
Abstract
Proton magnetic resonance spectroscopy (1H-MRS) allows the non-invasive measurement of several metabolites, including N-acetyl-aspartate (NAA), an amino acid exclusively synthesized in the mitochondria of neurons, and glutamate, an amino acid involved in excitatory neurotransmission and metabolism. In view of recent postmortem studies in schizophrenia (SZ) revealing mitochondrial abnormalities as well as perturbed expression of the enzymes regulating the glutamate–glutamine cycle, we hypothesized that a disruption in the homeostasis of NAA and glutamate in SZ is present. Fifty subjects with SZ and 48 matched healthy controls (HC) were enrolled in this 1H-MRS study. Voxels were placed in the anterior cingulate cortex (ACC) and hippocampus; NAA/Cr and glutamate + glutamine (Glx)/Cr ratios were obtained. We did not find any significant differences between the groups in metabolite levels in both the ACC and hippocampus. In the hippocampus we found that NAA/Cr and Glx/Cr ratios were significantly correlated in HC (r=0.40, p<0.01 (corrected p=0.048)) but not in SZ (r=−0.06; p=0.71), a difference that was statistically significant (z=2.22, p=0.02). Although no differences in neurometabolites between SZ and HC were apparent, correlations between NAA/Cr and Glx/Cr in healthy subjects in the hippocampus were found, and this correlation was lost in subjects with SZ. To our knowledge, this is the first study to suggest decoupling of these metabolites, a pathophysiological change that may be unique to SZ. However, these results warrant replication and further exploration before definite conclusions can be drawn.
Similar content being viewed by others

INTRODUCTION
Proton magnetic resonance spectroscopy (1H-MRS) allows the non-invasive measurement of several metabolites that have critical roles in cellular functions. As such, MRS studies have been of great interest for the study of the pathophysiology in schizophrenia (SZ). N-acetyl-aspartate (NAA) is an amino acid thought to be primarily synthesized in the mitochondria of neurons that can be measured relatively easily with MRS (Bates et al, 1996). The physiological function of NAA remains controversial; it is often conceptualized as a marker of neuronal integrity and thought to reflect mitochondrial function (Moffett et al, 2007). A number of MRS studies in SZ, but not all, have reported decreased NAA levels in several brain regions, including the anterior cingulate cortex (ACC) and the hippocampus (Kraguljac et al, 2012). Glutamate is an amino acid involved in excitatory neurotransmission and metabolism (Magistretti and Pellerin, 1996; Moffett et al, 2007), and glutamine is proposed to be the major metabolite of synaptic glutamate and an index of glutamate neurotransmitter release (Magistretti and Pellerin, 1996). The quantification of glutamate and glutamine with MRS is difficult. Glutamate, glutamine and gamma-aminobutyric acid (GABA) have overlapping peaks that are difficult to resolve at low magnetic field strength. A single peak termed glutamate + glutamine (Glx) containing the three peaks is often reported, although there are sequences that allow disentangling the overlapping peaks (Keltner et al, 1996; Weber et al, 1997). Compared with glutamate, GABA and glutamine make limited quantitative contributions to the Glx peak. Therefore, Glx abnormalities are thought to be primarily because of altered glutamate measurements, but this interpretation remains subject of controversy (Kondo et al, 2011). Because of the difficulty to quantify peaks, glutamatergic studies have been less common in SZ, and results have varied (Marsman et al, 2011).
NAA and glutamate are inherently linked through a series of biochemical reactions, mainly the tricarboxylic acid (TCA) and the glutamate–glutamine cycle (Moffett et al, 2007). Clark et al (2006) hypothesize NAA to be a reservoir for protecting the concentration of glutamate, maintaining low levels of glutamate and having the ability to rapidly produce it. They further speculate that NAA can not only be converted to glutamate, but glutamate can then be converted back into NAA, via a multi step, multi cell and multi compartment cycle (Clark et al, 2006). Several reports support a proposed connection between NAA and glutamatergic neurotransmission. A study conducted with 13C-MRS found that the rate of glutamate labeling from 13C-glucose infusions and NAA synthesis were tightly coupled (Moreno et al, 2001). In MRS studies conducted in healthy subjects, positive correlations between NAA and Glx have been identified in the dorsal ACC, cerebellum (Waddell et al, 2011), and pregenual ACC (Walter et al, 2009).
