
Abstract
In the cell division cycle of budding yeast, START refers to a set of tightly linked events that prepare a cell for budding and DNA replication, and FINISH denotes the interrelated events by which the cell exits from mitosis and divides into mother and daughter cells. On the basis of recent progress made by molecular biologists in characterizing the genes and proteins that control START and FINISH, we crafted a new mathematical model of cell cycle progression in yeast. Our model exploits a natural separation of time scales in the cell cycle control network to construct a system of differential-algebraic equations for protein synthesis and degradation, post-translational modifications, and rapid formation and dissociation of multimeric complexes. The model provides a unified account of the observed phenotypes of 257 mutant yeast strains (98% of the 263 strains in the data set used to constrain the model). We then use the model to predict the phenotypes of 30 novel combinations of mutant alleles. Our comprehensive model of the molecular events controlling cell cycle progression in budding yeast has both explanatory and predictive power. Future experimental tests of the model’s predictions will be useful to refine the underlying molecular mechanism, to constrain the adjustable parameters of the model, and to provide new insights into how the cell division cycle is regulated in budding yeast.
Similar content being viewed by others

INTRODUCTION
Cell growth and division in eukaryotes is controlled by a gene–protein regulatory network of immense complexity. To understand how this regulatory network governs the observed properties of proliferating cells requires mathematical models, which keep track of the myriad interactions controlling the process. Modeling approaches have proved helpful in unraveling the dynamics of cell proliferation in frog eggs,1 fruit fly embryos,2 fission yeast,3 and budding yeast.4–10 Mathematical models are not static objects; as new experimental data come to light, the models must be extended and improved.
In this paper we focus on cell cycle regulation in budding yeast, Saccharomyces cerevisiae. The molecular network regulating cell cycle progression in budding yeast (Figure 1), which is the foundation of our mathematical model, is the product of over 40 years of intensive study by molecular cell biologists starting in 1970 when Hartwell et al.11–13 used classical genetics to dissect the budding yeast cell cycle into a semiordered sequence of events. The earliest event of this process Hartwell called START, when a budding yeast cell initiates in rapid succession the onset of DNA replication, the emergence of a new bud, and duplication of the spindle pole body (centrosome). Later, it was determined that some ‘master’ transcription factors are activated at the START transition.14 After START, overlapping sequences of cell cycle events proceed simultaneously (DNA synthesis, migration of the nucleus to the bud neck, formation of an intranuclear spindle, congression of replicated chromosomes to the metaphase plate). Successful completion of these events is monitored at the metaphase checkpoint. When all systems are ‘go’, the yeast cell exits from mitosis (metaphase→anaphase→telophase→cell division). We subsume these processes under the title FINISH, because they return the mother and daughter cells to G1 phase, prepared to execute START of the next cell cycle.

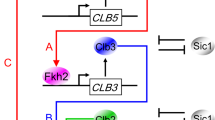
Proposed mechanism of cell cycle regulation in budding yeast: (top) overview; (bottom) mechanistic details of the ‘modules’. Solid arrows represent reactions of synthesis, degradation, complex formation, phosphorylation and dephosphorylation, and translocations. The rectangle+oval icon represents a protein complex, and the four small ovals represent end-products of proteolysis reactions. Dashed lines with arrowhead or bar head represent, respectively, activation or inhibition of a reaction, a substance, or a module by a substance or a module. Also a protein name written along a reaction arrow indicates the catalysis of the reaction by the protein. To simplify the model, we combine cyclins with similar functions: Cln1+Cln2=‘Cln2’, Clb5+Clb6=‘Clb5’, and Clb1+Clb2=‘Clb2’. The kinase partner, Cdk1, of each cyclin is not indicated in the diagram, because we assume (justifiably) that Cdk1 is in excess and that it combines rapidly and strongly with any available cyclin molecules. Cln2, Clb5, and Clb2 are primarily responsible for initiating bud formation, DNA synthesis and mitosis, respectively, although there is some overlap of function among these cyclin-dependent kinases. The cyclin-dependent kinase inhibitor, CKI, refers to both Sic1 and Cdc6, which are stoichiometric inhibitors of Clb-dependent kinases. For a full and precise description of the model, see the set of kinetic equations in Supplementary Table S2.
The major goals of this paper are: (a) to build a mathematical model of the wiring diagram; (b) to confirm that the model accurately reproduces known characteristics of wild-type and mutant strains of budding yeast; and (c) to predict the phenotypes of novel combinations of mutant alleles.
RESULTS
In the following subsections we discuss our model in light of the observed phenotypes of some selected strains carrying mutations in START and FINISH genes. Additional considerations are provided in the Supplementary Text. Unless otherwise stated, our model simulations are consistent with the observed phenotypes of these strains. Hence, all factual statements are confirmed by the references provided and by the simulations in the Supplementary Information.
