
Abstract
In budding yeast, synchronization of waves of mitotic cyclins that activate the Cdk1 kinase occur through Forkhead transcription factors. These molecules act as controllers of their sequential order and may account for the separation in time of incompatible processes. Here, a Forkhead-mediated design principle underlying the quantitative model of Cdk control is proposed for budding yeast. This design rationalizes timing of cell division, through progressive and coordinated cyclin/Cdk-mediated phosphorylation of Forkhead, and autonomous cyclin/Cdk oscillations. A “clock unit” incorporating this design that regulates timing of cell division is proposed for both yeast and mammals, and has a DRIVER operating the incompatible processes that is instructed by multiple CLOCKS. TIMERS determine whether the clocks are active, whereas CONTROLLERS determine how quickly the clocks shall function depending on external MODULATORS. This “clock unit” may coordinate temporal waves of cyclin/Cdk concentration/activity in the eukaryotic cell cycle making the driver operate the incompatible processes, at separate times.
Similar content being viewed by others

Introduction
Coordination of DNA replication (S-phase) and cell division (M-phase) is achieved by sequential activation of enzymatic activities that oscillate throughout the cell division cycle. These activities are realized by cyclin-dependent kinases or Cdks, formed by a catalytic (kinase) and a regulatory (cyclin) subunit. The cyclin determines the timing of Cdk activation, and a progressive activation and inactivation of a cyclin/Cdk complex is able to generate its sustained oscillations1,2.
Waves of multiple cyclin/Cdk activities raise and fall at a specific timing to guarantee cell cycle frequency, with mitotic (Clb) cyclins driving cell cycle events from S- through M-phase (Fig. 1a)3,4,5. Accumulation of cyclins occurs at definite temporal windows of transcriptional control. However, strikingly, molecular mechanisms responsible for the timely coordination of the “waves of cyclins” pattern6,7 remain elusive.

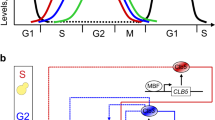
a Qualitative description of alternating waves of expression of mitotic cyclins throughout the cell cycle phases. In budding yeast, Clb indicates mitotic cyclins: Clb5,6 (red color) trigger DNA replication in S-phase; Clb3,4 (blue color) trigger completion of S-phase and early mitotic events in G2 phase; Clb1,2 (green color) trigger late mitotic events and cell division in M-phase. b Model for the transcriptional regulation of the mitotic Clb/Cdk1 complexes. A coherent type I feed-forward loop (FFL) may synchronize activation of mitotic Clb cyclins through the Fkh2 transcription factor: Clb5/Cdk1 promotes CLB3 transcription (arrow A), Clb3/Cdk1 promotes CLB2 transcription (arrow B) together with Clb5/Cdk1 (arrow C), and Clb2/Cdk1 promotes CLB2 transcription by a positive feedback loop (PFL, arrow D). For the sake of clarity, the Cdk1 subunit has been omitted. Arrows represent activating interactions among the Clb/Cdk1 complexes, whereas bar-headed black lines represent the mutual, inhibiting interactions between Clb/Cdk1 complexes and their stoichiometric inhibitor Sic1 (adapted from ref. 8). c Systems biology-driven design that rationalizes the quantitative model or “threshold model” of Cdk1 control: a progressive activation of Fkh2 is realized through a processive, multi-step phosphorylation mediated by different thresholds of Clb/Cdk1 activities determined by the accumulation of Clb cyclins (adapted from11).
We have recently demonstrated how the sequential order of waves of Clb cyclins is achieved by coupling Cdk with transcriptional activities in budding yeast8. Through mathematical modeling, we have predicted a Clb/Cdk-mediated regulation of an activator molecule that stimulates mitotic cyclin expression8. This prediction was validated experimentally, identifying the Forkhead (Fkh) transcription factor Fkh2—major activator of Clb2, which regulates the timing of cell division—as a pivotal molecule responsible for the sequential activation of mitotic CLB3 and CLB2 genes8. We discovered that Clb waves are temporally synchronized by Fkh2, and that a Clb/Cdk1-mediated regulation of Fkh2 modulates CLB expression. Thus, integrated computational and experimental analyses point to Fkh2 as a dynamical regulator of cyclin/Cdk complexes. In control engineering, elements that are designed for dynamic systems to behave in a desired manner are indicated as “controllers.” Similarly, Fkh2 is indicated here as a CONTROLLER molecule, which results in the desired synchronization of the temporal expression of mitotic Clb waves.
The findings reveal a principle of design that coordinates waves of Clb cyclins appearance, with Cdk and Fkh transcription activities being interlocked to guarantee a timely cell cycle. Intriguingly, within this design Clb3/Cdk1-centered regulations appear to drive self-sustained Clb/Cdk1 oscillations9. Through an extensive computational analysis that explores the full set of activatory and inhibitory regulations able to generate oscillations, we have recently shown that a minimal yeast cell cycle network involving Clb/Cdk1 complexes and their stoichiometric inhibitor exhibits transient and sustained oscillations in the form of limit cycles9. Specifically, we uncovered that a Clb3/Cdk1-mediated positive feedback loop (PFL) and a linear cascade of activation of mitotic Clb/Cdk1 complexes from S- through M-phase—through Clb3/Cdk1 (Clb5 → Clb3 → Clb2)—are recurrent network motifs that yield sustained, autonomous oscillations of Clb/Cdk1 waves9 that capture their sequential activation and inactivation.
Our evidence suggests that a Fkh-mediated design principle underlies Cdk control in budding yeast, specifically synchronizing the “waves of cyclins” pattern. However, at present, a molecular mechanism that rationalizes the coordinated appearance of Clb/Cdk complexes in eukaryotes is not known. Here, a “clock unit” that incorporates the Fkh-mediated design is proposed to regulate the timing of cell division in both yeast and mammals, to coordinate DNA replication and cell division through modulation of temporal waves of cyclin/Cdk activity.