Recent postmortem studies in SZ have revealed abnormalities in mitochondrial morphology, function and gene expression (Clay et al, 2011; Rosenfeld et al, 2011), as well as abnormal expression of some of the enzymes regulating the glutamate–glutamine cycle, such as the glutamine synthetase (Steffek et al, 2008), the excitatory amino-acid transporters (EAATs) that regualte the reuptake of glutamate from the synaptic cleft (Bauer et al, 2008), the glutaminase responsible for the conversion of glutamine to glutamate (Bruneau et al, 2005), and the gene that encodes the glutamate transporters EAAT1 and EAAT2 (Walsh et al, 2008). Further, a variant of the SZ risk gene GRM3, the gene that encodes for the type 3 metabotropic glutamate receptor, has been associated with reduced NAA levels in the prefrontal cortex (Marenco et al, 2006).
On the basis of the proposed cycle regulating glutamate availability via NAA, reported correlation between NAA and glutamate in the healthy brain, and related pathophysiological findings in SZ, we hypothesize that a disruption in the homeostasis of NAA and glutamate in SZ is present. To examine this hypothesis, we analyzed NAA and Glx MRS measurements in the ACC and hippocampus in subjects with SZ and matched healthy controls (HC) in vivo.
MATERIALS AND METHODS
Subjects
Subjects with SZ and schizoaffective disorder (SZ) were recruited from various outpatient psychiatry clinics at the University of Alabama at Birmingham. HC matched on age, sex, and parental occupation, were recruited by advertisements in flyers and the university's newspaper. Approval by the local Institutional Review Board was obtained; after explanation of the study to the subjects written informed consent to participate was obtained. Before signing consent, each SZ subject completed an Evaluation to Sign Consent form.
Diagnoses were established by review of medical records and consensus of two clinicians and then confirmed with the Diagnostic Interview for Genetic Studies (Nurnberger et al, 1994). Exclusion criteria were major medical conditions, substance abuse within 6 months before enrollment, pregnancy, and head injury with loss of consciousness for more than two minutes.
Fifty SZ subjects and 48 HC consented to participate. Of those, four had to be excluded from the analyses (two SZ and two HC). One subject revoked consent before the MRS scan, two subjects were found to have anatomical abnormalities on their MRI scan (hydrocephalus, calcification of falx close to voxel placement), and scan quality of one subject was poor (motion artefact), leaving 46 HC and 48 SZ subjects in the final analyses (Table 1). Five subjects were treated with first generation antipsychotics, 39 with second-generation antipsychotics, and four with a combination of both. Concomitant antidepressant or mood stabilizing treatment was permitted (two subjects were treated with mirtazapine, two with lithium and one with valproic acid). Subjects partially overlap with a previous report of 26 medicated subjects with SZ and 23 matched HC assessing function and biochemistry in the ACC by Reid et al (2010).
Clinical Instruments
Clinical assessments were obtained the day the scan was performed. The Brief Psychiatric Rating Scale (Flemenbaum and Zimmermann, 1973) and its positive and negative subscales were used to assess symptom severity. General cognitive function was characterized using the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) (Randolph et al, 1998).
1H-MRS
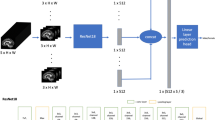
All imaging was performed on a 3T head-only scanner (Magnetom Allegra, Siemens Medical Solutions, Erlangen, Germany), equipped with a circularly polarized transmit/receive head coil. A series of sagittal, coronal, and axial T1-weighted anatomical scans (gradient-recalled echo sequence, TR/TE=250/3.48 ms, flip angle=70°, 5 mm slice thickness, 1.5 mm gap, 512 × 512 matrix) were acquired for spectroscopic voxel placement. Data were collected from a voxel in the ACC (2.7 × 2 × 1 cm3) and in the hippocampus (2.7 × 1.5 × 1 cm3) (Figure 1). To control for head tilt, slices were realigned to midline anatomical landmarks in transverse and coronal orthogonal plans. The MRS voxel was placed in a region of the bilateral dorsal ACC on the basis of the sagittal and coronal images, such that the amount of gray matter in the voxel as viewed on the T1-weighted images was maximized. In the sagittal view, the voxel was placed along the cingulate with the anterior edge of the voxel located approximately at the vertical line passing through the anterior commissure. In the coronal view, the center of the voxel was aligned approximately with the midline and rotated as needed to control for asymmetry across the midline. To facilitate prescription of the hippocampal volume, axial MR images were obtained in an orientation tilted along the long axis of the hippocampi, as viewed in the sagittal images (Figure 1). The voxel was placed in a region of the left hippocampus such that the amount of gray matter was maximized. Manual shimming was performed to optimize field homogeneity across the voxel, and chemical shift selective pulses were used to suppress the water signal. Spectra were acquired using the point-resolved spectroscopy sequence (PRESS; TR/TE=2000/80 ms to optimize the Glx signal (Schubert et al, 2004), number of averages: ACC=256 (scanning time=8 min 32 s); hippocampus=640 (21 min 20 s)). To limit possible effects of nicotine intoxication or withdrawal, patients were allowed, but not encouraged, to smoke up to 1 h before acquisition of images.

