenous RNA and that the cleavage products can then serve as endogenous ligands for RLRs.
To counteract this process, the SKIV2L RNA exosome degrades endogenous RNA, similar to the role of 3′ repair exonuclease 1 (TREX1; also known as DNase III) in the regulation of endogenous DNA (see below). The RNA exosome is a cellular RNA degradation machine in eukaryotes involved in the regulation of RNA turnover. Depletion of the SKIV2L RNA exosome resulted in the accumulation of endogenous RNA, which triggered the RIG-I pathway in an IRE1α-dependent manner100. Thus, the RNA degradation activity of SKIV2L RNA exosome appears to prevent accumulation of aberrant RNAs with immunostimulatory capacity. To date, the structural features of the 5′ end of IRE1α or RNase L cleavage products that lead to RIG-I activation have not been systematically analysed. Of note, both RNase L and IRE1α generate RNA molecules with 5′-hydroxyl and 3′-phosphate or 2′-3′-cyclic phosphate ends98,100. As viral infections trigger the IRE1α UPR, it makes sense that the IRE1α-dependent UPR pathway triggers the RIG-I pathway to achieve a full antiviral response.
Overall, RNA-sensing PRRs have evolved to sense RNA structures and modifications that typically occur during viral replication. Several unrelated PRRs share similar recognition motifs (for example base-paired or 5′-triphosphorylated RNA), which is suggestive of a convergent evolution towards efficient virus detection. Although there are hints that endogenous RNA cannot entirely avoid recognition by PRRs, additional tolerance mechanisms — including RNA modifications (especially 2′-O-methylation and A-to-I conversions) and RNA degradation of aberrant RNA structures — further prevent recognition of endogenous RNA. Viruses have established sophisticated mechanisms to exploit the endogenous tolerance mechanisms or to circumvent the generation of the typical structures recognized by PRRs.
DNA sensing
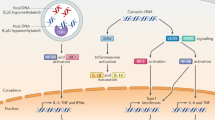
DNA-sensing receptors. TLR9 is responsible for the detection of DNA in the endolysosomal compartment101 and triggers type I IFN production in plasmacytoid dendritic cells and polyclonal activation of B cells via the myeloid differentiation primary response protein 88 (MYD88) and IRF7 signalling pathway. In contrast to the endolysosomal compartment, DNA is usually absent in the cytosol, and the mere presence of cytosolic DNA is indicative of non-self or 'dangerous' DNA. A number of DNA-sensing receptors and mechanisms have been proposed to promote inflammasome activation and the induction of type I IFN (Fig. 4).

Toll-like receptor 9 (TLR9) in the endolysosomal compartment detects CpG motif-containing DNA and RNA–DNA hybrids that have not been degraded by nucleases such as DNase I and DNase II. TLR9 signals via myeloid differentiation primary response protein 88 (MYD88), interferon (IFN)-regulatory factor 7 (IRF7) and nuclear factor-κB (NF-κB) to induce type I IFN and the inflammasome-related factors pro-interleukin-1β (pro-IL-1β) and NOD-, LRR- and pyrin domain-containing 3 (NLRP3). In the cytosol, cyclic GMP–AMP synthetase (cGAS), IFNγ-inducible protein 16 (IFI16) and absent in melanoma 2 (AIM2) detect DNA that has not been degraded by nucleases such as 3′ repair exonuclease 1 (TREX1). Knockout models reveal that cGAS signals via stimulator of IFN genes (STING) to stimulate type I IFN production. IFI16, DEAD box protein 41 (DDX41) and cGAS also induce apoptosis via STING, IRF3 and BCL-2-associated X protein (BAX) in response to double-stranded DNA (dsDNA). Polyglutamine binding protein 1 (PQBP1) is a co-receptor of cGAS that recognizes HIV reverse transcripts. AIM2 activation results in the formation of the AIM2 inflammasome, which induces IL-1β maturation and pyroptosis via ASC and caspase 1 (not shown). Recognition of dsDNA in the nucleus by RAD50 activates caspase recruitment domain 9 (CARD9)–BCL-10 to induce NF-κB, which upregulates pro-IL-1β transcription. DNA-sensing receptors implicated in the DNA damage response and nuclear recognition of viral nuclear DNA includes breast cancer type 1 susceptibility protein (BRCA1), IFI16, DNA-dependent serine/threonine protein kinase (DNA-PK) and MRE11, and these molecules also signal via STING. Ribonuclease H (RNase H) degrades DNA–RNA hybrids. ssDNA, single-stranded DNA.
The PYHIN (pyrin and HIN domain-containing protein) family member AIM2 is the major cytosolic dsDNA-sensing receptor responsible for inflammasome activation102,103,104,105. The AIM2-related PYHIN domain-containing IFNγ-inducible protein 16 (IFI16) has also been implicated in inflammasome activation in response to dsDNA and the death of HIV-infected CD4+ T cells106,107,108. In addition, the DNA-damage sensor RAD50 was reported to activate the CARD9–BCL-10 pathway leading to pro-IL-1β mRNA induction after stimulation with long dsDNA (such as, poly(dAdT), poly(dGdC) and calf thymus DNA)109 (Fig. 4).
Similar to poly(I:C), a self-complementary polymer of alternating AT (poly(dAdT)) that forms dsDNA, also termed 'B-DNA', served initially as a gold standard for investigating dsDNA-mediated immunostimulation110. However, it turned out that AT-rich DNA in the cytosol can also indirectly induce type I IFN via RNA-sensing receptors: cytosolic poly(dAdT) DNA is transcribed by RNA polymerase III into 5′-triphosphorylated RNA and folds into 5′-triphosphate dsRNA, which strongly activates RIG-I111,112. However, the biological relevance of this pathway is unclear, as AT-rich DNA is rather uncommon and self-complementarity is a prerequisite for RIG-I stimulation. Numerous candidate cytosolic dsDNA receptors — including ZBP1, DEAD box protein 41 (DDX41), IFI16, the DNA-PK complex (DNA-dependent serine/threonine protein kinase complex), MRE11, polyglutamine binding protein 1 (PQBP1) and cGAS — have been proposed to activate the IRF3 pathway (but not the RIG-I-dependent DNA sensing pathway) for type I IFN production (also known as the interferon stimulatory DNA pathway (ISD pathway))113,114.