The Fkh2/Clb3-centered design rationalizes the quantitative model of Cdk control in budding yeast
The mechanism uncovered for the progressive activation of the Fkh2 transcription factor, which activates—one after another—the mitotic Clb/Cdk1 complexes throughout the cell cycle, makes sense in the light of the well-known concept that unidirectional cell cycle progression is realized through the progressive increase in the Cdk activity10. This so-called “quantitative model” has been envisioned by the Nobel Prize 2001 recipient Sir Paul Nurse in 1996 and referred to as the “threshold model” by the Nobel Prize 2001 co-recipient Tim Hunt11, and has been subsequently demonstrated experimentally in fission yeast12,13, mammalian cells14, and budding yeast15. The quantitative model of Cdk control proposes that a progressive cyclin accumulation leads to an increase in the Cdk activity through different thresholds of activity that are required for a timely phosphorylation of targets10. Specifically, distinct thresholds of Cdk activity drive cell cycle progression through S-phase and M-phase, with M-phase requiring a higher threshold of cyclin level—thereby of Cdk activity—than S-phase10.
In line with the quantitative model of Cdk control, it has been shown in budding yeast that specificity of cyclins towards targets increases from G1 (Cln2) to S (Clb5) to G2 (Clb3) to M (Clb2) phase16. Specifically, a higher cyclin specificity in M-phase than in S-phase confers a higher Cdk activity in M-phase than in S-phase16,17. Moreover, inhibitory tyrosine phosphorylation of Clb/Cdk1 complexes—mediated by the Swe1 kinase—increases from S (Clb5/Cdk1) to M (Clb2/Cdk1) phase, thereby supporting their progressive activation throughout cell cycle progression18. In this scenario, the binding of Clb/Cdk1 complexes to Sic1—stoichiometric inhibitor of Clb/Cdk1 complexes19,20—protects the former from tyrosine phosphorylation, allowing accumulation of unphosphorylated kinase complexes18 that can promote DNA replication initiation dynamics at the G1/S transition upon Sic1 degradation21,22. These results are complementary to recent evidence that shed light on mechanistic details of phosphorylation events that are required to modulate targets at different thresholds of Cdk activity23,24.
Although the aforementioned studies support from different angles the concept underlying the quantitative model of Cdk control proposed by Sir Paul Nurse, a molecular mechanism that rationalizes the coordinated appearance of mitotic waves of Clb cyclins is currently not known. The molecular mechanism in place shall be able to temporally coordinate Clb waves such that these do appear one after another, at different times, and do disappear at the same time4,5.
A design principle may be proposed, which provides a mechanistic basis underlying the quantitative model of Cdk control for the budding yeast. The design explains the progressive cyclin accumulation from S- to M-phase, which leads to increased thresholds in the Cdk activity, through a coherent type I feed-forward loop (FFL) that incorporates the linear cascade (Clb5 → Clb3 → Clb2) aided by PFLs8 and the mutual inhibition of all Clb/Cdk1 complexes with Sic1 that we discovered25 (Fig. 1b). This design rationalizes the occurrence of staggered waves of Cdk1 activity and the progressive activation of Clb5, Clb3, and Clb2 mitotic cyclins—which are observed throughout a cell cycle round. Specifically, an increase in the extent of Fkh2 phosphorylation from S- to M-phase, mediated by the progressive accumulation first of Clb5/Cdk1, then of Clb3/Cdk1, and ultimately of Clb2/Cdk1 kinase activities, ensures the timely occurrence of Clb waves (Fig. 1c). An involvement of two different phosphorylation patterns mediated by various Clb/Cdk1 activities may be envisioned for the Fkh2-mediated transcription of CLB genes: (i) Clb5/Cdk1-mediated specific phosphorylation events on Fkh2 for CLB3 transcription, which may be reinforced by the Clb3/Cdk1-mediated PFL on CLB3 gene; and (ii) Clb3/Cdk1- and Clb5/Cdk1-mediated specific phosphorylation events on Fkh2 for CLB2 transcription, which is reinforced by the Clb2/Cdk1-mediated PFL on CLB2 gene.
The details of this sequential phosphorylation have been not yet elucidated and are currently under investigation in our laboratory. However, this hypothesis is supported by evidence from us and others that the Fkh2 phosphosites S683 and T697 are recognized by all Clb/Cdk1 kinase activities, and that their deletion leads to a reduction of Fkh2 phosphorylation8,26.
Many Cdk1 targets contain clusters of multiple phosphorylation sites27 and multisite phosphorylation of targets by cyclin/Cdk1 activities has been proposed to transform a graded protein kinase signal into an ultrasensitive switch-like response. Therefore, it can be speculated that dynamics and sequence of individual Clb/Cdk1-dependent phosphorylation events differ within the multisite phosphorylation patterns activating Fkh2, and that a potential cooperativity of the individual phosphorylation events is realized by a different specificity (binding affinity) of Clb5/Cdk1, Clb3/Cdk1, and Clb2/Cdk1 complexes to Fkh2. Thus, a mechanism of cooperativity among Clb/Cdk1-dependent phosphorylation events may promote the progressive activation of Fkh2 from S- to M-phase, to drive waves of CLB expression, thereby of Clb/Cdk1 waves of activity for a timely cell division, and ensure robust cell cycle oscillations.
The cooperativity that can be envisioned among Clb/Cdk1-dependent phosphorylation events on Fkh2 finds a parallel with studies that provided insights into the multisite phosphorylation mechanism that degrades Sic128,29,30,31. We have shown that, similarly to Fkh2, Sic1 interacts with all Clb cyclins32 and parallel studies have shown that a switch-like Sic1 destruction is dependent on a complex process in which both Cln2/Cdk1 (G1 phase) and Clb5/Cdk1 (S-phase) activities act in processive multi-phosphorylation steps29. Multisite phosphorylation patterns can act as timing signature that modulates substrate activity at different cyclin/Cdk1 thresholds23 and Fkh2 may be regulated by similar cooperative phosphorylation patterns.