Voxel placement and MRS spectra.
MRS Data Processing
To determine the volumes of gray matter, white matter, and cerebrospinal fluid in the MRS voxel, subjects’ structural scans were segmented in SPM8. MRS voxel images, created from the MRS raw data headers, were used to mask each of the tissue classes, and the volumes were calculated in MATLAB. MRS data were analyzed in jMRUI (version 3.0). The residual water peak was removed using the Hankel–Lanczos singular values decomposition filter. Spectra were quantified in the time domain using the AMARES (advanced method for accurate, robust, and efficient spectral fitting) algorithm (Vanhamme et al, 1997). Prior knowledge derived from in vitro and in vivo metabolite spectra was included in the model. A phantom solution of 20 mM glutamate in buffer (30 mM NaHCO3 and 30 mM Na2CO3, pH 7.1) was imaged using the MRS parameters from the in vivo study. The resulting spectrum was quantified in jMRUI, and this model was used to fit the in vivo data. The model consisted of peaks for NAA, choline, creatine (Cr and Cr-2), and three peaks for Glx, which correspond to the H-4 resonance of Glx. The quantification of Glx was based on the H-4 signal only. Amplitude, line width, and chemical shift were optimized for each peak. Cramer–Rao lower bounds (CRLB), used as an estimate of uncertainty, were calculated for each peak. Exclusion criteria were (1) line width of water >25 Hz at FWHM during manual shimming, (2) CRLB >30%, and (3) failure of the fitting algorithm. NAA, Glx, and choline were quantified with respect to creatine (creatine amplitudes of spectra differed between groups in the ACC (t=3.081; p=0.04) but not HIP (t=−0.076; p=0.94)). To assess the reproducibility of the MRS measurements, one HC was scanned on five consecutive days using the methods described above, and the coefficient of variation was calculated for each metabolite ratio. These coefficients of variation for NAA/Cr and Glx/Cr were 4.0 and 4.5% in the ACC, and 2.9 and 12% in hippocampus.
Statistical Analyses
Statistical analyses were performed using SPSS 12.0 for Microsoft Windows (SPSS, Chicago, IL, USA). For analyses of group differences in demographic and clinical variables independent t-tests and χ2-square analyses were performed. Differences in metabolite ratios were investigated with multivariate analyses of covariance, using metabolites as within-group factors, disease state as between-group factor, and age and smoking status as covariates. To examine intra- and interregional correlations of NAA and Glx partial correlations were used, with age and smoking status as covariates. A p-value of <0.05 was considered statistically significant. For correlation data we chose to correct for multiple comparisons with Holm's sequential Bonferroni procedure (Holm, 1979), and α level was set at 0.05.
RESULTS
Demographics
The SZ and HC groups did not differ in gender, age, or parental occupation (Table 1). As expected, HC subjects scored significantly higher than SZ subjects on all RBANS scores.
Neurometabolites
There were no significant differences in gray or white matter content of voxel as well as NAA/Cr and Glx/Cr measurements between SZ and HC subjects in either the ACC or the hippocampus (Tables 2 and 3). When only smokers (HC n=25; SZ n=33) or only non-smokers (HC n=11; SZ n=21) in these groups were compared, neurometabolite levels did not differ either.
