
Abstract
Although bacteria within the Verrucomicrobia phylum are pervasive in soils around the world, they are under-represented in both isolate collections and genomic databases. Here, we describe a single verrucomicrobial group within the class Spartobacteria that is not closely related to any previously described taxa. We examined more than 1,000 soils and found this spartobacterial phylotype to be ubiquitous and consistently one of the most abundant soil bacterial phylotypes, particularly in grasslands, where it was typically the most abundant. We reconstructed a nearly complete genome of this phylotype from a soil metagenome for which we propose the provisional name ‘Candidatus Udaeobacter copiosus’. The Ca. U. copiosus genome is unusually small for a cosmopolitan soil bacterium, estimated by one measure to be only 2.81 Mbp, compared to the predicted effective mean genome size of 4.74 Mbp for soil bacteria. Metabolic reconstruction suggests that Ca. U. copiosus is an aerobic heterotroph with numerous putative amino acid and vitamin auxotrophies. The large population size, relatively small genome and multiple putative auxotrophies characteristic of Ca. U. copiosus suggest that it may be undergoing streamlining selection to minimize cellular architecture, a phenomenon previously thought to be restricted to aquatic bacteria. Although many soil bacteria need relatively large, complex genomes to be successful in soil, Ca. U. copiosus appears to use an alternative strategy, sacrificing metabolic versatility for efficiency to become dominant in the soil environment.
Similar content being viewed by others


Soils harbour massive amounts of undescribed microbial diversity. For example, more than 120,000 unique bacterial and archaeal taxa were found in surface soils of Central Park in New York City, of which only ∼15% had 16S rRNA gene sequences matching those contained in reference databases and <1% had representative genome sequence information1. This undescribed soil microbial diversity is not evenly distributed across the tree of life. For example, Acidobacteria and Verrucomicrobia, two of the more abundant bacterial phyla found in soil2,3, represent only 0.08 and 0.06% of all cultured bacterial isolates in the Ribosomal Database Project (RDP)4 and only 0.08 and 0.14% of publicly available bacterial genomes found in Integrated Microbial Genomes (IMG)5, respectively. Although the ecology and genomic attributes of abundant soil taxa are beginning to be described6, we still lack basic information about the vast majority of soil microbes. These knowledge gaps highlight that a huge fraction of living biomass in terrestrial systems remains enigmatic7 and that we are only beginning to identify the influence of specific microbes on soil biogeochemistry and fertility.
In this study we focus our exploration of undescribed microbial diversity on the Verrucomicrobia phylum. Although Verrucomicrobia are generally recognized as being among the most numerically abundant taxa in soil2,3, we know very little about the ecological or genomic attributes that contribute to their success. The phylum Verrucomicrobia is highly diverse and its members possess a broad range of metabolic capabilities. For example, members of the class Methylacidiphilae are nitrogen-fixing acidophiles capable of methane oxidation8, while Akkermansia muciniphila of the class Verrucomicrobiae is a mucin-degrading resident of the human gut9. However, the dominant Verrucomicrobia found in soil typically belong to the class Spartobacteria. Although Verrucomicrobia accounted for >50% of all bacterial 16S rRNA gene sequences in tallgrass prairie soils in the USA, >75% of these sequences were assigned to the class Spartobacteria10. Currently, the class Spartobacteria contains only a single described and sequenced isolate, Chthoniobacter flavus, a slow-growing aerobic heterotroph capable of using common components of plant biomass for growth11,12. Although Spartobacteria are prevalent in soils, they have also been observed in marine systems (‘Spartobacteria baltica’)13 and as nematode symbionts (genus Xiphinematobacter)14.
Here, we report the distribution of a dominant Spartobacteria lineage, compiling data from both amplicon and shotgun metagenomic 16S rRNA gene surveys to quantify its relative abundance across >1,000 unique soils. We assembled a near-complete genome of this lineage from a single soil where it was exceptionally abundant. These results provide our first glimpse into the phylogeny, ecology and potential physiological traits of a dominant soil Verrucomicrobia and suggest that members of this group are efficient at growing and persisting in the low-resource conditions common in many soil microenvironments.