Clb/Cdk1-mediated phosphorylation patterns on Fkh transcription factors may control a timely gene expression through diverse mechanisms: (i) regulation of transcriptional elongation and termination33, (ii) regulation of a repressive chromatin structure in the coding region of CLB2 together with chromatin-remodeling ATPases34, (iii) regulation of Sir2-4 silencing proteins35, and/or (iv) regulation of metabolic genes that are crucial for cell growth and division36. It is apparent that Fkh are hubs that have the ability to control gene expression by connecting intracellular pathways that operate at different but specific times. However, the coordination of these mechanisms with the staggering waves of Clb cyclins is currently unexplored.
Finally, in addition to the cyclin/Cdk1-mediated (cooperative) phosphorylation of targets, further mechanisms may be involved in the quantitative model of Cdk control to modulate the timing of target’s phosphorylation. In budding yeast, the protein phosphatase Cdc14 has been proposed to be involved in this process by imposing Cdk thresholds through antagonization of Clb2/Cdk1-mediated phosphorylation, thus contributing to the correct order of cell cycle events37,38,39,40. Further investigations are required to disentangle the delicate balance between Clb/Cdk1 and phosphatases in the quantitative model of Cdk control.
A Forkhead CONTROLLER-based “clock unit” for cell cycle timing in yeast
Where the common view of cell division is that of a single cycle, a more sophisticated design may be recognized in it of multiple overlapping “oscillators” within a cycle. These oscillators are quasi-independent molecular-network “clocks” that, independently, contribute to the timing and optimal function of the cycle as a whole. Each oscillator emerges as a time-wave in the concentration of one out of a group of the regulatory cyclins.
Each CLOCK, i.e., each of the cyclins, determines the times at which molecular activities are (in)activated. It does this by binding to the catalytic Cdk, the actual DRIVER. The driver, Cdk, controls the cell cycle but not its temporal dynamics, which are instead controlled by the cyclins, the clocks. This organization repeats itself for each cell cycle phase: different cyclins progressively bind to Cdk as defined by successive waves of cyclins. The resulting cyclin/Cdk complexes define the timing of the cell cycle phase(s) in a unidirectional and irreversible manner; here, the concept of multiple overlapping “oscillators” within a cell cycle connects with the existing understanding of cell cycle regulation.
In budding yeast, there are nine distinct cyclins grouped in four subgroups. These subgroups, together, clock four phases of the cell cycle (these phases do not correspond precisely to the classically recognized G1, S, G2, and M, although this is often presented as a simplification). Here, the focus is on three clocks, i.e., cyclins Clb5,6 (CLOCK1), Clb3,4 (CLOCK2), and Clb1,2 (CLOCK3), their oscillations being responsible for the alternation of the incompatible processes of DNA replication, chromosome segregation, and cell division from S- through M-phase, respectively. Although the number of clocks should equal the number of functional phases, some cell cycle phases could be regulated by more than one clock and some cyclins may begin to regulate long before the beginning or ending of the phase they trigger. Of note, G1 cyclins are not part of the clocks, because the quantitative model for Cdk control has been proposed to describe the Cdk requirement for S-phase and mitosis10,11.
The investigations conducted in the last 20 years in our laboratory enable to propose candidate molecules that form and regulate the “clock unit” underlying the quantitative model of Cdk control through waves of Clb activities, i.e., of the clocks, and therewith the temporal coordination of the Clb/Cdk1 complexes: CLOCKS (Clb cyclins), DRIVER (Cdk1 kinase), TIMER (Sic1 inhibitor), CONTROLLER (Fkh2 transcription factor), and MODULATOR (Sir2 histone deacetylase) (Fig. 2a). One is (i) a TIMER of the activity of the clock/driver (Clb/Cdk1) complex: this is the inhibitor of the Clb/Cdk1 activity through cyclin-mediated recruitment of the Clb/Cdk1 inhibitor (Cki) Sic119,20. The other two molecular mechanisms control the concentration waves of Clb cyclins: (ii) a CONTROLLER of transcription of each CLB gene promoted by the previous Clb/Cdk1 complex: this is the Fkh transcription factor8 and (iii) MODULATOR(S) of the activity of the Fkh transcription factor through inhibition by chromatin (epigenetic) factor(s): these are the histone deacetylases such as Sir241 and Sin3/Rpd342.

a A “clock unit” of the cell cycle is formed by (i) a DRIVER (Cdk1 kinase) that, together with the CLOCK (cyclin: Clb cyclin; red color), drives cell cycle events through various phases; (ii) a TIMER (Cki, cyclin-dependent kinase inhibitor: Sic1; blue color) that inhibits the DRIVER; (iii) a CONTROLLER (TF, transcription factor: Fkh2) that activates the CLOCK (cyclin: CLB gene); and (iv) a MODULATOR (histone deacetylase: Sir2) that modulates the activity of the CONTROLLER. b “Clock unit” that integrates CLOCKS 1–3 (Clb cyclins; red color), CONTROLLER (Fkh2 transcription factor), MODULATOR (Sir2 histone deacetylase), CLOCK4 (TF TIMER, Ace2–Swi5 transcription factors; blue color), and TIMER (Cki, Clb/Cdk1 kinase inhibitor Sic1; blue color) together with the known regulations occurring among them.
Of note, the Anaphase-Promoting Complex (APC)-mediated mechanism of degradation of each cyclin within a Clb/Cdk1 complex—promoted by the subsequent Clb/Cdk1 complex (see refs. 25,43 and references therein)—is not explicitly considered in the scheme of Fig. 2a. We and others have shown, computationally and experimentally, that this mechanism is less relevant for oscillations of the Clb/Cdk1 activity25,44,45,46, despite its relevance to modulate Cdk1 activity through abolishment of Clb levels before the start of a new cell cycle. In fact, Sic1-mediated feed-forward regulations are required to maintain an oscillation-like behavior of Clb/Cdk1 activities and to prevent mitotic cyclin synthesis25,44. Thus, we did propose early that Sic1, rather than Clb degradation, acts as a TIMER of the temporal waves of mitotic Clb cyclins25.