Currently, cGAS is the most widely accepted dsDNA sensor; however, it cannot be excluded that the other candidates may have a role in influencing the cGAS pathway or in sensing dsDNA within specific tissues. It is broadly accepted that the mitochondrial adaptor protein stimulator of IFN genes (STING; also known as MITA, ERIS and MPYS) is downstream of the DNA receptor and is essential for the activation of the IRF3-dependent pathway115,116 (Fig. 4). STING was found to sense both cyclic dinucleotides secreted by intracellular bacteria117,118 and the endogenous cyclic dinucleotide cGAMP (cyclic GMP–AMP), which contains one 2′-5′-phosphodiester linkage and a canonical 3′5′ linkage (c[G(2′-5′)pA(3′-5′)p]). cGAMP functions as a second messenger and is generated by cGAS following binding of cytosolic DNA119,120,121,122. Crystal structures of cGAS and in vitro activation assays demonstrate that dsDNA directly activates the catalytic activity of cGAS123,124,125,126,127,128. In vivo, the DNA receptor cGAS is essential for the detection of dsDNA genome-based viruses such as herpes simplex virus (HSV), vaccinia virus and adenovirus119,129,130, as well as for retroviruses126,131,132,133.
In addition to cGAS, IFI16 was reported to colocalize with STING upon DNA stimulation134 and to contribute to IFN responses during infection with HSV, human cytomegalovirus, HIV and Listeria monocytogenes114,135. However, to date, a substantial contribution for IFI16 in the DNA-induced type I IFN response has not been confirmed in in vivo studies.
Using knockout and reconstituted cell lines, the DNA damage sensor protein BRCA1 (breast cancer type 1 susceptibility protein) was shown to be crucial for IL-1β and type I IFN induction following Kaposi's sarcoma-associated herpesvirus, Epstein–Barr virus and HSV1 infection; BRCA1 bound to nuclear IFI16 complexes and enabled IFI16 nuclear export, which resulted in the formation of an IFI16-containing inflammasome complex and the IFI16–STING complex136.
Furthermore, genetic deficiency in single DNA-PK components (DNA-PKcs, Ku70 and Ku80) abrogated dsDNA-induced type I IFN response in mouse embryonic fibroblasts (MEFs) in vitro137 but not in mouse bone marrow-derived macrophages113, suggesting tissue- or maturation-specific differences in the role of the DNA-PK complex in dsDNA sensing, which are not fully understood. Although cGAS expression was reported to be essential for the type I IFN response induced by genomic or plasmid DNA or by dsDNA viruses in lung fibroblasts, macrophages and dendritic cells, cGAS expression in MEFs is low, and the response to dsDNA in cGAS-deficient MEFs has not been analysed119. Therefore, the possibility remains that the recognition of dsDNA in MEFs differs from other cell types and might depend on receptors other than cGAS.
Altogether, it is possible that some of the proposed dsDNA receptors function upstream of or as enhancers for the cGAS–STING–IRF3 pathway. Indeed, one cGAS co-receptor, PQBP1, has recently been shown to be a specific enhancer of cGAS-dependent detection of HIV1 cDNA but not detection of plasmid or genomic DNA138.
Structural requirements for DNA recognition. Functional analysis of the cGAS–STING–IRF3 pathway and the AIM2 inflammasome pathway revealed length-dependent but not sequence-dependent requirements for DNA recognition, which is consistent with structural data showing that the DNA phosphate backbone binds to the HIN domain of AIM2 and the carboxyl terminus of cGAS121,123,125,139 (Table 3). For the cGAS–STING–IRF3 pathway, a minimum DNA length of 25 bp in mouse myeloid cells and of 40–60 bp for human myeloid cells was found111,133,134. By contrast, AIM2 was not activated by the so-called ISD sequence (which is a 45mer dsDNA strand) in mouse macrophages, and the observed minimum DNA length for AIM2 activation in human myeloid cells was 50–80 bp102,139. Unexpectedly, stem-loop ssDNA structures, which are characteristic of retroviruses and retroviral elements such as ssDNA reverse transcripts from HIV1, potently activates cGAS, although the stems in such stem-loop structures are shorter than 40 bp108,133. Further evidence showed that the presence of guanosines in the loops or in short single-stranded 3′- and 5′-flanking sequences was required for cGAS activation and that the sequence of the dsDNA stem regions had no impact on activity133. The minimal length of such Y-form DNA with G-containing overhangs that potently activates cGAS was 12 bp in length (Table 3). G-ended Y-form DNA with a 20mer dsDNA stem was as potent as plasmid or genomic DNA in activating cGAS133.
During lentiviral infection, the primary reverse transcription of the RNA genome to ssDNA occurs in the cytosol, whereas the second strand is typically synthesized in the nucleus. Most of the template RNA in the cytosol is rapidly degraded by RNase H. Using a mutated HIV1 reverse transcriptase, which impaired dsDNA but not ssDNA synthesis, the induction of type I IFN by lentiviral particles correlated with the presence of cytosolic ssDNA rather than the presence of dsDNA133. This finding is corroborated by the observation that elite controllers of HIV1 infection produce more type I IFN because of the cytosolic accumulation of HIV1 ssDNA reverse transcripts140. Here, the observed recognition of guanosines in unpaired DNA by cGAS may be essential for ssDNA recognition in the cytosol. In conclusion, primary ssDNA reverse transcripts appear to be the crucial DNA species recognized by cGAS during HIV1 infection and a special mechanism that allows for sensing of guanosine-rich Y-form DNA structures enables the sensitive detection of ssDNA.