In the ACC, there were trend-level positive correlations between NAA/Cr and Glx/Cr in both HC (r=0.26, p=0.08) and SZ (r=0.26, p=0.09) when data were corrected for age. When additional corrections for smoking status were performed, correlations were still at trend level in HC (r=0.28, p=0.08) but not in SZ (r=0.22, p=0.17). In the hippocampus, these metabolites were significantly positively correlated in HC when corrected for age only (r=0.61; p<0.01) as well as age and smoking status (r=0.40, p=0.048). In SZ correlations between these metabolites were absent when corrected for age only (r=0.30; p=0.25), or for age and smoking status (r=−0.02; p=0.89) (between-group comparison: z=2.01, p=0.02) (Figure 2).

Correlations between NAA/Cr and Glx/Cr. Partial correlations (controlling for age and smoking status) between NAA/Cr and Glx/Cr in the ACC (HC: r=0.28, p=0.08; SZ: r=0.22, p=0.17) and HIP (HC: r=0.40, p<0.01 (corrected p=0.048); SZ: r=−0.02; p=0.89). Residuals were obtained by regressing each metabolite ratio onto age and smoking status.
We found significant correlations of NAA/Cr between the ACC and hippocampus in both HC (r=0.60; p<0.01 (corrected for age and smoking status)) and SZ (r=0.54; p<0.01 (corrected for age and smoking status)). Glx/Cr ratios did not correlate between regions in HC (r=0.20; p=0.21) or SZ (r=0.30; p=0.25).
In addition, we found that NAA/Cr and Glx/Cr were significantly higher in the ACC than hippocampus (NAA/Cr: F=7.524, p<0.01; Glx/Cr: F=4.535, p=0.04). These findings were independent of group (SZ vs HC) (NAA/Cr: F=0.02 p=0.97; Glx/Cr: F=0.000, p=0.99), age (NAA/Cr: F=0.905, p=0.34; Glx/Cr: F=0.169, p=0.68).
DISCUSSION
Our study reports several key findings. There were no significant differences in the metabolite ratios of NAA/Cr or Glx/Cr between subjects with SZ and healthy controls. In the ACC, NAA/Cr and Glx/Cr ratios were trend-level positively correlated in both subjects with SZ and healthy controls. In the hippocampus, a significant positive correlation was seen between NAA/Cr and Glx/Cr in HC but not in subjects with SZ, a difference that was statistically significant. In both groups, NAA/Cr and Glx/Cr ratios were higher in the ACC than the hippocampus.
Several studies have now reported positive correlations between the metabolites of NAA/Cr and Glx/Cr in HC (Waddell et al, 2011; Walter et al, 2009). Concurring with these results, our study identified significant or trend-level correlations between these metabolites in healthy controls. In subjects with SZ, the same trend-level correlation seen in HC was identified in the ACC; however, this correlation did not exist in the hippocampus. Contrastingly, in the ACC Bustillo et al (2010) did find inverse correlation of Gln/Glu and NAA in SZ but not healthy controls. Furthermore, Kegeles et al (2012) found positive correlations between Glx and NAA in unmedicated patients in the medial prefrontal cortex and dorsolateral prefrontal cortex but did not find correlations of these metabolites in medicated patients or healthy controls.
Our findings imply that the coupling between NAA and glutamate in SZ is disrupted in the hippocampus. NAA and glutamate are inherently linked through a series of biochemical reactions, mainly the TCA and the glutamate–glutamine cycles, that occur in the mitochondria and between neurons and glia, respectively (Moffett et al, 2007). The altered correlation seen in the SZ group could be driven by a variety of underlying pathologies. Abnormal expression of some of the enzymes regulating the glutamate–glutamine cycle, such as glutamine synthetase, responsible for the conversion of glutamate to glutamine (Steffek et al, 2008), EAATs, which regualte the reuptake of glutamate form the synaptic cleft (Bauer et al, 2008), glutaminase responsible for the conversion of glutamine to glutamate (Bruneau et al, 2005) and the gene that encodes the glutamate transporters EAAT1 and EAAT2 (Walsh et al, 2008) have been reported in SZ. In addition, structural, functional, and gene expression of mitochondrial alterations have been observed in SZ (Clay et al, 2011; Rosenfeld et al, 2011), including in the hippocmapus (Altar et al, 2005). Furthermore, an imbalance of glutamatergic neurotransmission has been proposed as key mechanism for the pathophysiology of SZ (Lahti et al, 1995), and postmortem research has found prominent evidence of alterations of the glutamatergic signaling pathway in hippocampus (Beneyto et al, 2007; Healy and Meador-Woodruff, 2000; Tamminga et al, 2010). Taken together, these findings support the notion that the regulation between NAA and glutamate could be altered in SZ. Using the ratio of NAA to Glx in the hippocampus may prove to be a more sensitive and specific illness biomarker than either metabolite in isolation.