A design principle that can be therefore proposed has the DRIVER (Cdk1 kinase) operating the incompatible processes that is instructed by multiple CLOCKS (Clb cyclins). A TIMER (Sic1 inhibitor) determines whether the clocks are active, whereas a CONTROLLER (Fkh2 transcription factor) determines how quickly the clocks proceed depending on the external signal(s) or MODULATOR (Sir2 histone deacetylase) (Fig. 2b). This “clock unit” may interlock, i.e., coordinate together, the three clocks—cyclins Clb5,6 (CLOCK1), Clb3,4 (CLOCK2), and Clb1,2 (CLOCK3) (Fig. 3). Within this scenario, an additional clock may be envisioned with the DRIVER (Cdk1 kinase) and TIMER (Sic1 inhibitor) being interlocked in a fine balance between mutual activation and inhibition. A design principle proposed for this clock (CLOCK4) has the DRIVER (Cdk1) that activates the CONTROLLER (Fkh2), which in turn regulates the transcription of CLOCK4 (the ACE2 and SWI5 transcription factors) (Fig. 2b)36,47,48,49,50,51) that is responsible for the expression of the TIMER (Sic1)52,53,54. Beside the documented mutual inhibition of DRIVER (Cdk1) and TIMER (Sic1) at the protein level, the DRIVER (Cdk1) inhibits CLOCK4 (Ace2–Swi5)55, determining how quickly the timer is inactive and thereby whether the driver is active (Figs. 2b and 3).

Interaction scheme of the “clock units” Clb5,6 (CLOCK1), Clb3,4 (CLOCK2), and Clb1,2 (CLOCK3), which interlock one other based on the regulation core in Fig. 2a. In addition, CLOCK4 (TF TIMER, Ace2–Swi5 transcription factors) (i) is activated by the CONTROLLER, (ii) activates the TIMER (Cki), and (iii) is inhibited by the DRIVER. Red arrows and bar-headed red lines indicate CLOCKS 1–3-mediated reactions, whereas blue arrows and bar-headed blue lines indicate CLOCK4-mediated reactions. Blue crosses indicate inhibition of CLOCKS 1–3 by CLOCK4.
The three mechanisms act as timers that switch each clock ON and OFF, and determine how fast each clock is progressing. The clocks are among each other’s timers. Intriguingly, this coordination is such that waves of cyclin/Cdk activity occur sequentially, at different times, throughout the various cell cycle phases but disappear at the same time at cell division, as observed experimentally4. Although the processes around some cyclins individually have been investigated, neither the transcriptional mechanism inter-connecting all cyclin subgroups nor how timing of the cyclin waves is managed by the eukaryotic cell are understood. This is remarkable, being this timing of obvious importance to coordinate incompatible cell cycle phases. It is therefore critical to elucidate how the coordination among CLOCKS, DRIVER, TIMER, CONTROLLER, and MODULATOR is achieved such that waves of cyclin/Cdk activity can coordinate sequentially, possibly through the progressive, increasing Clb/Cdk1-mediated phosphorylation of the CONTROLLER (Fkh transcription factor). For this coordination to occur, it is critical to determine the molecular mechanisms that control—together—the timing of the cyclin waves and how the design(s) use any of three Clb clocks to prevent overlap of incompatible processes, thus guaranteeing a timely cell cycle.
By answering the questions above, why and how the Clb/Cdk activities are switched OFF simultaneously at cell division, and not progressively with the same temporal organization as that of their activation, will be elucidated. Switching OFF simultaneously the Clb/Cdk activities suggests that the three Clb clocks shall overlap; if they would not do so, the clocks would be independent and would switch OFF progressively, one after another, with a different temporality.
Therefore, a systems biology strategy of integrating appropriate computational modeling with a quantitative experimental investigation is the key to identify regulatory designs employed to control the timing of cellular proliferation.
A Forkhead CONTROLLER-based “clock unit” for cell cycle timing in mammals
A systems biology approach that integrates predictive modeling and dedicated biological experiments has proven to be pivotal, to uncover molecular mechanisms that address cellular timing, i.e., underlying the temporal regulation of mitotic (Clb) cyclins, and reveals a principle of design of cellular reproduction that may be conserved in eukaryotic organisms including humans. This design relies on cyclin/Cdk and transcription activities being interlocked to guarantee a timely completion of the cell cycle.
In humans, the question of how the temporal coordination of DNA replication and cell division occurs to prevent their overlap is unanswered. The understanding of the molecular mechanisms underlying this coordination would help to prevent an uncontrolled, enhanced cell division, which is a typical feature of human diseases such as cancer. It is thought that multiple Cdk and cyclins control the timing of this coordination by ensuring alternation with a definite temporal delay. However, the molecules involved in this process have not been pointed out yet.
One of these molecules may be p27Kip1, belonging to the Kip/Cip family of cyclin/Cdk inhibitors56,57, which binds to Cyclin E/Cdk2 (G1 phase) and to Cyclin A/Cdk2 (S-phase) that control the timing of the S-phase onset58. p27Kip1 is often mutated in human cancers59. On the one hand, we have shown earlier—for the first time—the structural and functional homology of the yeast Clb/Cdk1 inhibitor Sic1 to the mammalian p27Kip1 20. Similarly to the bimodal mechanism in place for the binding of Sic1 to Clb5/Cdk120,60, it was shown that p27Kip1 binds to Cyclin A/Cdk2 and blocks its activity through a mechanism where the inhibitor is first recognized by a hydrophobic pocket on the cyclin subunit and, subsequently, it extends on the Cdk subunit to reach and block the Cdk catalytic pocket61,62.