TLR9 in the endolysosomal compartment preferentially detects DNA containing unmethylated CpG dinucleotides. Unmethylated CpG dinucleotides are less frequent in eukaryotic self DNA compared with bacterial DNA101,141,142,143. Notably, the specificity of TLR9 for unmethylated CpG motifs is reduced when the CpG motif is placed in the context of phosphorothioate-stabilized ssDNA142.
TLR9 and cGAS can also recognize DNA–RNA hybrids144,145 (Table 3). The hybridization of a 30mer GU-repetitive RNA strand with a 30mer AC-repetitive DNA strand yields a potent TLR9 stimulus, yet abrogates the TLR7 stimulating activity of the GU-repetitive RNA sequence144. Of note, the AC-repetitive DNA strand that lacks CpG has no TLR9 activity as a single strand, underlining the RNA–DNA hybrid as a real recognition motif. Based on structural models, the mechanism through which RNA–DNA hybrids and dsDNA bind to cGAS is similar145. Indeed, RNA–DNA hybrids of the homopolymers poly(rA) and poly(dT) were found to activate cGAS but less robustly then dsDNA145. However, whether the accumulation of high levels of DNA–RNA hybrids in the cytosol is possible in the presence of RNase H activity is unclear. Altogether, the main dsDNA receptors (cGAS and TLR9) can also sense DNA–RNA hybrids. However, the biological relevance of DNA–RNA sensing during infections needs to be investigated.
The impact of endogenous DNA modifications. Two fundamental base modifications have been implicated in DNA sensing. One is endogenous methylation of the C5 carbon of cytosine within CpG motifs, which inhibits recognition by and activation of TLR9 (Refs 141,143). Another form of base modification occurs in the context of oxidative stress: the presence of reactive oxygen species as a result of UV radiation or cell stress leads to the incorporation of oxidative adducts, the most common of which is 8-hydroxyguanosine (8-OHG) (Fig. 5). Thus, the 8-OHG modification is indicative of oxidative damage. 8-OHG modifications have important consequences for the immune response: 8-OHG stabilizes DNA against degradation by the cytosolic DNA exonuclease TREX1, leading to the accumulation of cytosolic DNA and an increased cGAS activation146 (Fig. 5).

The cytosolic immune receptor cyclic GMP–AMP synthetase (cGAS) detects long double-stranded DNA (dsDNA) or short dsDNA with unpaired open ends containing guanosines, which is present in highly structured single-stranded DNA (ssDNA) of certain viruses, such as retroviruses. Cytosolic DNA is efficiently degraded by 3′ repair exonuclease 1 (TREX1) located in the cytosol. Oxidation of DNA, which occurs in situations of oxidative stress caused by UV radiation or cell stress, leads to oxidation of DNA, the most common of which is 8-hydroxyguanosine (8-OHG). This stabilizes DNA against degradation by TREX1, resulting in an accumulation of DNA in the cytosol. Oxidated DNA is recognized by cGAS, resulting in the formation of the cyclic dinucleotide cGAMP, which activates STING to induce type I interferon (IFN) production.
Endogenous DNases and autoinflammation. Cell death-derived DNA, DNA damage and endogenous retroviral elements are sources of self DNA that may accumulate in the circulation, in endolysosomes and in the cytosol of phagocytic cells. Three DNases continuously limit immune recognition of such DNA through degradation (Fig. 4). DNase I is secreted and degrades DNA in the circulation. The loss of DNase I causes a phenotype similar to systemic lupus erythematosus147. Lysosomal DNase II not only degrades phagocytosed DNA but is also crucial for the deletion of nuclear DNA that enters the autophagy pathway148. DNase II deficiency is fatal due to the expression of type I IFN during embryogenesis, but mice can be partially rescued via concurrent type I IFN receptor deficiency but develop a polyarthritis-like disease149,150. The tissue-specific activation of AIM2 and the TLR pathway by self DNA in DNase II-deficient mice has been associated with polyarthritis151,152. TREX1 degrades DNA in the cytosol (Fig. 4) and TREX1 deficiency in humans causes the autoinflammatory Aicardi–Goutières syndrome153. The fatal phenotypes of TREX1-deficient and DNase II-deficient mice, as well as associated polyarthritis, can be reversed by knockout of the gene encoding cGAS148,154,155,156. In contrast to its suppressive role on cytoplasmic DNA receptor activation, DNase II was reported to support the stimulation of endosomal TLR9 by enhancing the accessibility of ssDNA ligands from bacterial DNA in the endosome157.
Many of the cytosolic pathways proposed to trigger type I IFN production by dsDNA recognition are still controversial. cGAS, which activates STING via the second messenger cGAMP, is the most accepted DNA sensor. Recent research has revealed that the DNA sensing receptors cGAS and TLR9 can also be activated by DNA–RNA hybrids. In addition to length-dependent activation of cGAS by dsDNA, cGAS possesses an alternative mechanism for DNA recognition that detects G-rich Y-form DNA and appears to be critical for sensing ssDNA that is derived from exogenous or endogenous retroviruses.