Simulation of START mutants
SBF and MBF mutants
SBF is a master cell cycle transcription factor composed of two components, Swi4 and Swi6, and inhibited by Whi5.14,15 MBF is another important cell cycle transcription factor composed of Swi6 and Mbp1.16 Mutant strains with a single-gene deletion of either SWI4 or MBP1 are viable, indicating that either SBF or MBF is sufficient for budding yeast cells to execute START.15,17–19
Suppression of both SBF and MBF (e.g., in the strains swi4Δ swi6Δ or swi4Δ mbp1Δ) causes budding yeast cells to arrest before the START transition.15 whi5Δ cells initiate SBF transcription prematurely; hence, they are smaller than wild-type cells.20 WHI5OP cells overexpress Whi5 and delay the START transition, dividing at a larger size than wild-type cells.18 Deletion of SWI6, which encodes a common subunit of SBF and MBF, turns out to be viable and large, and swi6Δ bck2Δ cells are inviable.14,19 We explain these observations by assuming that Swi4, with the help by Bck2, can function as SBF, although with less ability to activate transcription.
Targets of Cln3-dependent kinase include Swi6 and Whi5. Phosphorylation of either of these proteins activates SBF and initiates START.17–19 Consequently, cln3Δ cells have an extended G1 phase and are larger than wild-type cells (1.85 times larger in our simulations). cln3Δ whi5Δ cells are as small as whi5Δ,18 confirming that the major role of Cln3:Cdk1 in promoting START is phosphorylating Whi5. Additional deletion of MBP1 in cln3Δ mutant cells has little effect on their phenotype; however, cln3Δ swi4Δ cells are inviable.21 Because swi4Δ cells (which have only MBF activity) are viable, lethality of the double mutant strain, swi4Δ cln3Δ, indicates that Cln3:Cdk1 has a significant role in triggering both SBF and MBF activity.
Bck2 protein is another important activator of SBF and MBF transcription factors. Because cln3Δ bck2Δ cells arrest in early G1,22,23 it appears that Cln3 and Bck2 perform some essential overlapping function(s) at the START transition. Viability of the triple deletion strain cln3Δ bck2Δ whi5Δ (ref. 17) suggests that this function involves removing Whi5 from SBF complexes. Model simulations agree with these experimental observations. These results confirm that Whi5 is a chief inhibitor of the START transition, and the main and essential target of both Cln3-dependent kinase and Bck2. The triple deletion strain cln3Δ bck2Δ whi5Δ is 1.48 times larger than wild-type cells because transcription of SBF-controlled promoters relies on non-phosphorylated Swi6:Swi4 complex, which is only moderately active in driving the synthesis of Cln1,2.
In cells lacking both CLN3 and BCK2, Mbp1 is not phosphorylated and when bound to promoter it represses the background synthesis of Clb5,6. This assumption is supported by the observation that Mbp1 may function as a gene repressor.24 That explains why the model predicts that the quadruple deletion strain, cln3Δ bck2Δ whi5Δ mbp1Δ, is slightly smaller than cln3Δ bck2Δ whi5Δ and only 30% larger than wild-type cells; see Supplementary Table S8. Other interesting predictions relate to the swi4Δ mbp1Δ strain, lacking both MBF and SBF, which is inviable even when either CLN3 or BCK2 are overexpressed. It is also not surprising that overexpression of CLB5 rescues the lethality of swi4Δ mbp1Δ cells. The model also predicts that lethality of swi4Δ mbp1Δ cannot be rescued by deletion of either SIC1 or CDH1. The SIC1-related prediction is indirectly supported by the fact that swi4Δ swi6Δ sic1Δ and bck2Δ swi6Δ sic1Δ strains (lacking both MBF and SBF activities) are inviable.22,25
Rescue of the inviable triple-cln deletion strain
The activation of SBF and MBF at START initiates the synthesis of Cln1,2 and Clb5,6 cyclins. These newly synthesized cyclins (in conjunction with Cdk1) enhance phosphorylation processes begun by Cln3-dependent kinase and/or Bck2 protein, thereby making the START transition irreversible. Once past START, the yeast cell is committed to DNA replication and budding. Another important role of Cln1,2-dependent kinases is to rid the cell of CKI, our designation for the stoichiometric inhibitors, Sic1 and Cdc6, of B-type cyclin-dependent kinases (Clb1–6:Cdk1). As CKI levels drop, Clb5,6-dependent kinase activities rise, initiating DNA replication and phosphorylating (inactivating) Cdh1. Inactivation of APC:Cdh1 allows the mitotic cyclins, Clb1 and Clb2, to accumulate, thereby preparing the cell for mitosis. This description is confirmed by the fact that cells lacking all three G1 cyclins, cln1Δ cln2Δ cln3Δ, arrest in G1,26 but lethality of the triple-cln deletion strain can be rescued by additional deletion of SIC1 (see Supplementary Figure S2).27,28 Viability of cln1Δ cln2Δ cln3Δ cells can also be restored by overexpressing CLB5,29,30 indicating high levels of Clb5 can overcome repression by Sic1. Interestingly, SIC1 deletion is not sufficient to rescue cln3Δ bck2Δ cells.22 Compared with triple-cln deletion cells, cln3Δ bck2Δ cells have significantly lower levels of Clb5,6 cyclins, as well as low levels of Cln1,2 cyclins. Hence, cln3Δ bck2Δ sic1Δ cells are unable to deactivate APC:Cdh1 (see Supplementary Figure S2).
We have tested whether deletion of CDH1 can rescue the triple-cln deletion strain. Model simulations agree with observations that cln1Δ cln2Δ cln3Δ cdh1Δ cells pass START but arrest in telophase.31 The model also agrees with the experimental observation that cln1Δ cln2Δ cdh1Δ cells are viable.32 In addition, the model predicts that deletion of CDH1 restores cln1Δ cln2Δ cells33 almost to wild-type cell size. The model also predicts that cln1Δ cln2Δ bck2Δ cdh1Δ cells are viable and that cln3Δ bck2Δ cdh1Δ cells arrest in telophase.