On the other hand, through computer modeling, we have provided a rationale for the role of p27Kip1 to set the timing of DNA replication dynamics at the S-phase onset63, following the same mechanism that we did propose early for Sic121,22. Both yeast and mammalian studies have been inspired by experimental findings showing that the Cdk inhibitors are responsible for the activation of cyclin/Cdk complexes64,65, possibly translocating them from the cytoplasm to the nucleus where they exploit their function.
It is also in the nucleus where another molecule or, more precisely, a class of molecules exploit their functions. These are the Fkh, highly conserved transcription factors in eukaryotes, from yeast to human, with roles in physiological processes and diseases. Human Fkh molecules have been intensively studied due to their crucial function in cellular processes such as cell cycle regulation66,67, genome replication and stability68, aging and oxidative stress69,70, metabolism71,72, cancer73,74,75,76, and neurodegeneration77. The human Fkh family comprises 18 subfamilies78,79, with two of them named Forkhead box O (FoxO) and M (FoxM) being the closest functional counterpart of the budding yeast Fkh1 and Fkh2.
Although the molecular mechanism(s) that synchronizes cyclin/Cdk complexes with Fox proteins is at present unknown, the FFL + PFLs motif that it is proposed here to control the waves of mitotic Clb/Cdk1 activities in budding yeast may be transposed to the mammalian system. Intriguingly, in the latter, a number of available experimental evidence has not been connected yet in a systems view. These data can be considered—together—to speculate that the yeast “clock unit” (Fig. 4a, black color) may hold true also in mammalian cells (Fig. 4b, black color). In Table 1, the regulatory interactions involved in the “clock unit” are reported for the two organisms. Of note, the homologous of the budding yeast Ndd1 is lacking in mammalian cells.

a, b Comparison between the integrated “clock units” in budding yeast (a) and in mammalian cells (b). Homologous molecules and regulations are indicated in black color; regulations that are currently known in one organism but not in the other, and vice versa, are indicated in red color; additional regulations that occur in mammalian cells are indicated in dotted gray color (see text for details). For simplicity, the DRIVER (Cdk) has been omitted.
Currently, unexplored regulatory interactions in budding yeast are recognized that may provide an additional level of control among the molecules forming the “clock unit” in mammalian cells (Figs. 4a and 4c, red color and Table 2). Furthermore, molecular mechanisms may act on top of the “clock unit” (Fig. 4b, dotted gray color and Table 2), to confer robustness to the mammalian complex system.
Altogether, the evidence presented here suggests that a “clock unit” regulating timing of cell division may be conserved from yeast to mammalian cells. The intricacy within the network of regulations among multiple Fox proteins, Sirtuin, mitotic cyclin/Cdk complexes, and their stoichiometric inhibitor reflects possible mechanisms through which timing and robustness of cell division is ensured for the more sophisticated living organisms.
Outlook
Deregulation of cell cycle timing, which speeds up or slows down the frequency of cyclin/Cdk oscillations, may result in disease development such as when mis-regulation of c-Myc and cyclin levels occur80,81. Because of the emergent role as hubs connecting intracellular pathways to control gene expression, Fkh may function as a building block that integrates regulatory modules to realize cell physiology. Within this scenario, molecular routes by which some cells (i) escape proper timing, (ii) alter dynamics of cell proliferation, and (iii) compromise viability potentially resulting in cellular dysfunctions and disease development in humans may be suggested to counter such escapes.
In humans, FoxM1 and FoxP transcription factors are the closest homologs of the yeast Fkh1 and Fkh267,82, and, of note, FoxM1 regulates the expression of the mitotic Cyclin B83 similarly to the mechanism through which Fkh2 regulates Clb2 expression26. The awareness of emerging roles of FoxM1 and FoxO transcription factors as prognostic and predictive markers for the diagnosis and precise screening of cancer patients84,85 suggests that a multi-scale, systems biology-driven understanding of the complex regulation between cyclin/Cdk activities and Fkh-centered transcriptional network may reveal new molecular mechanisms through which these factors act in the context of human physiology and its deregulation.
Data availability
Data sharing not applicable to this article, as no datasets were generated or analyzed during the current study.
References
Goldbeter, A. A minimal cascade model for the mitotic oscillator involving cyclin and cdc2 kinase. Proc. Natl Acad. Sci. USA 88, 9107–9111 (1991).
Tyson, J. J. Modeling the cell division cycle: cdc2 and cyclin interactions. Proc. Natl Acad. Sci. USA 88, 7328–7332 (1991).
Nasmyth, K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr. Opin. Cell Biol. 5, 166–179 (1993).
Nasmyth, K. At the heart of the budding yeast cell cycle. Trends Genet. 12, 405–412 (1996).
Futcher, B. Cyclins and the wiring of the yeast cell cycle. Yeast 12, 1635–1646 (1996).
Fitch, I. et al. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell 3, 805–818 (1992).
Richardson, H., Lew, D. J., Henze, M., Sugimoto, K. & Reed, S. I. Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes Dev. 6, 2021–2034 (1992).
Linke, C. et al. A Clb/Cdk1-mediated regulation of Fkh2 synchronizes CLB expression in the budding yeast cell cycle. NPJ Syst. Biol. Appl. 3, 7 (2017).
Mondeel, T. D. G. A., Ivanov, O., Westerhoff, H. V., Liebermeister, W. & Barberis, M. Clb3-centered regulations are recurrent across distinct parameter regions in minimal autonomous cell cycle oscillator designs. NPJ Syst. Biol. Appl. 6, 8 (2020).
Stern, B. & Nurse, P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 12, 345–350 (1996).
Hochegger, H., Takeda, S. & Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat. Rev. Mol. Cell Biol. 9, 910–916 (2008).
Coudreuse, D. & Nurse, P. Driving the cell cycle with a minimal CDK control network. Nature 468, 1074–1079 (2010).