Concluding remarks
Innate immune cells have established a powerful system to specifically detect the presence of foreign, potentially harmful nucleic acids. Several virus-associated nucleic acid structures have been identified, such as long dsRNA, 5′-triphosphate or 5′-diphosphate dsRNA, CpG motifs in ssDNA, and short dsDNA with guanosine-containing overhangs that resemble highly structured viral ssDNA. In addition, cells actively label their self nucleic acids in order to distinguish them from non-self molecules. The 2′-O-methylation at the N1 position of capped RNA completely eliminates recognition by RIG-I and IFIT1, the incorporation of 2′-O-methyl groups or other modifications (such as pseudouridine) at internal positions of RNA impairs recognition by TLR7, TLR8 and MDA5, and C5 methylation of CpG motifs in DNA abolishes TLR9 recognition.
However, pattern sensing of nucleic acids extends beyond the detection of specific molecular structures at the receptor–ligand level. To achieve sensitivity and specificity for viral nucleic acids among an abundance of self nucleic acids, this system further integrates the enzymatic function of extracellular and intracellular nucleases, allowing the detectability of nucleic acids to be determined by their stability against nucleases. Thus, potentially dangerous nucleic acids, such as those associated with viral particles or released during oxidative stress, are particularly potent immune activators.
The nucleic acid-sensing system further integrates information about the localization of detected nucleic acids in order to select the most appropriate response: TLRs localized in the endosomal membrane induce different signalling pathways to those induced by receptors localized in the cytosol. The distinct function of nucleic acid-sensing receptors is also regulated by their differential expression in functionally distinct immune cell subsets and even non-immune cells. An important example is the restrictive expression of TLR7 and TLR9 to plasmacytoid dendritic cells, which, upon stimulation, respond with very high levels of type I IFN production. In contrast, TLR8-mediated stimulation of myeloid cells leads to the release of IL-12.
Overall, the main responses induced by nucleic acid-sensing receptors comprises: production of cytokines (for example, type I IFNs) and chemokines to activate neighbouring cells and to recruit immune cells to the site of infection; and direct antiviral responses, such as cell autonomous antiviral mechanisms, that target viral replication, translation of viral proteins and virus assembly, and that induce cell death, including apoptosis, necroptosis and pyroptosis (reviewed in Ref. 158).
It is evident that the sensitivity of the nucleic acid-sensing system can differ according to the immunological context. In the presence of type I IFN or nucleic acid ligands, the sensitivity of this system is heightened, and, consequently, endogenous ligands can become more immunogenic. In addition, certain stressors can increase the recognition of endogenous ligands; for example exposure of DNA to reactive oxygen species results in DNA oxidation, thereby enhancing its stability against TREX1-mediated degradation and increasing its sensitivity to cGAS recognition.
Future perspectives
Although the major nucleic acid-sensing receptor pathways seem to have been identified, numerous urgent questions remain regarding the functional interactions of these receptors within and beyond the immune system. We still do not understand the nature of immune sensing of foreign extrachromosomal nucleic acids in the nucleus. Another pressing area of research is the link between nucleic acid sensing and the DNA damage response.
Within well-described nucleic acid-sensing pathways, we are still just beginning to understand the implications of genetic alterations for human disease. Genetic alterations can lead to enhanced or reduced expression or function of nucleic acid-sensing receptors, DNases, RNases and nucleic acid-modifying enzymes. Perturbations of the balance between self and non-self nucleic acid recognition are involved in the pathogenesis of several rare autoimmune diseases such as Aicardi–Goutières syndrome (which is associated with genetic defects in TREX1, RNase H components and SAMHD1 (Ref. 159)), Singleton–Merten syndrome (associated with mutations in the ATPase domain of RIG-I, leading to constitutive RIG-I activation by self RNA)160 and STING-associated vasculopathy with onset in infancy (SAVI)161. These perturbations can also contribute to the pathogenesis of type I diabetes (owing to genetic variants of RIG-I and MDA5 (Refs 2,162,163)). Furthermore, recent work indicates that disrupted nucleic acid sensing is involved in systemic lupus erythematosus, rheumatoid arthritis and primary Sjögren syndrome2,164. Several studies also implicate the RIG-I–MAVS signalling axis in gut homeostasis165,166,167. A better understanding of nucleic acid sensing and its role in disease may also pave the way for new therapeutic interventions involving therapeutic oligonucleotides10 or other molecules that specifically block certain nucleic acid receptor pathways165,166,167.
Finally, it will be important to better understand the relationship between RNAi and the innate immune sensing of nucleic acids. From an evolutionary perspective, Dicer, the central endoribonuclease of the RNAi pathway, and RIG-I helicases are closely related. Moreover, several recent studies suggest that many pathogenic viruses disable Dicer11. Whether viruses disable Dicer to affect the function of microRNAs or to escape antiviral RNAi or both merits further investigation.
References
Isaacs, A., Cox, R. A. & Rotem, Z. Foreign nucleic acids as the stimulus to make interferon. Lancet 2, 113–116 (1963).
Junt, T. & Barchet, W. Translating nucleic acid-sensing pathways into therapies. Nat. Rev. Immunol. 15, 529–544 (2015).
Ablasser, A., Hertrich, C., Wassermann, R. & Hornung, V. Nucleic acid driven sterile inflammation. Clin. Immunol. 147, 207–215 (2013).
Wu, J. & Chen, Z. J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488 (2014).
Kawasaki, T., Kawai, T. & Akira, S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol. Rev. 243, 61–73 (2011).
Rathinam, V. A. & Fitzgerald, K. A. Cytosolic surveillance and antiviral immunity. Curr. Opin. Virol. 1, 455–462 (2012).
Desmet, C. J. & Ishii, K. J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 12, 479–491 (2012).
Yoneyama, M. & Fujita, T. Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20, 4–22 (2010).
Rehwinkel, J. & Reis e Sousa, C. Targeting the viral Achilles' heel: recognition of 5′-triphosphate RNA in innate anti-viral defence. Curr. Opin. Microbiol. 16, 485–492 (2013).
van den Boorn, J. G. & Hartmann, G. Turning tumors into vaccines: co-opting the innate immune system. Immunity 39, 27–37 (2013).