Results and discussion
Distribution of the dominant Verrucomicrobia in soil
A single spartobacterial clade dominates bacterial communities found in a wide range of soil types across the globe. One phylotype from this group of Spartobacteria represented up to 31% of total 16S rRNA gene sequences recovered from prairie soils10. This phylotype shares 99% 16S rRNA gene sequence identity with a ribosomal clone named ‘DA101’, first described in 1998 as a particularly abundant 16S rRNA sequence recovered from grassland soils in the Netherlands15. To determine if the DA101 phylotype (termed ‘DA101’ herein) is abundant in other soils, we re-analysed amplicon 16S rRNA gene sequence data obtained from >1,000 soils representing a wide range of soil and site characteristics (Supplementary Table 1). We found that DA101 was on average ranked within the top two most abundant bacterial phylotypes in each study (Fig. 1). In over 70% of the soils analysed, DA101 was within the top ten most abundant phylotypes. Interestingly, other phylotypes belonging to the same family as DA101 (Chthoniobacteraceae) were also found within the top five most abundant phylotypes of several studies (Fig. 1).

DA101 phylotype data are indicated in blue and other abundant taxa in grey. Taxa are listed on the x axis in order of their median rank abundance (taxa on the left are the most abundant). The top and bottom of each box represent the 25th and 75th percentiles, the mid line represents the 50th percentile/median and the whiskers represent the range of points excluding outliers. Data sets from previously published studies are indicated. Further details on each of these studies are provided in Supplementary Table 1.
As some 16S rRNA gene PCR primer sets can misestimate the relative abundance of Verrucomicrobia3,16, we investigated whether the apparent numerical dominance of DA101 in amplicon data sets was a product of PCR primer biases. To do so, we quantified the abundance of DA101 16S rRNA genes within previously published soil shotgun metagenomes17,18. The relative abundance of DA101 in amplicon data was well correlated with the relative abundance of DA101 in shotgun metagenomic data (P < 0.0001, ρ = 0.50, n = 102). Confirming the amplicon-based results (Fig. 1), we found that DA101 was also among the most abundant phylotypes observed in the soil bacterial communities characterized via shotgun metagenomic sequencing (Supplementary Fig. 1). Therefore, the numerical dominance of DA101 in soils is not simply a product of primer bias.
Despite DA101 being one of the most abundant phylotypes found in soil, its proportional abundance can vary significantly across soil types (Fig. 1 and Supplementary Fig. 1). We used metadata associated with each soil sample to determine which of the measured soil and site characteristics best predicted the relative abundance of DA101. We found that DA101 was significantly more abundant in grassland soils than in forest soils (P < 0.0001, n = 64, Mann–Whitney test, Supplementary Fig. 2); on average, DA101 is six times more abundant in grassland soils. These findings indicate that the soils in which DA101 excels do not overlap with those forest soils dominated by non-symbiotic Bradyrhizobium taxa, another ubiquitous and abundant group of soil bacteria6. Across the grassland soils included in our meta-analysis, the relative abundance of DA101 was positively correlated with both soil microbial biomass (P < 0.0001, ρ = 0.57, n = 31, Spearman, Supplementary Fig. 3) and aboveground plant biomass (P < 0.0001, ρ = 0.47, n = 366, Spearman, Supplementary Fig. 3). Together, these results suggest that DA101 prefers soils receiving elevated amounts of labile carbon inputs. We did not identify any consistently significant correlations between the abundance of DA101 and other prokaryotic or eukaryotic taxa, suggesting that DA101 is unlikely to be a part of an obligate pathogenic or symbiotic relationship.