Simulation of FINISH mutants
Exit from mitosis and return to G1 phase involves degradation of all B-type cyclins, Clb1–6, and activation of a phosphatase, Cdc14, that reverses the phosphorylations carried out by Clb-dependent kinases during S/G2/M.34 CDC14 is an essential gene in budding yeast, and cdc14Δ cells arrest in telophase. The activity of Cdc14 is tightly controlled by association with its inhibitory binding partner, Net1, which is located in the nucleolus.35,36 Successful exit from mitosis requires that Cdc14 be released from the nucleolus to do its jobs in the nucleus and cytoplasm. Hence, genes involved in the FINISH transition are often identified by their effects on Cdc14 release from the nucleolus, which occurs in two consecutive steps: Cdc14 early anaphase release (FEAR)9,37–40 and the mitotic exit network (MEN).36,38,40–43
FEAR mutants
In the absence of essential MEN components, cells exhibit only an early, partial release of Cdc14, enabled by the FEAR pathway: APC:Cdc20 ⊣ Pds1⊣ Esp1 ⊣ PPA2:Cdc55→Net1 ⊣ Cdc14. The FEAR pathway can be observed in isolation in strains in which MEN effectors, Cdc15 or Tem1, are deleted or substantially inhibited.44,45 In Figure 2 we simulate exit from mitosis, by cdc20 block and release, in a wild-type genetic background (Figure 2a) and in a cdc15Δ cell (Figure 2b). The Cdc15-deficient cell arrests in telophase with high Clb2 level, because Cdc14 is only partially and transiently released from the nucleolus, in agreement with ref. 45. The FEAR pathway stifles itself by partially degrading mitotic cyclins and thereby quenching the phosphorylation of Net1 by Cdc5 and Clb-dependent kinase activities, leading to resequestration of Cdc14. MEN activity is needed to maintain Net1 in its phosphorylated state and Cdc14 released.

Cdc14 early anaphase release (FEAR) in cdc15Δ cells. (a) Control cells (cdc20Δ GAL-CDC20) are synchronized in metaphase by pre-simulation for 135 min in the absence of Cdc20, then Cdc20 is added back at t=0. These cells exit mitosis normally. (b) When cells lacking Cdc15 are treated the same way, Cdc14 is released only for a short time. Without MEN support, Cdc14 quickly returns to the nucleolus because, as Clb2 drops, Net1 is reactivated and Cdc14 becomes resequestered. This transient release of Cdc14 agrees well with observations shown on Figure 1b of ref. 45. MEN, mitotic exit network.
In the FEAR pathway, APC:Cdc20 activates Esp1 by degrading its stoichiometric inhibitor Pds1. If PDS1 is deleted, then Esp1 is active, promoting early Cdc14 release and exit from mitosis.46,47 Similarly, deletion of CDC55 promotes early Cdc14 release, although cdc55Δ cells are viable.48 Unsurprisingly, cdc20Δ cells arrest in metaphase with Cdc14 sequestered in the nucleus, but it is noteworthy that PDS1 deletion does not rescue cdc20Δ cells.49 To rescue cdc20Δ cells requires additional deletion of both PDS1 and CLB5.50 Viability of the cdc20Δ pds1Δ clb5Δ strain depends on the CDH1 gene product.50 The lethality of the cdc20Δ pds1Δ and cdc20Δ pds1Δ clb5Δ cdh1Δ strains50 can be rescued by adding ~10 copies of the SIC1 gene.51 Our model simulations are consistent with all these phenotypes (Supplementary Table S7).
Despite the fact that early release of Cdc14 activates the MEN, it seems that the FEAR pathway is not essential for exit from mitosis. The FEAR pathway is blocked in esp1Δ cells and in PDS1-dbΔ cells (expressing a non-degradable form of Pds1, the stoichiometric inhibitor of Esp1).45,52 Mutants lacking separase (Esp1) activity are inviable (or mitotic exit is substantially delayed) mainly because of Esp1’s role as a protease to cleave cohesin rings, thereby initiating sister chromatid separation. This role of Esp1 can be replaced by TEV protease, which can be used to cleave cohesin molecules that have been engineered to carry the TEV protease recognition sequence. In this case, anaphase proceeds normally, activating the MEN while the FEAR pathway remains blocked.42 In agreement with Lu and Cross,42 our simulations (Figure 3) show that MEN activity is absolutely needed for mitotic exit, and FEAR is dispensable. The control (Figure 3a) shows how wild-type cells exit from mitosis after cdc20 block and release, whereas GAL-PDS1-dbΔ cells (no FEAR, no MEN) arrest in metaphase (Figure 3b). By contrast, cells with functional MEN but no FEAR, GAL-PDS1-dbΔ GAL-TEV, are able to exit mitosis (Figure 3c). Whereas cdc20-blocked cells are not able to exit mitosis after expression of TEV protease (Figure 3e) because both FEAR and MEN pathways are inactive, overexpression of non-degradable Sic1 from a GAL-SIC1-4A gene promotes mitotic exit (Figure 3f) by the pathway Sic1 ⊣ Clb2 ⊣ Cdc15→MEN.