Svoboda, P. Renaissance of mammalian endogenous RNAi. FEBS Lett. 588, 2550–2556 (2014).
Seo, G. J. et al. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14, 435–445 (2013). This is the first study indicating reciprocal roles of RNAi and antiviral signalling in host defence.
Sato, S. et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 (2015).
Weber, M. et al. Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17, 309–319 (2015).
McAllister, C. S., Taghavi, N. & Samuel, C. E. Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2α-mediated translational control. J. Biol. Chem. 287, 36384–36392 (2012).
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 (2001). The identification of TLR3 as a dsRNA-sensing receptor.
Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).
Heil, F. et al. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303, 1526–1529 (2004). References 17 and 18 identify TLR7 as an ssRNA receptor.
Hornung, V. et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 11, 263–270 (2005). This study describes the sequence-dependent recognition of siRNA by TLR7 and provides an explanation for sequence-dependent off-target effects caused by TLR7 activation. It is also the first introduction to RNA backbone modifications that prevent TLR7-mediated off-target effects.
Matsumoto, M. et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 171, 3154–3162 (2003).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004). This paper details the discovery of the RLR family.
Ahmad, S. & Hur, S. Helicases in antiviral immunity: dual properties as sensors and effectors. Trends Biochem. Sci. 40, 576–585 (2015).
Schlee, M. Master sensors of pathogenic RNA — RIG-I like receptors. Immunobiology 218, 1322–1335 (2013).
El Maadidi, S. et al. A novel mitochondrial MAVS/caspase-8 platform links RNA virus-induced innate antiviral signaling to Bax/Bak-independent apoptosis. J. Immunol. 192, 1171–1183 (2014).
Besch, R. et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J. Clin. Invest. 119, 2399–2411 (2009).
Kumar, S. et al. IPS-1 differentially induces TRAIL, BCL2, BIRC3 and PRKCE in type I interferons-dependent and -independent anticancer activity. Cell Death Dis. 6, e1758 (2015).
Glas, M. et al. Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem Cells 31, 1064–1074 (2013).
Chattopadhyay, S. et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 29, 1762–1773 (2010).
Park, S. et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366 (2013).
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z. & Germain, R. N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013).
Franchi, L. et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 193, 4214–4222 (2014).
Poeck, H. et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat. Immunol. 11, 63–69 (2010).
Pothlichet, J. et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 9, e1003256 (2013).
Levin, D. & London, I. M. Regulation of protein synthesis: activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc. Natl Acad. Sci. USA 75, 1121–1125 (1978).
Daffis, S. et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468, 452–456 (2010).
Pichlmair, A. et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 12, 624–630 (2011). References 35 and 36 report the discovery of IFIT proteins as 5′-triphosphate RNA-binding antiviral effector molecules.
Abbas, Y. M., Pichlmair, A., Gorna, M. W., Superti-Furga, G. & Nagar, B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494, 60–64 (2013).
Habjan, M. et al. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 9, e1003663 (2013).
Kumar, P. et al. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 42, 3228–3245 (2014).
Zilberstein, A., Kimchi, A., Schmidt, A. & Revel, M. Isolation of two interferon-induced translational inhibitors: a protein kinase and an oligo-isoadenylate synthetase. Proc. Natl Acad. Sci. USA 75, 4734–4738 (1978).
Hovanessian, A. G., Brown, R. E. & Kerr, I. M. Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 268, 537–540 (1977).
Zhou, A., Hassel, B. A. & Silverman, R. H. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72, 753–765 (1993).
George, C. X., John, L. & Samuel, C. E. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interferon Cytokine Res. 34, 437–446 (2014).
Gelinas, J. F., Clerzius, G., Shaw, E. & Gatignol, A. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J. Virol. 85, 8460–8466 (2011).
Weber, F., Wagner, V., Rasmussen, S. B., Hartmann, R. & Paludan, S. R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80, 5059–5064 (2006).
Gitlin, L. et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl Acad. Sci. USA 103, 8459–8464 (2006).
Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Kato, H. et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 (2008).
Goubau, D. et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514, 372–375 (2014).
Leonard, J. N. et al. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl Acad. Sci. USA 105, 258–263 (2008).
Kleinman, M. E. et al. Short-interfering RNAs induce retinal degeneration via TLR3 and IRF3. Mol. Ther. 20, 101–108 (2012).
Weber, C. et al. Toll-like receptor (TLR) 3 immune modulation by unformulated small interfering RNA or DNA and the role of CD14 (in TLR-mediated effects). Immunology 136, 64–77 (2012).
Tatematsu, M., Nishikawa, F., Seya, T. & Matsumoto, M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat. Commun. 4, 1833 (2013).
Wu, B. et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289 (2013).
Liddicoat, B. J. et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015).
Pestal, K. et al. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944 (2015).
Luthra, P., Sun, D., Silverman, R. H. & He, B. Activation of IFN-β; expression by a viral mRNA through RNase L and MDA5. Proc. Natl Acad. Sci. USA 108, 2118–2123 (2011).
Runge, S. et al. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 10, e1004081 (2014).
Childs, K. et al. MDA-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359, 190–200 (2007).
Benitez, A. A. et al. In vivo RNAi screening identifies MDA5 as a significant contributor to the cellular defense against influenza A virus. Cell Rep. 11, 1714–1726 (2015).
Binder, M. et al. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 286, 27278–27287 (2011).
Minks, M. A., West, D. K., Benvin, S. & Baglioni, C. Structural requirements of double-stranded RNA for the activation of 2′,5′-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells. J. Biol. Chem. 254, 10180–10183 (1979).