Diversity of soil Verrucomicrobia
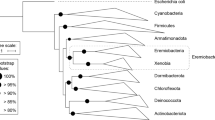
We determined the phylogenetic placement of DA101 and other soil Verrucomicrobia by assembling near full-length 16S rRNA gene sequences from six distinct grassland soils collected from multiple continents (Fig. 2 and Supplementary Table 2). Although we were able to assemble representative 16S rRNA gene sequences from all verrucomicrobial classes except Methylacidiphilae, 93% of verrucomicrobial sequences fell within the Spartobacteria class and 87% of these fell within the DA101 clade. These phylogenetic analyses confirm that DA101 belongs to the class Spartobacteria (Fig. 2). However, within the Spartobacteria class, the DA101 clade is clearly distinct from the clade containing Chthoniobacter flavus11,12, as DA101 shares only 92% 16S rRNA gene sequence identity with C. flavus. These findings indicate that DA101 is a representative of a new verrucomicrobial genus. We propose the candidate genus name ‘Candidatus Udaeobacter’ for the DA101 clade; the proposed name combines Udaeus (‘of the earth’, Greek) with bacter (‘rod’ or ‘staff’, Greek) and like Chthoniobacter refers to one of the Spartoi of the Cadmus myth. We recommend the provisional name ‘Candidatus Udaeobacter copiosus’ for the DA101 phylotype, which refers to its numerical dominance in soil.

Stars denote 16S rRNA gene sequences of named isolates and circles represent environmental 16S rRNA gene sequences assembled from six soils using EMIRGE56 (Supplementary Table 2). The uncultivated verrucomicrobial phylotype DA101 falls within a cluster distinct from cultivated Spartobacteria. The UPGMA phylogenetic tree was constructed using 1,200 bp verrucomicrobial 16S rRNA gene sequences and is rooted with a 16S rRNA gene sequence from Escherichia coli K-12. Notable verrucomicrobial isolates and genera are labelled. Colours indicate verrucomicrobial classes.
Draft genome of ‘Candidatus Udaeobacter copiosus’ recovered from metagenomic data
Despite their ubiquity and abundance in soil, there is no genomic data currently available for any representative of the ‘Candidatus Udaeobacter’ clade. Typically, soil hyperdiversity confounds the assembly of genomes from metagenomes19, requiring single-cell analysis or laboratory isolation to produce an assembled genome. However, we leveraged the sheer abundance of Ca. U. copiosus in an individual soil to obtain a nearly complete genome from metagenomic data. We deeply sequenced a soil where Ca. U. copiosus accounted for >30% of 16S rRNA gene sequences and assembled a draft genome from the resulting metagenome. We used GC content, coverage, tetranucleotide frequencies and the phylogenetic affiliation of predicted proteins to bin assembled contigs, resulting in a draft Ca. U. copiosus genome with 238 contigs. The draft genome is 2.65 Mbp in size, has a GC content of 54%, and encodes for 3,042 predicted proteins, 67% of which could be assigned to Pfam protein families20 by the IMG annotation pipeline5.
The Ca. U. copiosus genome shares only 69.3% average nucleotide identity21 with the genome of its closest sequenced relative C. flavus, further supporting its proposed placement in the distinct genus ‘Candidatus Udaeobacter’. Although no 16S rRNA gene was assembled within the Ca. U copiosus genome, we used Metaxa2 (ref. 22) to extract fragments of a single DA101-like 16S rRNA gene from the raw metagenomic sequences we used for assembly. This 16S rRNA gene has 100% identity to the DA101 amplicon sequence and has the same average coverage (23–29×) as the Ca. U copiosus genome (27×), suggesting this genome belongs to a representative of the DA101 clade. As a second measure to verify this genome is a representative of the DA101 clade, we compared the abundance of three housekeeping genes assembled within the Ca. U. copiosus genome (dnaK, rpoB and secY) to the abundance of the DA101 16S rRNA gene in >100 metagenomic samples from two separate studies17,18. All three genes show a very strong significant correlation with the DA101 16S rRNA gene (P < 0.0001, ρ > 0.87, n = 102, Pearson correlation, Supplementary Fig. 4), further evidence that this genome represents the DA101 clade and that this lineage is as abundant in soil as our analyses based on the 16S rRNA gene suggest.