Cdc14 release in strains carrying GAL-PDS1-dbΔ or GAL-SIC1-4A. These simulations are comparable to Figure 1 of ref. 42. (a) Control; same as Figure 2a. (b) In the mutant strain expressing a non-degradable version of Pds1 (securin), when Cdc20 is activated, Clb2 is only partially degraded, and the FEAR pathway (Pds1⊣ Esp1 ⊣ PPA2:Cdc55→Net1 ⊣ Cdc14) is inhibited. Consequently, Cdc14 remains sequestered in nucleolus, Cdh1:APC is not fully activated, and the cell cannot exit from mitosis. (c) Nonetheless, if cohesin molecules are cleaved by TEV protease, then the spindle position checkpoint can be turned off and the MEN pathway activated, promoting Cdc14 release and exit from mitosis, even though the FEAR pathway is blocked by non-degradable Pds1. (d,e) Cdc14 is not released in cells carrying either the GAL-SIC1-4A gene (encoding non-degradable Sic1) or the GAL-TEV construct, but (f) Cdc14 is released in cells carrying both GAL-SIC1-4A and GAL-TEV.
MEN mutants
The MEN promotes sustained Cdc14 release by activating kinases that continue to phosphorylate Net1 as the activity of Clb2-kinase drops due to its degradation by Cdc20. The MEN kinase Cdc15 and its partner Tem1 integrate spatial and temporal cues to promote mitotic exit when the spindle poles are properly positioned and the metaphase-to-anaphase transition is executed. Cdc15 activity is inhibited by phosphorylation by Clb2:Cdk153 and activated by dephosphorylation by Cdc14.54 Thus, partial degradation of Clb2 by APC:Cdc20 in anaphase and partial release of Cdc14 by the FEAR pathway in anaphase together promote activation of the MEN. Tem1 is kept inactive by Bfa1.55,56 Bfa1’s regulatory partner, Bub2, is inactivated by phosphorylation by Cdc5 and activated by dephosphorylation by Cdc14.39 Notice that, even as MEN promotes Cdc14 release, Cdc14 phosphatase feeds back on MEN activity by an incoherent signaling pathway: Cdc14 activates MEN effector kinases by dephosphorylating Cdc15, but Cdc14 inactivates MEN effector kinases by activating Bfa1:Bub2 (see the MEN module in Figure 1). The dynamical consequences of these complex interactions demand mathematical modeling.
Deletion of both BUB2 and BFA1 is not lethal,57 whereas deletion of CDC15 or CDC5 or TEM1 is lethal.44,58 Overexpression of CDC15 rescues the tem1Δ strain, but overexpression of TEM1 does not rescue cdc15Δ cells, suggesting that Cdc15 has some activity, independent of Tem1, in regulating Cdc14 release. To account for this fact in our model, Cdc15 phosphorylates Net1 both in conjunction with Tem1 (MEN variable) and independent of Tem1 (see the RENT module in Figure 1). Reducing the level of Net1, using temperature-sensitive alleles of the NET1 gene, rescues both tem1Δ and cdc15Δ strains.35 The cdc15Δ strain can also be rescued by overexpressing CDC14. Our model is consistent with all these phenotypes. Another interesting MEN mutant is net1ts cdc15Δ cdh1Δ, whose viability59 is accounted for by our model but not by others, to our knowledge.
Cdc5 is a key regulator of the FINISH transition because of the roles it has in activating the MEN and in the release of Cdc14 from its inhibitor, Net1, in the nucleolus (see the MEN and RENT modules in Figure 1). Notice that Cdc14 release requires the combined activity of two different kinases: Cdc5 and either MEN or Clb2:Cdk1. Successful exit from mitosis requires that Cdc14 be released from the nucleolus to do its jobs in the nucleus and cytoplasm. Not surprisingly, CDC5 deletion is lethal. CDC5 overexpression, on the other hand, promotes Cdc14 release from the nucleolus even in the absence of both MEN and FEAR functions (Figure 4). Simulations agree with experimental data37,60 that GAL-CDC5 rescues lethality of cdc15Δ strain (Figure 4a) and promotes Cdc14 release in GAL-PDS1-dbΔ cdc20Δ cells (Figure 4b), in cells treated with nocodazole (Figure 4c), and in hydroxyurea-arrested cells (Figure 4d).

The roles of Polo kinase (Cdc5) in mitotic exit. (a) Overexpression of Cdc5 rescues the lethality of cdc15Δ cells, which lack MEN-dependent kinase activity. (b,c) Overexpression of Cdc5 promotes Cdc14 release in cells with blocked FEAR pathway, either by non-degradable Pds1 or by nocodazole. (d) Overexpression of Cdc5 promotes Cdc14 release in hydroxyurea-treated cells. These simulations agree well with experimental observations.37 MEN, mitotic exit network.
Cdc14 Oscillations
Once Cdc14 is fully released, it dephosphorylates Cdh1P, thereby activating APC:Cdh1, which polyubiquitinates Cdc5, labeling it for destruction.61–63 The degradation of Cdc5 causes Cdc14 to be resequestered in the nucleolus.62 It has been observed by two groups independently that the release and resequestration of Cdc14 in budding yeast can cycle periodically when Clb2:Cdk1 activity is maintained at a high level by using a non-degradable form of Clb2.7,64 Cdc14 ‘endocycles’ are a consequence of a negative-feedback loop in the MEN (Cdc5 ⊣ Net1 ⊣ Cdc14→Cdh1 ⊣ Cdc5), as demonstrated by simple mathematical models.7,8,64 But is this simple picture consistent with everything else known about exit-from-mitosis in budding yeast? Can Cdc14 endocycles be reproduced by a more comprehensive model that is consistent with the observed phenotypes of all exit-from-mitosis mutants? In light of this question, reproducing Cdc14 endocycles becomes an important test for our model.