In both groups, NAA/Cr and Glx/Cr ratios in the ACC were higher than in the hippocampus. Several studies conducted in healthy subjects describe similar results; an anterior-to-posterior decrease in NAA in cortical gray matter and an anterior-to-posterior increase of gray and white matter choline has been reported (Baker et al, 2008; Pouwels and Frahm, 1997). In contrast, no regionally dependent changes in Glx levels in gray matter were observed (Pouwels and Frahm, 1998; Waddell et al, 2011).
In contrast to others, reviewed in Steen et al (2005), we did not find decreased NAA levels in the ACC or the hippocampus, but the direction was that of a reduction of about 2–2.5% in the ACC. A more recent meta-analysis found no significant decrease of NAA in the ACC but reported lower NAA levels in the hippocampus of patients with SZ (Kraguljac et al, 2012). However, several other studies report unchanged NAA levels in the hippocampus (Lutkenhoff et al, 2010; Olbrich et al, 2008). It is possible that our negative findings for alterations in NAA levels in SZ represent type-II statistical errors. Steen et al (2005) have recommended a sample size of 39 subjects and 39 HC to have adequate power to detect a 10% reduction of NAA levels. In their meta-analytic approach, the decrease of NAA in the hippocampus and frontal lobe of patients with SZ was estimated to be 5%; adequate power to detect this subtle decrease would require enrolling 103 subjects and 103 HC.
There have been relatively fewer MRS studies of glutamate in SZ because of the difficulty in quantifying glutamate in spectra obtained at low fields. We were able to quantify Glx/Cr in 46 SZ subjects, which is the largest group of patients studied thus far, and found no significant differences compared with controls. Comparison with other studies is challenging because of the use of various measures of glutamatergic function (glutamate, glutamine, or Glx), the study of different brain regions, and different populations. Like us, Wood et al (2007) reported no change in Glx in both the dorsal and rostral ACC in 15 chronic, medicated subjects with SZ. In the ACC, Théberge et al (2002) reported increased levels of glutamine in 21 antipsychotic-naïve SZ subjects and decreased glutamate and glutamine levels in 21 chronic, medicated SZ subjects (Théberge et al, 2003). Bustillo et al (2010) reported an increased ratio of glutamine/glutamate in the ACC measured at 4T in 14 minimally-treated SZ subjects. Only a few studies in SZ have reported glutamate measurements in the hippocampus. van Elst et al (2005) reported higher glutamate levels in a group of 21 medicated participants hospitalized for acute exacerbation of illness, while Lutkenhoff et al (2010) failed to identify such differences in medicated participants (n=9) or their unaffected co-twins. In addition, Olbrich et al (2008) did not find a glutamate difference in a small group of medicated, first-episode participants (n=9). Clearly, these results need to be replicated with larger and well-characterized groups of participants.
Various clinical parameters could have confounded our results. The subjects with SZ were medicated. Rodent studies have suggested that even short-term antipsychotic treatment can modulate glutamatergic neurotransmission (López-Gil et al, 2007). An upregulation of NAA with antipsychotic medication, even after a short period of treatment in human, has been repeatedly reported (Bertolino et al, 2001; McLoughlin et al, 2009; Szulc et al, 2005). Also, some of the subjects were treated with mood stabilizers or antidepressants in addition to antipsychotic medication. It has been suggested that valproic acid or lithium may alter glutamatergic neurotransmission (Garcia et al, 2009; Shibuya-Tayoshi et al, 2008). Smoking status is another factor to be considered potentially biasing outcomes in MRS studies. Patients with SZ have high rates of smoking (Dervaux and Laqueille, 2008). Decreased NAA and choline, but not glutamate levels in smokers have been reported (Durazzo et al, 2004; Gallinat and Schubert, 2007; Gallinat et al, 2007). To alleviate this bias we attempted to match subjects on smoking status, but overall smoking rates in our sample were higher in subjects with SZ than healthy controls. When smoking status was defined as additional covariate in statistical analyses, neurometabolite levels in both the ACC and hippocampus again did not differ between groups. Furthermore, when only smokers or non-smokers were included in analyses again, no differences in neurometabolites between subjects with SZ and HC were found. Trend level correlations between NAA/Cr and Glx/Cr were no longer present in the ACC in subjects with SZ; other correlations were not affected by smoking status. Our findings are in support of previous reports by Gallinat, as they did not see effects of cigarette smoking on cingulate or hippocampal glutamate concentration or NAA levels in the ACC, but we did not replicate their finding of lower NAA levels in the left hippocampus of smokers (Gallinat and Schubert, 2007; Gallinat et al, 2007).