We estimate that the full Ca. U copiosus genome will be ∼2.81 Mbp in length based on the recovery of 94% of domain-specific single-copy housekeeping genes commonly used to estimate genome completion23 (Supplementary Table 3). Based on this estimate, Ca. U. copiosus appears to have a particularly small genome size compared to C. flavus and other sequenced heterotrophic soil Verrucomicrobia (Supplementary Table 4). Indeed, the genome size of Ca. U. copiosus is much more similar to Verrucomicrobia of the class Methylacidiphilae24, thermophiles for whom genome size and growth temperature are negatively correlated25. To determine how the genome size of Ca. U. copiosus compares to other soil bacteria, we compiled data from 378 finished and permanent draft genomes in IMG whose 16S rRNA gene sequences matched the 16S rRNA gene amplicon sequences obtained by Leff et al.17 with at least 99% identity. Nearly all of these 378 bacterial genomes were from cultivated taxa (99%). We estimated the genome completeness for each of the 378 taxa using the same domain-specific marker genes as for Ca. U. copiosus and found the mean estimated genome size of these taxa to be 5.28 ± 2.15 Mbp (mean ± s.d.), which is similar to metagenomic-based estimates of mean genome size for soil microbes (4.74 ± 0.69)26. Strikingly, the estimated 2.81 Mbp genome of Ca. U. copiosus is ∼50% smaller than the mean genome size of these 378 taxa; only 48 (13%) of these genomes are smaller than Ca. U. copiosus. Furthermore, the majority (65%) of soil taxa with genomes smaller than Ca. U. copiosus originate from organisms with obligate intracellular or host-associated lifestyles (Fig. 3).

a, Points represent the estimated genome size and relative abundances of 378 bacterial genomes obtained by matching 16S rRNA gene sequences from Leff et al.17 to 16S rRNA gene sequences extracted from IMG genomes at 99% sequence identity. This data set focused on surface soils collected from grasslands across the globe. The average abundances shown here may not apply to other soil or ecosystem types. Only genomes classified as ‘permanent draft’ or ‘finished’ status were used. Bacteria with larger genomes tend to be more abundant (P < 0.0001, ρ = 0.368, n = 378, Spearman correlation), with Ca. U. copiosus (indicated in blue) being a notable exception to this pattern, as it has a high relative abundance (2.26% of 16S rRNA sequences) but a relatively small genome. The shaded region represents the 95% confidence interval of the trend line. b, Host-associated bacteria make up a majority of sequenced small genomes in soil. In the genome size range for Ca. U. copiosus (2.75–3.00 Mbp), 56% of soil taxa have a purely host-associated lifestyle.
Although soil bacteria with larger genomes tend to be more common in soil, Ca. U. copiosus seems to be a notable exception to this pattern. We linked the genome size of each of the matched IMG bacterial genomes with the average abundance of their corresponding amplicon sequence from Leff et al.17 and found that genome size is positively correlated with average relative abundance (P < 0.001, ρ = 0.37, n = 378, Spearman, Fig. 3). That is, sequenced bacteria with large genomes tend to comprise a significantly larger proportion of soil bacterial communities. On average, the genomes of soil prokaryotes are larger than those inhabiting aquatic ecosystems27 or the human gut28. These relatively large genomes are thought to provide soil-dwelling bacteria with a more diverse genetic inventory to enhance survival in conditions where resources are diverse, but sparse29,30. However, the Ca. U. copiosus genome has a conspicuously reduced genome given its abundance (Fig. 3). This suggests that Ca. U. copiosus occupies a niche space that does not require expansive functional diversity and points to an alternative route to success for soil bacteria. These results also suggest that abundant, uncultivated soil bacteria probably have smaller genomes than the cultivated taxa that represent the majority of available genomic data. A similar pattern has been observed in aquatic systems, where uncultivated taxa often have smaller genomes than cultivated taxa31. Because most genomic information is derived from cultivated bacterial taxa, the lack of genomic information from bacteria with compact genomes may stem from challenges associated with culturing taxa with reduced genomes27.