In Figure 5a we show that our wide-ranging model of exit from mitosis reproduces Cdc14 endocycles in the mutant strain, GAL-CLB2-dbΔ, where they were first observed. The period of oscillation is ~70 min, in agreement with experimental observations.7

Cdc14 endocycles. (a) When grown in galactose, GAL-CLB2-dbΔ cells express a mutant form of Clb2 that is not degraded by APC:Cdc20 and only weakly degraded by APC:Cdh1 (37% of that for the wild-type protein). Because these cells maintain a high level of Clb2-kinase activity, they cannot exit mitosis, and yet they exhibit regular oscillations of Cdc14 release and resequestration (period=73 min). (b) The frequency of Cdc14 endocycles depends linearly on Clb2 level. (c,d) Similar oscillations in Cdc14 release are exhibited by the GAL-CLB2nd strain. The CLB2nd allele encodes Clb2 protein that is not degraded by either APC:Cdc20 or APC:Cdh1. The simulations in this figure agree well with experimental observations in refs 7,64.
As expected for cells carrying the GAL-CLB2-dbΔ gene, Clb2 level is very high when the cells are growing in galactose, and the sustained high level of Clb2 arrests these cells in telophase. For GAL-CLB2-dbΔ we set the degradation rate constant of Clb2 by APC:Cdh1 to 37% of that for wild-type cells. In this case, periodic Cdc14 release causes oscillations of Clb2 as well as Cdh1 and Cdc5. Although Clb2 oscillates, its level remains high, because Clb2 is rapidly and constitutively synthesized and only partially degraded in GAL-CLB2-dbΔ cells. Our model also predicts that the frequency of Cdc14 endocycles (Figure 5b) increases linearly over a range of Clb2 levels: [Clb2] large enough to sustain mitotic arrest but not so large as to cause constitutive Cdc14 release.
Because Cdc14 oscillations are observed in GAL-CLB2nd cells, lacking both destruction and KEN boxes of Clb2, the degradation of Clb2 by APC:Cdh1 is not necessary for oscillations; see ref. 64 and Figure 5c. Although GAL-CLB2nd cells continue to synthesize endogenous Clb2, it is quickly degraded by the high level of APC:Cdh1. Hence, the endogenous fraction of Clb2 accounts for only a small amplitude oscillatory component on top of the constant high level of non-degradable Clb2. The simulated dependence of oscillation frequency on [Clb2] in this strain (Figure 5d) agrees well with observed trends.64
Using the model for predictive purposes, we find that Cdc14 endocycles are likely to be observed in strains carrying the GAL-CLB2-dbΔ gene in combination with: cdc15as1, swi5Δ, cdc20-3, or pds1Δ cdc20Δ clb5Δ (Supplementary Figures S3A–D); but they are absent when GAL-CLB2-dbΔ is combined with cdc5-1, CDC5-dbΔ, cdh1Δ, or cdc14-1 (Supplementary Figures S4A–D). Thus, we conclude that Cdc14 endocycles will be abolished in GAL-CLB2-dbΔ strains if the negative-feedback loop Cdc5 ⊣ Net1 ⊣ Cdc14→Cdh1 ⊣ Cdc5 is interrupted but persist when other genes of the FINISH transition are deleted (e.g., cdc15Δ, sic1Δ, swi5Δ, pds1Δ, cdc20Δ).
DISCUSSION
Using a new approach for modeling gene–protein regulatory networks in a modular manner, we have reformulated and extended earlier models of the START and FINISH transitions in the cell division cycle of Saccharomyces cerevisiae. Exploiting a natural separation of time scales in these networks, the new modeling approach classifies reactions into three basic types: (1) protein synthesis and degradation (slow); (2) protein modification, e.g., phosphorylation and dephosphorylation (moderate); and (3) multi-protein complex formation (fast). This classification allows considerable latitude in describing biochemically realistic networks, while limiting the number of adjustable parameters that must be estimated from the data. The modularity of the scheme allows the model to be quickly and easily extended to encompass new aspects of the control mechanism without drastically disturbing the ancestral model and its legacy parameter values. In our experience, the ‘standard component’ modeling strategy65 has all these advantages.
Our standard-component model of the budding yeast cell cycle successfully accounts for the observed phenotypes of 257 yeast strains carrying mutant alleles of cell cycle genes in various combinations (from a test collection of 263 strains). The good agreement of simulation results with experimental data is strong evidence confirming our hypothetical regulatory mechanism of cell cycle control in budding yeast (Figure 1). We have used the model to predict the phenotypes of 30 mutant strains that were not included in our test collection of 263 strains. Some of the most interesting predictions are listed in Table 1. Three of these predictions are already confirmed by published experiments, and two novel strains have been created and phenotyped by our collaborators (Adames & Peccoud, private communication). One prediction was not confirmed, suggesting that Whi5 has a role inhibiting MBF, a possibility not included in our model.