Swaffer, M. P., Jones, A. W., Flynn, H. R., Snijders, A. P. & Nurse, P. CDK substrate phosphorylation and ordering the cell cycle. Cell 167, 1750–1761 (2016).
Gavet, O. & Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533–543 (2010).
Oikonomou, C. & Cross, F. R. Rising cyclin-CDK levels order cell cycle events. PLoS ONE 6, e20788 (2011).
Kõivomägi, M. et al. Dynamics of Cdk1 substrate specificity during the cell cycle. Mol. Cell 42, 610–623 (2011).
Loog, M. & Morgan, D. O. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature 434, 104–108 (2005).
Keaton, M. A. et al. Differential susceptibility of yeast S and M phase CDK complexes to inhibitory tyrosine phosphorylation. Curr. Biol. 17, 1181–1189 (2007).
Schwob, E., Bohm, T., Mendenhall, M. D. & Nasmyth, K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79, 233–244 (1994).
Barberis, M. et al. The yeast cyclin-dependent kinase inhibitor Sic1 and mammalian p27Kip1 are functional homologues with a structurally conserved inhibitory domain. Biochem. J. 387, 639–647 (2005).
Barberis, M., Klipp, E., Vanoni, M. & Alberghina, L. Cell size at S phase initiation: an emergent property of the G1/S network. PLoS Comput. Biol. 3, e64 (2007).
Barberis, M. & Klipp, E. Insights into the network controlling the G1/S transition in budding yeast. Genome Inf. 18, 85–99 (2007).
Örd, M. et al. Multisite phosphorylation code of CDK. Nat. Struct. Mol. Biol. 26, 649–658 (2019).
Örd, M., Venta, R., Möll, K., Valk, E. & Loog, M. Cyclin-specific docking mechanisms reveal the complexity of M-CDK function in the cell cycle. Mol. Cell 75, 76–89 (2019).
Barberis, M. et al. Sic1 plays a role in timing and oscillatory behaviour of B-type cyclins. Biotechnol. Adv. 30, 108–130 (2012).
Pic-Taylor, A., Darieva, Z., Morgan, B. A. & Sharrocks, A. D. Regulation of cell cycle-specific gene expression through cyclin-dependent kinase-mediated phosphorylation of the forkhead transcription factor Fkh2p. Mol. Cell. Biol. 24, 10036–10046 (2004).
Holt, L. J. et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 (2009).
Nash, P. et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414, 514–521 (2001).
Kõivomägi, M. et al. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 480, 128–131 (2011).
Yang, X., Lau, K. Y., Sevim, V. & Tang, C. Design principles of the yeast G1/S switch. PLoS Biol. 11, e1001673 (2013).
Venta, R. et al. A processive phosphorylation circuit with multiple kinase inputs and mutually diversional routes controls G1/S decision. Nat. Commun. 11, 1836 (2020).
Schreiber, G. et al. Unraveling interactions of cell cycle-regulating proteins Sic1 and B-type cyclins in living yeast cells: a FLIM-FRET approach. FASEB J. 26, 546–554 (2012).
Morillon, A., O’Sullivan, J., Azad, A., Proudfoot, N. & Mellor, J. Regulation of elongating RNA polymerase II by forkhead transcription factors in yeast. Science 300, 492–495 (2003).
Sherriff, J. A., Kent, N. A. & Mellor, J. The Isw2 chromatin-remodeling ATPase cooperates with the Fkh2 transcription factor to repress transcription of the B-type cyclin gene CLB2. Mol. Cell. Biol. 27, 2848–2860 (2007).
Casey, L., Patterson, E. E., Müller, U. & Fox, C. A. Conversion of a replication origin to a silencer through a pathway shared by a Forkhead transcription factor and an S phase cyclin. Mol. Biol. Cell. 19, 608–622 (2008).
Mondeel, T. D. G. A., Holland, P., Nielsen, J. & Barberis, M. ChIP-exo analysis highlights Fkh1 and Fkh2 transcription factors as hubs that integrate multi-scale networks in budding yeast. Nucleic Acids Res. 47, 7825–7841 (2019).
Drapkin, B. J., Lu, Y., Procko, A. L., Timney, B. L. & Cross, F. R. Analysis of the mitotic exit control system using locked levels of stable mitotic cyclin. Mol. Syst. Biol. 5, 328 (2009).
Bouchoux, C. & Uhlmann, F. A quantitative model for ordered Cdk substrate dephosphorylation during mitotic exit. Cell 147, 803–814 (2011).
Uhlmann, F., Bouchoux, C. & López-Avilés, S. A quantitative model for cyclin-dependent kinase control of the cell cycle: revisited. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3572–3583 (2011).
Godfrey, M. et al. PP2ACdc55 phosphatase pmposes ordered cell-cycle phosphorylation by opposing threonine phosphorylation. Mol. Cell 65, 393–402 (2017).
Linke, C., Klipp, E., Lehrach, H., Barberis, M. & Krobitsch, S. Fkh1 and Fkh2 associate with Sir2 to control CLB2 transcription under normal and oxidative stress conditions. Front. Physiol. 4, 173 (2013).
Veis, J., Klug, H., Koranda, M. & Ammerer, G. Activation of the G2/M-specific gene CLB2 requires multiple cell cycle signals. Mol. Cell. Biol. 27, 8364–8373 (2007).
Yeong, F. M., Lim, H. H., Wang, Y. & Surana, U. Early expressed Clb proteins allow accumulation of mitotic cyclin by inactivating proteolytic machinery during S phase. Mol. Cell. Biol. 21, 5071–5081 (2001).
Thornton, B. R. & Toczyski, D. P. Securin and B-cyclin/CDK are the only essential targets of the APC. Nat. Cell Biol. 5, 1090–1094 (2003).
Thornton, B. R., Chen, K. C., Cross, F. R., Tyson, J. J. & Toczyski, D. P. Cycling without the cyclosome: modeling a yeast strain lacking the APC. Cell Cycle 3, 629–633 (2004).