Metabolic reconstruction of the Ca. U. copiosus genome points to an aerobic heterotrophic lifestyle with the capacity to use a limited range of carbon substrates for growth, including glucose, pyruvate and chitobiose. Glycogen/starch synthesis and utilization genes were identified (glgABCP and amyA), suggesting that Ca. U. copiosus has the capacity to store surplus carbon as glycogen or starch. Glycogen metabolism has been demonstrated in other Verrucomicrobia32. Genes encoding for the complete biosynthesis of vitamins B2, B3, B5 (from valine) and B6 were recovered, as well as full biosynthetic pathways for de novo synthesis of alanine, aspartate, asparginine, glutamate, glutamine, lysine, serine and proline. Nearly complete pathways were recovered for glycine, threonine and methionine biosynthesis (Supplementary Fig. 5). Genes encoding for the conversion of methionine to cysteine were present as the only apparent route to cysteine biosynthesis. Genes indicative of autotrophic metabolism (for example, RuBisCO, ATP citrate lyase) were not identified. Additionally, genes indicative of methanotrophy (pmo), methylotrophy (mxaF or xoxF), ammonia (amo) or nitrite oxidation (nxr) were not found.
Genes encoding for the biosynthesis of all branched-chain (isoleucine, leucine and valine) and aromatic (tryptophan, tyrosine and phenylalanine) amino acids were conspicuously underrepresented in the Ca. U copiosus genome. The biosynthetic pathways for arginine and histidine were also incomplete (Supplementary Fig. 5), along with the entire vitamin B12 synthesis pathway, despite the presence of three genes encoding vitamin B12-dependent proteins (methionine synthase, ribonucleotide reductase and methylmalonyl-CoA mutase). It is conceivable that genomic information encoding for these putative auxotrophies is present on genome fragments that were not recovered in our metagenome assembly, or is encoded on extrachromosomal elements that are commonly missed in metagenomic assemblies (for example, a plasmid33). Relative to C. flavus, 34 biosynthetic genes are needed for Ca. U. copiosus to be fully prototrophic for all amino acids. In C. flavus, these genes are not organized on operons12, meaning they are probably randomly distributed throughout the Ca. U. copiosus genome as well. Moreover, the absence of branched-chain amino acid and histidine synthesis pathways in the Ca. U. copiosus genome is consistent with previous observations that branched chain and histidine biosynthesis genes are underrepresented in native prairie populations of soil Verrucomicrobia10. Additionally, plasmids are uncommon within isolates of the Verrucomicrobia phylum, with only one species known to maintain a plasmid—Opitutaceae Bacterium Strain TAV534—a distant relative of Ca. U. copiosus.
Auxotrophy in free-living bacteria is not expected to be a rare phenomenon; one study estimated that 85% of free-living bacteria have at least one vitamin or amino acid auxotrophy35, and multiple studies have shown that auxotrophic mutants have a pronounced growth advantage over their wild-type counterparts when supplied with the compounds they cannot synthesize35,36. Vitamin B12 auxotrophies are relatively common in soil37, suggesting this metabolically expensive vitamin is generally available to many soil bacteria. Similarly, the eight amino acids for which we did not identify complete pathways in the Ca. U. copiosus genome are among the most energetically expensive to make38 (Supplementary Fig. 5). This suggests that if Ca. U. copiosus is auxotrophic for some of these metabolites, acquiring them from the environment would provide Ca. U copiosus with an energetic saving relative to taxa that synthesize them de novo.
Although Ca. U. copiosus appears to lack genes for several amino acid synthesis pathways, numerous genes encoding for peptide transport, degradation and recycling were identified. Indeed, when scaled for genome size, Ca. U. copiosus encodes four times as many putative peptide and amino acid transporters as C. flavus (1.5% of the genome, compared with 0.37%) and twice as many predicted proteases (6.5% of genome versus 3.2%). Ca. U. copiosus also encodes for all components of the bacterial proteasome. Proteasomal degradation is critical for amino acid recycling under starvation conditions in mycobacteria39. The enrichment of peptide transport and degradation systems in the Ca. U. copiosus genome suggest that at least some of the amino acids Ca. U. copiosus appears incapable of synthesizing are available directly from the soil environment or by associations with other soil biota.