Manche, L., Green, S. R., Schmedt, C. & Mathews, M. B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 12, 5238–5248 (1992).
Judge, A. D. et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 23, 457–462 (2005).
Ablasser, A. et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J. Immunol. 182, 6824–6833 (2009).
Tanji, H. et al. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 22, 109–115 (2015).
Shibata, T. et al. Guanosine and its modified derivatives are endogenous ligands for TLR7. Int. Immunol. 28, 211–222 (2016).
Saito, T., Owen, D. M., Jiang, F., Marcotrigiano, J. & Gale, M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527 (2008).
Atkinson, N. J., Witteveldt, J., Evans, D. J. & Simmonds, P. The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res. 42, 4527–4545 (2014).
Tulloch, F., Atkinson, N. J., Evans, D. J., Ryan, M. D. & Simmonds, P. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. eLife 3, e04531 (2014).
Marques, J. T. et al. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 24, 559–565 (2006).
Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Schlee, M. et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34 (2009). References 72 and 73 detail the discovery and characterization of RIG-I-recognition motifs.
Vela, A., Fedorova, O., Ding, S. C. & Pyle, A. M. The thermodynamic basis for viral RNA detection by the RIG-I innate immune sensor. J. Biol. Chem. 287, 42564–42573 (2012).
Marq, J. B., Kolakofsky, D. & Garcin, D. Unpaired 5′ ppp-nucleotides, as found in arenavirus double-stranded RNA panhandles, are not recognized by RIG-I. J. Biol. Chem. 285, 18208–18216 (2010).
Wang, Y. et al. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 17, 781–787 (2010). This study describes the interaction of the RIG-I RNA-binding pocket with its ligand (5′-triphosphate dsRNA) on a molecular level and details the requirements for RIG-I ligands.
Civril, F. et al. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 12, 1127–1134 (2011).
Jiang, F. et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011).
Luo, D. et al. Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 (2011).
Kowalinski, E. et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147, 423–435 (2011).
Kohlway, A., Luo, D., Rawling, D. C., Ding, S. C. & Pyle, A. M. Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep. 14, 772–779 (2013).
Monroe, K. M., McWhirter, S. M. & Vance, R. E. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665 (2009).
Abdullah, Z. et al. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J. 31, 4153–4164 (2012).
Hagmann, C. A. et al. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS ONE 8, e62872 (2013).
Strahle, L., Garcin, D. & Kolakofsky, D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351, 101–111 (2006).
Nallagatla, S. R. et al. 5′-Triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318, 1455–1458 (2007).
Eigenbrod, T. & Dalpke, A. H. Bacterial RNA: an underestimated stimulus for innate immune responses. J. Immunol. 195, 411–418 (2015).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Eberle, F. et al. Modifications in small interfering RNA that separate immunostimulation from RNA interference. J. Immunol. 180, 3229–3237 (2008).
Schuberth-Wagner, C. et al. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1-2′O-methylated self RNA. Immunity 43, 41–51 (2015). The paper describes Cap1 2′- O -methylation as a marker of self RNA and as a viral escape mechanism that prevents recognition by RIG-I; it details for the first time a feature in the binding pocket of an innate immune receptor that is dispensable for receptor activation but is essential for tolerance to endogenous RNA.
Devarkar, S. C. et al. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl Acad. Sci. USA 113, 596–601 (2016).
Zust, R. et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12, 137–143 (2011).
Hyde, J. L. & Diamond, M. S. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology 479–480, 66–74 (2015).
Hyde, J. L. et al. A viral RNA structural element alters host recognition of nonself RNA. Science 343, 783–787 (2014).
Habjan, M. et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 3, e2032 (2008).
Anchisi, S., Guerra, J., Mottet-Osman, G. & Garcin, D. Mismatches in the influenza A virus RNA panhandle prevent retinoic acid-inducible gene I (RIG-I) sensing by impairing RNA/RIG-I complex formation. J. Virol. 90, 586–590 (2015).
Yu, P. et al. Nucleic acid-sensing Toll-like receptors are essential for the control of endogenous retrovirus viremia and ERV-induced tumors. Immunity 37, 867–879 (2012).
Malathi, K., Dong, B., Gale, M. Jr & Silverman, R. H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819 (2007).
Cho, J. A. et al. The unfolded protein response element IRE1α senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 13, 558–569 (2013).
Eckard, S. C. et al. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 15, 839–845 (2014).
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000). This paper identifies TLR9 as a receptor for DNA.
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Burckstummer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Kerur, N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011).
Monroe, K. M. et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343, 428–432 (2014).
Jakobsen, M. R. et al. From the cover: IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl Acad. Sci. USA 110, E4571–E4580 (2013).
Roth, S. et al. Rad50–CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat. Immunol. 15, 538–545 (2014).
Ishii, K. J. et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48 (2006).
Ablasser, A. et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072 (2009).
Chiu, Y. H., Macmillan, J. B. & Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Stetson, D. B. & Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 (2006).
Paludan, S. R. Activation and regulation of DNA-driven immune responses. Microbiol. Mol. Biol. Rev. 79, 225–241 (2015).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Zhong, B. et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 (2008).
Sauer, J. D. et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79, 688–694 (2011).
Burdette, D. L. et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP–AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP–AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2012).
Gao, P. et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP–AMP synthase. Cell 153, 1094–1107 (2013). Identification of the actual second messenger structure of cGAMP as G(2′-5′)pA(3′-5′)p.
Ablasser, A. et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
Kato, K. et al. Structural and functional analyses of DNA-sensing and immune activation by human cGAS. PLoS ONE 8, e76983 (2013).
Kranzusch, P. J., Lee, A. S., Berger, J. M. & Doudna, J. A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368 (2013).
Gao, D. et al. Cyclic GMP–AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 (2013).
Zhang, X. et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 (2014).