López-Avilés, S., Kapuy, O., Novák, B. & Uhlmann, F. Irreversibility of mitotic exit is the consequence of systems-level feedback. Nature 459, 592–595 (2009).
Pic, A. et al. The forkhead protein Fkh2 is a component of the yeast cell cycle transcription factor SFF. EMBO J. 19, 3750–3761 (2000).
Kumar, R., Reynolds, D. M., Shevchenko, A., Goldstone, S. D. & Dalton, S. Forkhead transcription factors, Fkh1p and Fkh2p, collaborate with Mcm1p to control transcription required for M-phase. Curr. Biol. 10, 896–906 (2000).
Zhu, G. et al. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 406, 90–94 (2000).
Ostrow, A. Z. et al. Fkh1 and Fkh2 bind multiple chromosomal elements in the S. cerevisiae genome with distinct specificities and cell cycle dynamics. PLoS ONE 9, e87647 (2014).
Venters, B. J. et al. A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces. Mol. Cell 41, 480–492 (2011).
Knapp, D., Bhoite, L., Stillman, D. J. & Nasmyth, K. The transcription factor Swi5 regulates expression of the cyclin kinase inhibitor p40SIC1. Mol. Cell. Biol. 16, 5701–5707 (1996).
Toyn, J. H., Johnson, A. L., Donovan, J. D., Toone, W. M. & Johnston, L. H. The Swi5 transcription factor of Saccharomyces cerevisiae has a role in exit from mitosis through induction of the cdk-inhibitor Sic1 in telophase. Genetics 145, 85–96 (1997).
McBride, H. J., Yu, Y. & Stillman, D. J. Distinct regions of the Swi5 and Ace2 transcription factors are required for specific gene activation. J. Biol. Chem. 274, 21029–21036 (1999).
Kishi, T., Ikeda, A., Koyama, N., Fukada, J. & Nagao, R. A refined two-hybrid system reveals that SCF(Cdc4)-dependent degradation of Swi5 contributes to the regulatory mechanism of S-phase entry. Proc. Natl Acad. Sci. USA 105, 14497–14502 (2008).
Polyak, K. et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78, 59–66 (1994).
Toyoshima, H. & Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78, 67–74 (1994).
Barr, A. R., Heldt, F. S., Zhang, T., Bakal, C. & Novák, B. A dynamical framework for the all-or-none G1/S transition. Cell Syst. 2, 27–37 (2016).
Weinberg, R. A. in The Biology of Cancer (ed. Weinberg, R. A.) 2nd edn, p. 320 (Garland Science, Taylor & Francis Group, 2014).
Barberis, M. Sic1 as a timer of Clb cyclin waves in the yeast cell cycle–design principle of not just an inhibitor. FEBS J. 279, 3386–3410 (2012).
Russo, A. A., Jeffrey, P. D., Patten, A. K., Massagué, J. & Pavletich, N. P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 382, 325–331 (1996).
Lacy, E. R. et al. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 11, 358–364 (2004).
Alfieri, R. et al. Towards a systems biology approach to mammalian cell cycle: modeling the entrance into S phase of quiescent fibroblasts after serum stimulation. BMC Bioinformatics 10, S16 (2009).
Cheng, M. et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 18, 1571–1583 (1999).
Rossi, R. L., Zinzalla, V., Mastriani, A., Vanoni, M. & Alberghina, L. Subcellular localization of the cyclin dependent kinase inhibitor Sic1 is modulated by the carbon source in budding yeast. Cell Cycle 4, 1798–1807 (2005).
Huang, H. & Tindall, D. J. Dynamic FoxO transcription factors. J. Cell Sci. 120, 2479–2487 (2007).
Murakami, H., Aiba, H., Nakanishi, M. & Murakami-Tonami, Y. Regulation of yeast forkhead transcription factors and FoxM1 by cyclin-dependent and polo-like kinases. Cell Cycle 9, 3233–3242 (2010).
Jin, Y., Liang, Z. & Lou, H. The emerging roles of Fox family transcription factors in chromosome replication, organization, and genome stability. Cells 9, 258 (2020).
Tia, N. et al. Role of Forkhead Box O (FOXO) transcription factor in aging and diseases. Gene 648, 97–105 (2018).
Jiang, Y., Yan, F., Feng, Z., Lazarovici, P. & Zheng, W. Signaling network of Forkhead family of transcription factors (FOXO) in dietary restriction. Cells 9, 100 (2019).
Unterman, T. G. Regulation of hepatic glucose metabolism by FoxO proteins, an integrated approach. Curr. Top. Dev. Biol. 127, 119–147 (2018).
Kodani, N. & Nakae, J. Tissue-specific metabolic regulation of FOXO-binding protein: FOXO does not act alone. Cells 9, 702 (2020).
Yadav, R. K., Chauhan, A. S., Zhuang, L. & Gan, B. FoxO transcription factors in cancer metabolism. Semin. Cancer Biol. 50, 65–76 (2018).
Ma, J., Matkar, S., He, X. & Hua, X. FOXO family in regulating cancer and metabolism. Semin. Cancer Biol. 50, 32–41 (2018).
Liao, G. B. et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 16, 57 (2018).
Laissue, P. The forkhead-box family of transcription factors: key molecular players in colorectal cancer pathogenesis. Mol. Cancer 18, 5 (2019).
Hu, W. et al. Roles of forkhead box O (FoxO) transcription factors in neurodegenerative diseases: a panoramic view. Prog. Neurobiol. 181, 101645 (2019).
Tuteja, G. & Kaestner, K. H. SnapShot: Forkhead transcription factors I. Cell 130, 1160 (2007).
Tuteja, G. & Kaestner, K. H. SnapShot: Forkhead transcription factors II. Cell 131, 192 (2007).