Other factors could have confounded our results. Several technical limitations have to be considered. With methods used in this study, it is impossible to differentiate between glutamate, glutamine, and GABA. The Glx peak therefore is difficult to interpret. Certainly this peak cannot be simply equated with glutamatergic neurotransmission, as extracellular and vesicular glutamates represent only a small fraction of total brain glutamate. Also, the lower data quality of Glx in the hippocampus for subjects with SZ (CRLB of 11% compared with 9% for healthy controls) may explain the absence of the correlation with NAA. Metabolite quantification is affected by high variability as the signal has low sensitivity and multiple processing steps are performed. To take these variations in account, a reference signal is obtained. Data acquisition parameters allowed for only calculating metabolite ratio data, not absolute metabolite concentrations. In this study, amplitudes of creatine spectra that were used as reference were higher in HC than subjects with SZ in the ACC; this could have confounded group mean comparisons of the ratios to creatine and may partially explain the absence of group differences of NAA/Cr and Glx/Cr in the ACC. Using creatine as reference theoretically could also explain the correlations between metabolite levels; however, this is unlikely as intra- and interregional correlations were not consistently observed for metabolites referenced to creatine.
In summary, the hippocampus integrates projections from multiple neocortical areas, regulating learning and memory function via glutamatergic neurotransmission. Different lines of research implicate the hippocampus as a central brain structure affected in SZ. Decoupling of NAA and glutamate within the hippocampus resulting in disruption of glutamatergic neurotransmission may be part of the underlying pathophysiology. However, these results warrant replication and further exploration before definite conclusions can be drawn.
References
Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y et al (2005). Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry 58: 85–96.
Baker EH, Basso G, Barker PB, Smith MA, Bonekamp D, Horská A (2008). Regional apparent metabolite concentrations in young adult brain measured by (1)H MR spectroscopy at 3 Tesla. J Magn Reson Imaging 27: 489–499.
Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB (1996). Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport 7: 1397–1400.
Bauer D, Gupta D, Harotunian V, Meador-Woodruff JH, McCullumsmith RE (2008). Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr Res 104: 108–120.
Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH (2007). Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 32: 1888–1902.
Bertolino A, Callicott JH, Mattay VS, Weidenhammer KM, Rakow R, Egan MF et al (2001). The effect of treatment with antipsychotic drugs on brain N-acetylaspartate measures in patients with schizophrenia. Biol Psychiatry 49: 39–46.
Bruneau EG, McCullumsmith RE, Haroutunian V, Davis KL, Meador-Woodruff JH (2005). Increased expression of glutaminase and glutamine synthetase mRNA in the thalamus in schizophrenia. Schizophr Res 75: 27–34.
Bustillo JR, Rowland LM, Mullins P, Jung R, Chen H, Qualls C et al (2010). 1H-MRS at 4 tesla in minimally treated early schizophrenia. Mol Psychiatry 15: 629–636.
Clark JF, Doepke A, Filosa JA, Wardle RL, Lu A, Meeker TJ et al (2006). N-acetylaspartate as a reservor for glutamate. Med Hypotheses 67: 506–512.
Clay HB, Sillivan S, Konradi C (2011). Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci 29: 311–324.
Dervaux A, Laqueille X (2008). [Smoking and schizophrenia: epidemiological and clinical features]. L’Encéphale 34: 299–305.
Durazzo TC, Gazdzinski S, Banys P, Meyerhoff DJ (2004). Cigarette smoking exacerbates chronic alcohol-induced brain damage: a preliminary metabolite imaging study. Alcohol Clin Exp Res 28: 1849–1860.
Flemenbaum A, Zimmermann RL (1973). Inter- and intra-rater reliability of the Brief Psychiatric Rating Scale. Psychol Rep 32: 783–792.
Gallinat J, Lang UE, Jacobsen LK, Bajbouj M, Kalus P, von Haebler D et al (2007). Abnormal hippocampal neurochemistry in smokers: evidence from proton magnetic resonance spectroscopy at 3 T. J Clin Pychopharmacol 27: 80–84.