In putting forward this mechanism, we focused our attention on the genes and proteins affecting the START and FINISH transitions, because recent experimental results on these transitions have necessitated revisions of our previous model of the yeast cell cycle.5 In particular, our revised model captures the appearance of Cdc14 endocycles in mutant strains that are arrested in metaphase by high levels of the mitotic cyclin Clb2. Although Cdc14 endocycles have been simulated by simplified models of the FEAR pathway,7,8,64 they have not appeared (until now) in models that attempt a more comprehensive account of the entire cell cycle of budding yeast. Using our model, we have predicted the presence or absence of Cdc14 endocycles in mutant strains that combine Clb2 overexpression with the deletion of other genetic elements of the FINISH transition. These simulations support a ‘two-hit’ mechanism, whereby Cdc14 release requires the combined activity of two different kinases: Cdc5 and either MEN kinase or mitotic cyclin-dependent kinase.
Although our model is consistent with the diverse phenotypes of >250 experimentally characterized mutant strains, only a small fraction of the possible combinations of mutant alleles have been experimentally characterized to date. Hence, our model, to the extent that it is an accurate portrayal of the cell cycle control system in yeast, could be a useful tool to predict the phenotypes of an enormous number of yeast strains that are yet to be studied experimentally. Just to simulate strains with combinations of deletion and overexpression alleles of up-to-five genetic loci will provide predictions of >20 million phenotypes, of which only about 300 have been studied experimentally. We are currently undertaking this calculation, and we will sift through the predictions for the most interesting and informative strains that can be tested experimentally. For example, strains lacking many genes but retaining viability may be especially helpful in understanding the molecular mechanism of cell cycle control. Also, by classifying predictions as robust or fragile (see Supplementary Text and Supplementary Figure S5), we will provide a further criterion for prioritizing experimental tests of the model.
MATERIALS AND METHODS
In the Supplementary Text, we describe the modeling approach65 used to formulate the differential-algebraic equations set forth in Supplementary Tables S1–S3. Supplementary Table S4 lists the 293 mutant strains simulated in this study, and Supplementary Table S5 records the parameter changes used to simulate these mutations. Supplementary Tables S6 and S7 summarize the observed phenotypes of the START and FINISH mutant strains discussed in the main text, and Supplementary Table S8 summarizes the model’s predicted phenotypes of novel mutant strains.
The model and all simulations reported in this paper are implemented in the Virtual Cell and in our lab’s custom-built software, PET.66 In the Virtual Cell Database (VCell DB) our model is found under MathModels: PublicMathModels: pavelkr: BuddingYeastCellCycle_2015. PET software can be downloaded freely from http://mpf.biol.vt.edu/pet/, and the pet file needed to run our model can be downloaded from the site http://mpf.biol.vt.edu/research/start_to_finish/. That site also provides a pdf containing simulations of wild-type cells and all 293 mutant strains. The model can be explored with an online simulator at http://sbmlsimulator.org/simulator/2. In Figure 6 we present, for reference, the simulation of a wild-type daughter cell growing in glucose, and in Supplementary Figure S1 we present simulations of three inviable mutant strains.

The cell cycle of a wild-type daughter cell in glucose medium. Cell size (mass) increases exponentially (mass-doubling time=90 min). The cell divides at a size of 2.37 (arbitrary units) into a smaller daughter cell (size=1.09) and a larger mother cell (size=1.28). Following the time-course of daughter cells over three generations, we plot the rise and fall of certain key cell cycle regulators in a, b. The cycle time of daughter cells in this simulation is 100 min. One arbitrary unit of cell size=33pg=30 fL.
References
Novak, B. & Tyson, J. J. Modeling the cell division cycle: M-phase trigger, oscillations and size control. J. Theor. Biol. 165: 101–134 (1993).
Calzone, L., Thieffry, D., Tyson, J. J. & Novak, B. Dynamical modeling of syncytial mitotic cycles in Drosophila embryos. Mol. Syst. Biol. 3, 131–141 (2007).
Sveiczer, A., Csikasz-Nagy, A., Gyorffy, B., Tyson, J. J. & Novak, B. Modeling the fission yeast cell cycle: quantized cycle times in wee1–cdc25Δ mutant cells. Proc. Natl Acad. Sci. USA 97, 7865–7870 (2000).
Chen, K. C. et al. Kinetic analysis of a molecular model of the budding yeast cell cycle. Mol. Biol. Cell 11, 369–391 (2000).
Chen, K. C. et al. Integrative analysis of cell cycle control in budding yeast. Mol. Biol. Cell 15, 3841–3862 (2004).
Toth, A., Queralt, E., Uhlmann, F. & Novak, B. Mitotic exit in two dimensions. J. Theor. Biol. 248, 560–573 (2007).
Manzoni, R., Visintin, C., Montani, F., Ciliberto, A. & Visintin, R. Oscillation in Cdc14 release and sequestration reveal a circuit underlying mitotic exit. J. Cell. Biol. 180, 210–222 (2010).
Vinod, P. K. et al. Computational modeling of mitotic exit in budding yeast; the role of separase and Cdc14 endocycles. J. R. Soc. Interface 8, 1128–1141 (2011).
Queralt, E., Lehane, C., Novak, B. & Uhlmann, F. Downregulation of PP2 ACdc55 phosphatase by separase initiates mitotic exit in budding yeast. Cell 125, 1–14 (2006).