Rangarajan, N., Fox, Z., Singh, A., Kulkarni, P. & Rangarajan, G. Disorder, oscillatory dynamics and state switching: the role of c-Myc. J. Theor. Biol. 386, 105–114 (2015).
Moore, J. D. In the wrong place at the wrong time: does cyclin mislocalization drive oncogenic transformation? Nat. Rev. Cancer 13, 201–208 (2013).
Ostrow, A. Z. et al. Conserved forkhead dimerization motif controls DNA replication timing and spatial organization of chromosomes in S. cerevisiae. Proc. Natl Acad. Sci. USA 114, E2411–E2419 (2017).
Laoukili, J. et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 7, 126–136 (2005).
Nandi, D., Cheema, P. S., Jaiswal, N. & Nag, A. FoxM1: repurposing an oncogene as a biomarker. Semin. Cancer Biol. 52, 74–84 (2018).
Yao, S., Fan, L. Y. & Lam, E. W. The FOXO3-FOXM1 axis: a key cancer drug target and a modulator of cancer drug resistance. Semin. Cancer Biol. 50, 77–89 (2018).
Ghaemmaghami, S. et al. Global analysis of protein expression in yeast. Nature 425, 737–741 (2003).
Bailly, E., Cabantous, S., Sondaz, D., Bernadac, A. & Simon, M. N. Differential cellular localization among mitotic cyclins from Saccharomyces cerevisiae: a new role for the axial budding protein Bud3 in targeting Clb2 to the mother-bud neck. J. Cell Sci. 116, 4119–4230 (2003).
Pines, J. & Hunter, T. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 13, 3772–3781 (1994).
Lüscher-Firzlaff, J. M., Lilischkis, R. & Lüscher, B. Regulation of the transcription factor FOXM1c by Cyclin E/CDK2. FEBS Lett. 580, 1716–1722 (2006).
Wierstra, I. & Alves, J. FOXM1c is activated by cyclin E/Cdk2, cyclin A/Cdk2, and cyclin A/Cdk1, but repressed by GSK-3alpha. Biochem. Biophys. Res. Commun. 348, 99–108 (2006).
Laoukili, J. et al. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell. Biol. 28, 3076–3087 (2008).
Ma, R. Y. et al. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J. Cell Sci. 118, 795–806 (2005).
Obaya, A. J. & Sedivy, J. M. Regulation of cyclin-Cdk activity in mammalian cells. Cell. Mol. Life Sci. 59, 126–142 (2002).
Amon, A., Tyers, M., Futcher, B. & Nasmyth, K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell 74, 993–1007 (1993).
Major, M. L., Lepe, R. & Costa, R. H. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 24, 2649–2661 (2004).
Park, T. J. et al. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology 47, 1533–1543 (2008).
Sherr, C. J. & Roberts, J. M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512 (1999).
Lv, C. et al. Acetylation of FOXM1 is essential for its transactivation and tumor growth stimulation. Oncotarget 7, 60366–60382 (2016).
Sasaki, T. et al. Phosphorylation regulates SIRT1 function. PLoS ONE 3, e4020 (2008).
Sasaki, T., Maier, B., Bartke, A. & Scrable, H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell 5, 413–422 (2006).
Deota, S. et al. Allosteric regulation of Cyclin-B binding by the charge state of catalytic lysine in CDK1 is essential for cell-cycle progression. J. Mol. Biol. 431, 2127–2142 (2019).
Pinton, G. et al. SIRT1 at the crossroads of AKT1 and ERβ in malignant pleural mesothelioma cells. Oncotarget 7, 14366–14379 (2016).
Zhu, G. Y., Shi, B. Z. & Li, Y. FoxM1 regulates Sirt1 expression in glioma cells. Eur. Rev. Med. Pharmacol. Sci. 18, 205–211 (2014).
Zeng, J. et al. FoxM1 is up-regulated in gastric cancer and its inhibition leads to cellular senescence, partially dependent on p27 kip1. J. Pathol. 218, 419–427 (2009).
Huang, H., Regan, K. M., Lou, Z., Chen, J. & Tindall, D. J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 314, 294–297 (2006).
Yang, Y., Hou, H., Haller, E. M., Nicosia, S. V. & Bai, W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 24, 1021–1032 (2005).
Frescas, D., Valenti, L. & Accili, D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 280, 20589–20595 (2005).
Motta, M. C. et al. Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551–563 (2004).
Brunet, A. et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 (2004).
van der Horst, A. et al. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 279, 28873–28879 (2004).
McGovern, U. B. et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol. Cancer Ther. 8, 582–591 (2009).
Yung, M. M., Chan, D. W., Liu, V. W., Yao, K. M. & Ngan, H. Y. Activation of AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer 13, 327 (2013).
Gomes, A. R., Zhao, F. & Lam, E. W. Role and regulation of the forkhead transcription factors FOXO3a and FOXM1 in carcinogenesis and drug resistance. Chin. J. Cancer 32, 365–370 (2013).
Stahl, M. et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J. Immunol. 168, 5024–5031 (2002).
Medema, R. H., Kops, G. J., Bos, J. L. & Burgering, B. M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787 (2000).
Acknowledgements
M.B. thanks Hans Westerhoff for stimulating discussions about the yeast model system and Paul Verbruggen for help with the final layout of the figures. This work was supported by the Systems Biology Grant of the University of Surrey. The corresponding author can also be contacted at matteo@barberislab.com.
Author information
Authors and Affiliations
Contributions
M.B. conceived and formulated the ideas and hypotheses, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barberis, M. Quantitative model of eukaryotic Cdk control through the Forkhead CONTROLLER. npj Syst Biol Appl 7, 28 (2021). https://doi.org/10.1038/s41540-021-00187-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-021-00187-5
This article is cited by
-
Cyclin/Forkhead-mediated coordination of cyclin waves: an autonomous oscillator rationalizing the quantitative model of Cdk control for budding yeast
npj Systems Biology and Applications (2021)