Gallinat J, Schubert F (2007). Regional cerebral glutamate concentrations and chronic tobacco consumption. Pharmacopsychiatry 40: 64–67.
Garcia M, Huppertz HJ, Ziyeh S, Buechert M, Schumacher M, Mader I (2009). Valproate-induced metabolic changes in patients with epilepsy: assessment with H-MRS. Epilepsia 50: 486–492.
Healy DJ, Meador-Woodruff JH (2000). Ionotropic glutamate receptor modulation preferentially affects NMDA receptor expression in rat hippocampus. Synapse 38: 294–304.
Holm S (1979). A simple sequentially rejective multiple test procedure. Scan J Statist 6: 65–70.
Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X et al (2012). Elevated prefrontal cortex Y-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry 69: 449–459.
Keltner JR, Wald LL, Christensen JD, Maas LC, Moore CM, Cohen BM et al (1996). A technique for detecting GABA in the human brain with PRESS localization and optimized refocusing spectral editing radiofrequency pulses. Magn Reson Med 36: 458–461.
Kondo DG, Hellem TL, Sung YH, Kim N, Jeong EK, Delmastro KK et al (2011). Review: magnetic resonance spectroscopy studies of pediatric major depressive disorder. Depress Res Treat 2011: 650450.
Kraguljac NV, Reid MA, White D, Jones R, den Hollander J, Lowman D et al (2012). Neurometabolites in schizophrenia and bipolar disorder—a systematic review and metaanalysis. Psychiatry Res; e-pub ahead of print. doi:10.1016/j.psychresns.2012.02.003.
Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13: 9–19.
López-Gil X, Babot Z, Amargós-Bosch M, Suñol C, Artigas F, Adell A (2007). Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 32: 2087–2097.
Lutkenhoff ES, van Erp TG, Thomas MA, Therman S, Manninen M, Huttunen MO et al (2010). Proton MRS in twin pairs discordant for schizophrenia. Mol Psychiatry 15: 308–318.
Magistretti PJ, Pellerin L (1996). Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann N Y Acad Sci 777: 380–387.
Marenco S, Steele SU, Egan MF, Goldberg TE, Straub RE, Sharrief AZ (2006). Effect of metabotropic glutamate receptor 3 genotype on N-acetylaspartate measures in the dorsolateral prefrontal cortex. Am J Psychiatry 163: 740–742.
Marsman A, van den Heuvel MP, Klomp DWJ, Kahn RS, Luijten PR, Hulshoff Pol HE (2011). Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophrenia Bulletin; e-pub ahead of print 11 July 2011. doi:10.1093/schbul/sbr069.
McLoughlin G, Ma D, Tsamg TM, Jones D, Cilia J, Hill M et al (2009). Analysing the effects of psychotropic drugs on metabolite profiles in rat brain using 1H NMR spectroscopy. J Proteome Res 8: 1943–1952.
Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AMA (2007). N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 81: 89–131.
Moreno A, Ross BD, Blüml S (2001). Direct determination of the N-acetyl-L-aspartate synthesis rate in the human brain by (13)C MRS and [1-(13)C]glucose infusion. J Neurochem 77: 347–350.
Nurnberger JI, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J et al (1994). Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry 51: 849–859.
Olbrich HM, Valerius G, Rüsch N, Buchert M, Thiel T, Hennig J et al (2008). Frontolimbic glutamate alterations in first episode schizophrenia: evidence from a magnetic resonance spectroscopy study. World J Biol Psychiatry 9: 59–63.
Pouwels PJ, Frahm J (1997). Differential distribution of NAA and NAAG in human brain as determined by quantitative localized proton MRS. NMR Biomed 10: 73–78.
Pouwels PJ, Frahm J (1998). Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med 39: 53–60.
Randolph C, Tierney MC, Mohr E, Chase TN (1998). The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol 20: 310–319.
Reid MA, Stoeckel LE, White DM, Avsar KB, Bolding MS, Akella NS et al (2010). Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psychiatry 68: 625–633.
Rosenfeld M, Brenner-Lavie H, Ari SGB, Kavushansky A, Ben-Shachar D (2011). Perturbation in mitochondrial network dynamics and in complex I dependent cellular respiration in schizophrenia. Biol Psychiatry 69: 980–988.