Ca. U. copiosus clearly has a reduced genome size compared to other soil bacteria (Fig. 3) and other Verrucomicrobia with similar lifestyles (Supplementary Table 4). Bacterial genome reduction is thought to occur through two main mechanisms, genetic drift and streamlining selection, both mediated by extremes in effective population sizes (Ne, reviewed in ref. 40). The effect of genetic drift on microbes with a small Ne and low recombination rates, such as endosymbiotic bacteria, leads to the accumulation of deleterious mutations and subsequent loss of genetic material, which is typically identifiable in genomes by the presence of numerous pseudogenes and large noncoding intergenic regions40. In contrast, free-living organisms with a large Ne are thought to undergo ‘streamlining’ selection to minimize genome size27,40. The genome-streamlining hypothesis proposes that, in large bacterial populations, reduced genome complexity is a trait under natural selection, especially in environments where nutrients can be sparse and periodically limit growth27.
The abundance, putative auxotrophies (Supplementary Fig. 5) and cosmopolitan distribution of Ca. U. copiosus (Fig. 1), together with its small genome size relative to other soil microbes (Fig. 3) and Verrucomicrobia with similar lifestyles (Supplementary Table 4), suggest that its small genome is a product of streamlining selection. Although it is difficult to accurately measure Ne in wild populations of bacteria, evidence of drift-mediated genome reduction was not present in the Ca. U. copiosus genome (such as large numbers of pseudogenes or unusually large intergenic spaces). Although most contemporary free-living organisms with streamlined genomes inhabit aquatic environments27,41, compared to these aquatic environments, soil is more heterogeneous42, has greater overall microbial diversity43 and slower carbon turnover44. Therefore, the functional complexity required by soil microbes to succeed within a given niche is probably large relative to that required by aquatic microbes. This means that the effects of genome streamlining are likely to be most evident (that is, result in smaller genomes) in aquatic environments. This expectation is reflected in the fact that, on average, the genomes of aquatic microbes are smaller than their terrestrial counterparts27. However, the small genome and numerous putative pathways missing from Ca. U. copiosus suggest that genome streamlining may not be unique to aquatic organisms and that genome streamlining may also confer a selective growth advantage in the soil environment.
The probable effects of genome streamlining in Ca. U. copiosus seem to have resulted in reduced catabolic and biosynthetic capacity and thus an apparent loss of metabolic versatility. The under-representation of multiple costly amino acid and vitamin biosynthetic pathways in the Ca. U. copiosus genome implies that these compounds can be acquired from the soil environment. Several studies have shown that free amino acids and oligopeptides are present in soil45,46. The enrichment of proteases and amino acid and peptide importers in the Ca. U. copiosus genome suggests that it is well equipped to assimilate this fraction of soil organic matter. Dispensing the capacity to synthesize costly amino acids and vitamins would provide Ca. U copiosus with a growth advantage in resource-limiting conditions when competition for labile carbon is high. Furthermore, many of the amino acids and vitamins Ca. U copiosus appears unable to synthesize are involved in synergistic growth47 and may be supplied by other microbes as common community goods48. Based on the few spartobacterial isolates that have been cultivated11, culture-independent studies10,49 and the genomic data presented here, we speculate that Ca. U. copiosus is a small, oligotrophic soil bacterium that reduces its requirement for soil organic carbon by acquiring costly amino acids and vitamins from the environment.
Conclusions
Whereas successful soil microbes are predicted to have large genomes29,30 (Fig. 3), Ca. U. copiosus has a small genome, indicating that, similar to some aquatic microbes, minimization of cellular architecture can also represent a successful strategy for soil microbes. We do not know if other uncultivated abundant soil taxa also contain reduced genomes, because pre-existing genome databases are preferentially biased towards cultivated isolates. For example, only 4.5% of bacterial genomes in IMG are from uncultivated taxa (accessed April 2016). Bacteria encoding for greater metabolic versatility have larger genomes and therefore may be easier to cultivate in the laboratory31. On the other hand, specific and combinatorial nutrient requirements such as those described for Ca. U. copiosus present a complex problem for researchers attempting to cultivate microbes with reduced genomes50. Although Ca. U. copiosus has not yet been grown in the laboratory, cultivation is clearly a crucial next step to describing this organism, using the information described here to ‘tailor’ a growth medium specifically for Ca. U. copiosus and related microbes. Such an approach could improve our ability to describe and study the majority of soil microbes, even dominant soil microbes like Ca. U. copiosus, which remain difficult to cultivate under laboratory conditions.