Tyson, J. J. & Novak, B. Control of cell growth, division and death: information processing in living cells. Interface Focus 4, 20130070 (2014).
Hartwell, L. H. Saccharomyces cerevisiae cell cycle. Bacteriol. Rev. 38, 164–198 (1974).
Hartwell, L. H., Culotti, J., Pringle, J. R. & Reid, B. J. Genetic control of the cell division cycle in yeast. Science 183, 46–51 (1974).
Johnston, G. C., Pringle, J. R. & Hartwell, L. H. Coordination of growth with cell division in the yeast Saccharomyces cerevisiae . Exp. Cell Res. 105, 79–98 (1977).
Nasmyth, K. & Dirick, L. The Role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell 66, 995–1013 (1991).
Koch, C., Moll, T., Neuberg, M., Ahorn, H. & Nasmyth, K. A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science 261, 1551–1557 (1993).
Dirick, L., Moll, T., Auer, H. & Nasmyth, K. A central role for SWI6 in modulating cell cycle start-specific transcription in yeast. Nature 357, 508–513 (1992).
de Bruin, R. A. M., McDonald, W. H., Kalashnikova, T. I., Yates, J. III & Wittenberg, C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117, 887–898 (2004).
Costanzo, M. et al. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117, 899–913 (2004).
Wijnen, H., Landman, A. & Futcher, B. The G1 cyclin Cln3 promotes cell cycle entry via the transcription factor Swi6. Mol. Cell Biol. 22, 4402–4418 (2002).
Jorgensen, P., Nishikawa, J. L., Breitkreutz, B. -J. & Tyers, M. Systematic identification of pathways that couple cell growth and division in yeast. Science 297, 395–400 (2002).
Ferrezuelo, F., Aldea, M. & Futcher, B. Bck2 is a phase-independent activator of cell cycle-regulated genes in yeast. Cell Cycle 8, 239–252 (2009).
Wijnen, H. & Futcher, B. Genetic analysis of the shared role of CLN3 and BCK2 at the G1-S transition in Saccharomyces cerevisiae . Genetics 153, 1131–1143 (1999).
Di Como, C. J., Chang, H. & Arndt, K. T. Activation of CLN1 and CLN2 G1 cyclin gene expression by BCK2. Mol. Cell Biol. 15, 1835–1846 (1995).
de Bruin, R. A. M. et al. Constraining G1-specific transcription to late G1 phase: the MPF associated corepressor Nrm1 acts via negative feedback. Mol. Cell 23, 483–496 (2006).
Flick, K. & Wittenberg, C. Multiple pathways for suppression of mutants affecting G1-specific transcription in Saccharomyces cerevisiae . Genetics 169: 37–49 (2005).
Richardson, H. E., Wittenberg, C., Cross, F. & Reed, S. I. An essential G1 function for cyclin-like proteins in yeast. Cell 59, 1127–1133 (1989).
Tyers, M. The cyclin-dependent kinase inhibitor p40sic1imposes the requirement for Cln G1 cyclin function at Start. Proc. Natl Acad. Sci. USA 93, 7772–7776 (1996).
Schneider, B. L., Yang, Q. -H. & Futcher, A. B. Linkage of replication to Start by the Cdk inhibitor Sic1. Science 272, 560–562 (1996).
Epstein, C. B. & Cross, F. R. CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev. 6, 1695–1706 (1992).
Schwob, E. & Nasmyth, K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae . Genes Dev. 7, 1160–1175 (1993).
Schwab, M., Lutum, A. S. & Seufert, W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90, 683–693 (1997).
Cross, F. R., Archambault, V., Miller, M. & Klovstad, M. Testing a mathematical model for the yeast cell cycle. Mol. Biol. Cell 13, 52–70 (2002).
Dirick, L., Bohm, T. & Nasmyth, K. Roles and regulation of Cln/Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae . EMBO J. 14, 4803–4813 (1995).
Visintin, R. et al. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell 2, 709–718 (1998).
Visintin, R., Hwang, E. S. & Amon, A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature 398, 818–823 (1999).
Shou, W. et al. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97, 233–244 (1999).
Visintin, R., Stegmeier, F. & Amon, A. The role of the polo kinase Cdc5 in controlling Cdc14 localization. Mol. Biol. Cell 14, 4486–4498 (2003).
Yoshida, S. & Toh-e, A. Budding yeast Cdc5 phosphorylates Net1 and assists Cdc14 release from the nucleolus. Biochim. Biophys. Res. Commun. 294, 687–691 (2002).
Pereira, G., Manson, C., Grindlay, J. & Schiebel, E. Regulation of the Bfa1p-Bub2p complex at spindle pole bodies by the cell cycle phosphatase Cdc14p. J. Cell Biol. 157, 367–379 (2002).
Tinker-Kulberg, R. L. & Morgan, D. O. Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev. 13, 1936–1949 (1999).
Rock, J. M. & Amon, A. Cdc15 integrates Tem1 GTP-ase mediated spatical signals with Polo kinase-mediated temporal cues to activate mitotic exit. Genes Dev. 25, 1943–1954 (2012).
Lu, Y. & Cross, F. R. Mitotic exit in the absence of separase activity. Mol. Biol. Cell 20, 1576–1591 (2009).