Schubert F, Galliant J, Seifert F, Rinneberg H (2004). Glutamate concentrations in human brain using single voxel proton magnetic resonance spectroscopy at 3 Tesla. Neuroimage 21: 1762–1771.
Shibuya-Tayoshi S, Tayoshi S, Sumitani S, Ueno S, Harada M, Ohmori T (2008). Lithium effects on brain glutamatergic and GABAergic systems of healthy volunteers as measured by proton magnetic resonance spectroscopy. Prog Neuro-psychopharmacol Biol Psychiatry 32: 249–256.
Steen RG, Hamer RM, Lieberman JA (2005). Measurement of brain metabolites by 1H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology 30: 1949–1962.
Steffek AE, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH (2008). Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophr Res 103: 71–82.
Szulc A, Galinska B, Tarasow E, Dzienis W, Kubas B, Konarzewska B et al (2005). The effect of risperidone on metabolite measures in the frontal lobe, temporal lobe, and thalamus in schizophrenic patients. A proton magnetic resonance spectroscopy (1H MRS). Pharmacopsychiatry 38: 214–219.
Tamminga CA, Stan AD, Wagner AD (2010). The hippocampal formation in schizophrenia. Am J Psychiatry 167: 1178–1193.
Théberge J, Al-Semaan Y, Williamson PC, Menon RS, Neufeld RWJ, Rajakumar N et al (2003). Glutamate and glutamine in the anterior cingulate and thalamus of medicated patients with chronic schizophrenia and healthy comparison subjects measured with 4.0-T proton MRS. Am J Psychiatry 160: 2231–2233.
Théberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J et al (2002). Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry 159: 1944–1946.
van Elst LT, Valerius G, Büchert M, Thiel T, Rüsch N, Bubl E et al (2005). Increased prefrontal and hippocampal glutamate concentration in schizophrenia: evidence from a magnetic resonance spectroscopy study. Biol Psychiatry 58: 724–730.
Vanhamme L, van den Boogaart A, Van Huffel S (1997). Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson 129: 35–43.
Waddell KW, Zanjanipour P, Pradhan S, Xu L, Welch EB, Joers JM et al (2011). Anterior cingulate and cerebellar GABA and Glu correlations measured by 1H J-difference spectroscopy. Magn Reson Imaging 29: 19–24.
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM et al (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320: 539–543.
Walter M, Henning A, Grimm S, Schulte RF, Beck J, Dydak U et al (2009). The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch Gen Psychiatry 66: 478–486.
Weber OM, Trabesinger AH, Duc CO, Meier D, Boesiger P (1997). Detection of hidden metabolites by localized proton magnetic resonance spectroscopy in vivo. Technol Healthcare 5: 471–491.
Wood SJ, Yücel M, Wellard RM, Harrison BJ, Clarke K, Fornito A et al (2007). Evidence for neuronal dysfunction in the anterior cingulate of patients with schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. Schizophr Res 94: 328–331.
Acknowledgements
This work was supported by the National Institute of Health (Grant RO1 MH081014).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Over the past 2 years, Dr Lahti has received research funds from the National Institute of Health (Grant RO1 MH081014), and an investigator-initiated research grant from Pfizer. Dr Kraguljac has received a research grant from the University of Alabama at Birmingham. Ms Reid, Mr White, and Dr den Hollander declare no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Kraguljac, N., Reid, M., White, D. et al. Regional Decoupling of N-acetyl-aspartate and Glutamate in Schizophrenia. Neuropsychopharmacol 37, 2635–2642 (2012). https://doi.org/10.1038/npp.2012.126
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.126
Keywords
This article is cited by
-
Higher-order functional brain networks and anterior cingulate glutamate + glutamine (Glx) in antipsychotic-naïve first episode psychosis patients
Translational Psychiatry (2024)
-
Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: a meta-analysis of 1H-magnetic resonance spectroscopy studies
Molecular Psychiatry (2022)
-
Glutamatergic dysfunction is associated with phenotypes of VGF-overexpressing mice
Experimental Brain Research (2022)
-
The relationship between synaptic density marker SV2A, glutamate and N-acetyl aspartate levels in healthy volunteers and schizophrenia: a multimodal PET and magnetic resonance spectroscopy brain imaging study
Translational Psychiatry (2021)
-
Altered anterior cingulate glutamatergic metabolism in depressed adolescents with current suicidal ideation
Translational Psychiatry (2020)