Methods
Estimating the abundance and distribution of Verrucomicrobia in soil
Five abundant Verrucomicrobia phylotypes were described in Fierer et al.10, but a single phylotype with 99% identity to the clone DA101 (ref. 15) was clearly dominant. We searched previously published soil data sets for representative sequences with 100% identity to this DA101 phylotype, including 31 soils from US native tallgrass prairies10, 64 soils from matched forest and grassland sites across North America51, 595 soils collected from Central Park in New York City1, 367 grassland soils collected from North America, Europe, Australia and Africa17, and a cross-biome collection of 15 desert and non-desert soils from across the globe18. We also included a data set from a grassland terrace near Boulder, Colorado (105.23W, 40.12N, Table Mountain) where 29 soils were collected from a depth of 25 cm within a 100 m2 area on 28 January 2015. Collectively, these data sets represent 1,101 unique soil samples collected from a wide range of ecosystem and soil types.
For all samples, DNA was extracted with the MoBio PowerSoil kit and the V4 region of the 16S rRNA gene was amplified in triplicate with the 515f/806r primer pair. After normalization to equimolar concentrations, amplicons were sequenced on an Illumina MiSeq (151 bp paired end) at the University of Colorado BioFrontiers Institute Next-Gen Sequencing Facility. Sequences were processed as described previously17. In brief, we used a combination of QIIME (ref. 52) and UPARSE (ref. 53) to quality-filter, remove singletons and merge paired reads. Sequences were assembled into phylotypes at the 97% identity level using UCLUST (ref. 54). Taxonomy was assigned using the Greengenes 13_8 database55 and the Ribosomal Database Project classifier4 and each data set was rarefied independently (Supplementary Table 1).
As PCR primer biases can misestimate the relative abundances of Verrucomicrobia3,16, we also estimated the abundances of the DA101 phylotype directly from shotgun metagenomic data. We used Metaxa2 with default settings22 to extract bacterial 16S rRNA gene sequences from shotgun metagenomic data compiled from previous analyses of 75 different soils after rarefaction17,18. Extracted 16S rRNA gene fragments were matched to Greengenes full-length sequences at 99% ID using the usearch7 command usearch_global. The matched Greengenes sequences were then clustered and assigned taxonomy as described above. All statistical tests were carried out in R and ggplot2 was used for all plots unless specifically mentioned. Variances between groups tested were within one order of magnitude.
Describing the phylogenetic diversity of soil Verrucomicrobia
We reconstructed near full-length 16S rRNA gene sequences to build a phylogeny of soil Verrucomicrobia from six soil samples (Supplementary Table 2) that were selected to represent geographically distinct grasslands with a range of verrucomicrobial abundances. We extracted DNA from each of these soils as described previously17 and used the 27f/1392r primer pair to amplify near full-length 16S rRNA genes as described in ref. 56. The amplicons were sheared using the Covaris M220, and 16S rRNA gene libraries were prepared using TruSeq DNA LT library preparation kits (Illumina). Samples were pooled and sequenced on an Illumina MiSeq (2 × 300 bp) at the University of Colorado Next Generation Sequencing Facility.
After quality-filtering of sequences, near full-length SSU sequences were reconstructed using EMIRGE (ref. 56). After 40 iterations, sequences were merged into phylotypes with ≥97% similarity. Reconstructed sequences were trimmed to 1,200 bp and all sequences were further clustered at 95% identity due to gaps in some assemblies. Full-length 16S rRNA sequences from named verrucomicrobial isolates were aligned along with the reconstructed sequences using PyNAST (ref. 57). A UPGMA tree was constructed using the R packages seqnir, phangorn and ape and visualized with GraPhlAn (R 3.2.2, version 0.9.7)58.