Stegmeier, F. & Amon, A. Closing mitosis: the function of the Cdc14 phosphatase and its regulation. Annu. Rev. Genet. 38, 203–232 (2004).
Shirayama, M., Matsui, Y. & Toh-e, A. The yeast TEM1 gene, which encodes a GTP-binding protein, is involved in termination of M phase. Mol. Cell Biol. 14, 7476–7482 (1994).
Stegmeier, F., Visintin, R. & Amon, A. Separase polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell 108, 207–220 (2002).
Yamamoto, A., Guacci, V. & Koshland, D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J. Cell Biol. 133, 99–110 (1996).
Yamamoto, A., Guacci, V. & Koshland, D. Pds1p is required for faithful execution of anaphase in the yeast Saccharomyces cerevisiae . J. Cell Biol. 133, 85–97 (1996).
Wang, Y. & Burke, D. J. Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae . Mol. Cell Biol. 17, 620–626 (1997).
Lim, H. H., Goh, P. -Y. & Surana, U. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr. Biol. 8, 231–234 (1998).
Shirayama, M., Toth, A., Galova, M. & Nasmyth, K. A. P. C. (CDC20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 402, 203–207 (1999).
Thornton, B. R. & Toczyski, D. P. Securin and B-cyclin/CDK are the only essential targets of the APC. Nat. Cell Biol. 5, 1090–1094 (2003).
Cohen-Fix, O., Peters, J. -M., Kirschner, M. W. & Koshland, D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 10, 3081–3093 (1996).
Jaspersen, S. L. & Morgan, D. O. Cdc14 activates Cdc15 to promote mitotic exit in budding yeast. Curr. Biol. 10, 615–618 (2000).
Menssen, R., Neutzner, A. & Seufert, W. Asymmetric spindle pole localization of yeast Cdc15 kinase links mitotic exit and cytokinesis. Curr. Biol. 11: 345–350 (2001).
Ro, H. S., Song, S. & Lee, K. S. Bfa1 can regulate Tem1 function independently of Bub2 in the mitotic exit network of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 99, 5436–5441 (2002).
Kim, J., Jang, S. S. & Song, K. Different levels of Bfa1/Bub2 GAP activity are required to prevent mitotic exit of budding yeast depending on the type of perturbations. Mol. Biol. Cell 19: 4328–4340 (2008).
Alexandru, G., Zachariae, W., Schleiffer, A. & Nasmyth, K. Sister chromatid separation and chromosome reduplication are regulated by different mechanisms in response to spindle damage. EMBO J. 18, 2707–2721 (1999).
Hu, F. et al. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 107, 655–665 (2001).
Shou, W. & Deshaies, R. J. Multiple telophase arrest bypassed (tab) mutants alleviate the essential requirement for Cdc15 in exit from mitosis in S. cerevisiae . BMC Genet. 3, 4 (2002).
Sullivan, M. & Uhlmann, F. A non-proteolytic function of separase links the onset of anaphase to mitotic exit. Nat. Cell Biol. 5, 249–254 (2003).
Visintin, R., Prinz, S. & Amon, A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278, 460–463 (1997).
Visintin, C. et al. APC/Cdh1 mediated degradation of the polo kinase Cdc5 promotes the return of Cdc14 into the nucleolus. Genes Dev. 22, 79–90 (2008).
Charles, J. F. et al. The polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr. Biol. 8, 497–507 (1998).
Lu, Y. & Cross, F. R. Periodic cyclin-Cdk activity entrains an autonomous Cdc14 release oscillator. Cell 141, 268–279 (2010).
Laomettachit, T., Chen, K. C., Baumann, W. T. & Tyson, J. J. Modeling protein regulatory networks using standardized components with application to the START transition in the budding yeast cell cycle. In submission (2015).
Shaffer, C. A., Zwolak, J. W., Randhawa, R. & Tyson, J. J. Modeling molecular regulatory networks with JigCell and PET. Methods Mol. Biol. 500, 81–111 (2009).
Acknowledgements
This work was supported by a grant from the US National Institutes of Health, 5R01GM095955-01, to TMM and JJT. Jason Zwolak created the online simulator for our model. We are grateful to the Virtual Cell project, supported by the US National Institutes of Health, for hosting our model on their server. We thank our collaborators, Jean Peccoud, Neil Adames, Anna Ritz and Chris Poirel, for helpful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplemental Information accompanies the paper on the Systems Biology and Applications website (http://www.nature.com/npjsba)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kraikivski, P., Chen, K., Laomettachit, T. et al. From START to FINISH: computational analysis of cell cycle control in budding yeast. npj Syst Biol Appl 1, 15016 (2015). https://doi.org/10.1038/npjsba.2015.16
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/npjsba.2015.16
This article is cited by
-
A continuous-time stochastic Boolean model provides a quantitative description of the budding yeast cell cycle
Scientific Reports (2022)
-
Boolean factor graph model for biological systems: the yeast cell-cycle network
BMC Bioinformatics (2021)
-
Crosstalk between Plk1, p53, cell cycle, and G2/M DNA damage checkpoint regulation in cancer: computational modeling and analysis
npj Systems Biology and Applications (2021)
-
Towards a synthetic cell cycle
Nature Communications (2021)
-
A hybrid stochastic model of the budding yeast cell cycle
npj Systems Biology and Applications (2020)