Li, X. et al. Cyclic GMP–AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 (2013).
Li, X. D. et al. Pivotal roles of cGAS–cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Lam, E., Stein, S. & Falck-Pedersen, E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol. 88, 974–981 (2014).
Lahaye, X. et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 39, 1132–1142 (2013).
Rasaiyaah, J. et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405 (2013).
Herzner, A. M. et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 16, 1025–1033 (2015). The first evidence for the sequence-dependent recognition of cytosolic ssDNA by cGAS.
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010).
Jakobsen, M. R. & Paludan, S. R. IFI16: at the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev. 25, 649–655 (2014).
Dutta, D. et al. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PLoS Pathog. 11, e1005030 (2015).
Ferguson, B. J., Mansur, D. S., Peters, N. E., Ren, H. & Smith, G. L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 1, e00047 (2012).
Yoh, S. M. et al. PQBP1 Is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 161, 1293–1305 (2015).
Jin, T. et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36, 561–571 (2012).
Martin-Gayo, E. et al. Potent cell-intrinsic immune responses in dendritic cells facilitate HIV-1-specific T cell immunity in HIV-1 elite controllers. PLoS Pathog. 11, e1004930 (2015).
Krieg, A. M. et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374, 546–549 (1995).
Hartmann, G. & Krieg, A. M. Mechanism and function of a newly identified CpG DNA motif in human primary B cells. J. Immunol. 164, 944–953 (2000). This paper shows the identification of the human CpG recognition motif.
Coch, C. et al. Higher activation of TLR9 in plasmacytoid dendritic cells by microbial DNA compared with self-DNA based on CpG-specific recognition of phosphodiester DNA. J. Leukoc. Biol. 86, 663–670 (2009).
Rigby, R. E. et al. RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 33, 542–558 (2014).
Mankan, A. K. et al. Cytosolic RNA:DNA hybrids activate the cGAS–STING axis. EMBO J. 33, 2937–2946 (2014).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 (2013). The first evidence for DNA damage-induced cGAS activation by TREX1-resistant oxidized DNA.
Napirei, M. et al. Features of systemic lupus erythematosus in DNase1-deficient mice. Nat. Genet. 25, 177–181 (2000).
Lan, Y. Y., Londono, D., Bouley, R., Rooney, M. S. & Hacohen, N. DNase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 9, 180–192 (2014).
Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2005).
Kawane, K. et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 (2006).
Baum, R. et al. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. J. Immunol. 194, 873–877 (2015).
Jakobs, C., Perner, S. & Hornung, V. AIM2 drives joint inflammation in a self-DNA triggered model of chronic polyarthritis. PLoS ONE 10, e0131702 (2015).
Stetson, D. B., Ko, J. S., Heidmann, T. & Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008).
Gao, D. et al. Activation of cyclic GMP–AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl Acad. Sci. USA 112, E5699–E5705 (2015). This study describes the impact of cytosolic DNA receptor stimulation by endogenous DNases on immune function and disease.
Ablasser, A. et al. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 192, 5993–5997 (2014).
Gray, E. E., Treuting, P. M., Woodward, J. J. & Stetson, D. B. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi–Goutieres syndrome. J. Immunol. 195, 1939–1943 (2015).
Chan, M. P. et al. DNase II-dependent DNA digestion is required for DNA sensing by TLR9. Nat. Commun. 6, 5853 (2015).
Kono, H., Kimura, Y. & Latz, E. Inflammasome activation in response to dead cells and their metabolites. Curr. Opin. Immunol. 30, 91–98 (2014).
Crow, Y. J. et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 167, 296–312 (2015).
Lassig, C. et al. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. eLife 4, e10859 (2015).
Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014).
Shigemoto, T. et al. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J. Biol. Chem. 284, 13348–13354 (2009).
Cen, H. et al. Association of IFIH1 rs1990760 polymorphism with susceptibility to autoimmune diseases: a meta-analysis. Autoimmunity 46, 455–462 (2013).
Smith, S. & Jefferies, C. Role of DNA/RNA sensors and contribution to autoimmunity. Cytokine Growth Factor Rev. 25, 745–757 (2014).
Li, X. D. et al. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc. Natl Acad. Sci. USA 108, 17390–17395 (2011).
Wang, Y. et al. Rig-I−/− mice develop colitis associated with downregulation of Gαi2. Cell Res. 17, 858–868 (2007).
Kernbauer, E., Ding, Y. & Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98 (2014).
Acknowledgements
The authors would like to thank E. Bartok for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
M.S. and G.H. are supported by the Deutsche Forschungsgemeinschaft (www.dfg.de); SFB670 to M.S. and G.H.; DFG SCHL1930/1–1 to M.S.; SFB704 to G.H.; SFB832 and KFO177 to G.H. G.H. and M.S. are supported by the DFG Excellence Cluster ImmunoSensation. G.H. is supported by the German Center of Infectious Disease (DZIF). G.H. is a co-founder and shareholder of Rigontec GmbH; M.S. and G.H. are inventors on a patent covering structures described in a manuscript that is cited in this Review.
Glossary
- RNA interference
-
(RNAi). The use of double-stranded RNA molecules containing sequences that match a given gene to 'knockdown' the expression of that gene by inhibiting translation of the targeted mRNA or by directing RNA-degrading enzymes to destroy the encoded mRNA transcript.
- Cyclic GMP–AMP synthetase
-
(cGAS). A cellular protein that binds to cytosolic DNA species and generates stimulator of interferon genes (STING)-activating cyclic dinucleotides.
- Inflammasome
-
A molecular complex that upon assembly activates inflammatory caspases, which in turn promote the maturation of pro-inflammatory cytokines and induce pyroptosis.
- Cap-dependent translation
-
A mechanism for initiation of translation. It involves the binding of the eIF4F complex — which is composed of the mRNA helicase eIF4A (eukaryotic translation initiation factor 4A), eIF4E and the scaffolding protein eIF4G — to mRNA transcripts that have a 5′ 7-methylguanosine cap, resulting in recruitment of the 43S ribosome. The ribosome then scans the mRNA until it reaches the first AUG codon, and it initiates translation following the GTP-dependent release of eIF2B.
- Ribonuclease L
-
(RNaseL). A constitutively expressed, latent, nonspecific endoribonuclease that is activated by 2′-5′-oligoadenylate. Type I interferons induce RNaseL expression as part of the host antiviral response.
- A-form dsRNA
-
Double-stranded RNA (dsRNA) molecules usually assemble into A-form helices within the cell. A-form helices are right-handed with 11 base pairs per helical turn, and the bases are not completely perpendicular to the helical axis. In A-form dsRNA, the major groove is deep and narrow.
- Z-form dsRNA
-
Certain conditions (high ionic strength and/or CG-rich sequences) force the transition of A-form dsRNA helices into Z-form helices. Z-form helices are left-handed with 12.4 base pairs per helical turn. In Z-form dsRNA, the minor groove is deep and narrow.
- Panhandle structures
-
Double-stranded stem structures with bulges that are formed by the partially complementary 5′- and 3′-ends of some (-)ssRNA viruses (for example, influenza virus) and are used as a replication control platform.
- Defective interfering RNA genomes
-
Virus genome mutants that are spontaneously generated owing to erroneous replication, most probably because of suboptimal replication conditions. They have lost a crucial portion of the genome. 'Snap back' defective interfering genomes occur when the replicase transcribes one strand and (without clearing the replication complex) immediately uses this new strand as a template.
- m7G cap
-
(7-methylguanosine cap). A type of cap that is linked by a triphosphate bridge to the first transcribed nucleotide at the 5′ end of eukaryotic mRNA. Recognition of the m7G cap by the cap-binding protein eIF4E is the initiation step of cap-dependent translation.
- Cap 1
-
In addition to 7-methylguanosine, the mRNA of higher eukaryotes has a 2′-O-methylation at the penultimate nucleotide (N1). This structure is called cap 1.
- Cap-snatching mechanism
-
The process by which some viruses (for example, influenza virus) cleave a 10–14 nt fragment from the capped 5′ end of cellular mRNAs and use it as a primer (leader) for their own mRNA transcription of a cap 1 structure.
- Endogenous retroviruses
-
(ERVs). 8% of the human genomic DNA contains sequences of retroviral origin that were acquired from ancestral retroviral infections and inherited into the germline.
- Unfolded protein response
-
(UPR). A response that increases the ability of the endoplasmic reticulum to fold and translocate proteins, decreases the synthesis of proteins and causes the arrest of the cell cycle and apoptosis.
- SKIV2L RNA exosome
-
A multiprotein cellular complex in eukaryotes that degrades various types of RNA. In this way, the cytosolic 3′-to-5′ RNA exosome complex, defined by the SKIV2L RNA helicase subunit, is a crucial negative regulator that prevents the stimulation of RIG-I-like receptors by endogenous RNA.
- DNA-PK complex
-
The nuclear DNA-dependent serine/threonine protein kinase (DNA-PK) complex is composed of the proteins DNA-PKcs, Ku70 and Ku80. It is involved in DNA non-homologous end-joining during DNA double-strand break repair.
- Interferon stimulatory DNA pathway
-
(ISD pathway). A cytosolic dsDNA recognition pathway that involves the second messenger receptor STING, but not RIG-I, and leads to type I IFN production. The ISD pathway is triggered by any random dsDNA sequence of a certain minimum length (40–60 bp).
- Y-form DNA
-
DNA that contains a region of base-paired DNA at one end and a region of unpaired DNA strands at the opposite end.
- Elite controllers of HIV1 infection
-
HIV1-infected individuals with an immune system that can limit HIV1 infection without antiviral treatment.
- Autophagy
-
An evolutionarily conserved process in which acidic double-membrane vacuoles sequester intracellular contents (such as damaged organelles and macromolecules) and target them for degradation through fusion to secondary lysosomes.
- Aicardi–Goutières syndrome
-
(AGS). A neurodegenerative disorder that can be caused by stimulator of IFN genes (STING)-dependent cytokine hyperproduction owing to mutations in genes such as TREX1.
- Necroptosis
-
A type of necrosis and a form of non-apoptotic cell death driven by receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 under conditions in which caspase 8 is inhibited.
- Pyroptosis
-
An inflammatory and lytic form of programmed cell death mediated by inflammatory caspases.
- MicroRNAs
-
Small, RNA molecules that regulate the expression of genes by binding to the 3′-untranslated regions (3′-UTR) of specific mRNAs.
Rights and permissions
About this article

Cite this article
Schlee, M., Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 16, 566–580 (2016). https://doi.org/10.1038/nri.2016.78
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri.2016.78
This article is cited by
-
Chronic endoplasmic reticulum stress in myotonic dystrophy type 2 promotes autoimmunity via mitochondrial DNA release
Nature Communications (2024)
-
NAP-seq reveals multiple classes of structured noncoding RNAs with regulatory functions
Nature Communications (2024)
-
Epigenetic priming improves salvage chemotherapy in diffuse large B-cell lymphoma via endogenous retrovirus-induced cGAS-STING activation
Clinical Epigenetics (2023)
-
In vitro culture of bovine fibroblasts using select serum-free media supplemented with Chlorella vulgaris extract
BMC Biotechnology (2023)
-
Cell-free DNA as diagnostic and prognostic biomarkers for adult sepsis: a systematic review and meta-analysis
Scientific Reports (2023)