Assembly and annotation of a genome from the dominant soil verrucomicrobial phylotype
We assembled the genome of ‘Ca. Udaeobacter copiosus’ from a metagenome of a US prairie soil sample (NTP21, Hayden, IA) estimated to have particularly high abundances of bacteria within the DA101 clade10. Fragmented DNA extracted from this soil was prepared for sequencing using WaferGen's PrepX ILM DNA library Kit (WaferGen Biosystems) and the Apollo 324 Automated Library Prep System for library generation. The library was sequenced on one Illumina HiSeq2000 lane (2 × 101 bp), yielding 17 Gb of sequence with an average paired-end insert size of 345 bp. Low-quality reads were trimmed using Sickle v. 1.29 with a quality score threshold of Q = 3, or removed if trimmed to <80 bp long (https://github.com/najoshi/sickle). The sequences were assembled using IDBA_ud v. 1.1.0 (ref. 59) with a kmer range of 40–70 and step size of 15. To improve recovery of the most abundant Verrucomicrobia, the genome was selectively re-assembled using Velvet with a kmer size of 59 and expected kmer coverage of 11.5 (range 7.5–15.5). To bin contigs ≥2 kb long, genes and protein sequences were predicted using Prodigal v. 2.60 in metagenomics mode60. For each contig, we determined the GC content, coverage and the phylogenetic affiliation based on the best hit for each predicted protein in the Uniref90 database61 (Sept. 2013) following ublast searches. We also constructed emergent self-organizing maps (ESOM)62 using tetranucleotide frequencies of 5 kb DNA fragments. A combination of these approaches was used to identify the genome. The draft genome was uploaded to IMG for annotation under the taxon ID 2651869889.
We estimate that the Ca. U copiosus genome is ∼94% complete, based on domain-specific single-copy housekeeping genes commonly used to estimate genome completion23 (Supplementary Table 3). This list of single-copy genes has been used to estimate genome completeness in several recent studies13,63. When we analysed the genome using another metric of genome completeness (checkM64), the results suggested that the genome was 80% complete with 4% contamination, a level categorized as a ‘substantially complete draft with low contamination’. This level of completeness is similar to several other recent genomes assembled from metagenomes65,66. However, because checkM relies on lineage-specific marker genes, the completeness of genomes without lineage representation can often be underestimated64. As there is only one complete genome for the entire class Spartobacteria (C. flavus), the checkM genome completeness estimate for Ca. U. copiosus may likewise be underestimated. Simply put, there are limitations and caveats associated with any genome completeness measure and the true completeness of the Ca. U. copiosus genome probably lies somewhere between these estimates.
No rRNA genes were annotated by IMG, so we used Metaxa2 with default settings on the unassembled sequences to extract any 16S rRNA genes. Metaxa2 recovered two ∼500 bp 16S rRNA gene fragments at 23–29× coverage that aligned to separate regions of the full-length 16S rRNA gene from the closest related verrucomicrobial genome (C. flavus). Because these two rRNA gene fragments have the same coverage as the genome (27×) and align to separate regions of one 16S rRNA gene, it is likely that Ca. U. copiosus encodes a single rRNA operon, similar to its closest relative C. flavus11,12 and all other sequenced heterotrophic soil Verrucomicrobia (Supplementary Table 4).
Data availability
The draft genome of ‘Candidatus Udaeobacter copiosus’ is publicly available in the Integrated Microbial Genomes (IMG) database under IMG genome ID 2651869889. Raw sequences from which the Ca. U. copiosus genome was assembled are available at the Sequence Read Archive (SRA) under bioproject ID PRJNA342239. Amplicon sequences and associated metadata generated exclusively for this study are available at figshare at http://dx.doi.org/10.6084/m9.figshare.3363505.v3. Accession numbers for all other amplicon data sets have been published previously. The raw sequences used for EMIRGE near full-length 16S amplicon reconstruction are also available at figshare at http://dx.doi.org/10.6084/m9.figshare.3799422.v1. All other data sets supporting these findings are available from the corresponding author upon request.
Change history
14 July 2017
In the PDF version of this article previously published, the year of publication provided in the footer of each page and in the 'How to cite' section was erroneously given as 2017, it should have been 2016. This error has now been corrected. The HTML version of the article was not affected.
References
Ramirez, K. S. et al. Biogeographic patterns in below-ground diversity in New York City's Central Park are similar to those observed globally. Proc. R. Soc. B. 281, 20141988 (2014).
Janssen, P. H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728 (2006).
Bergmann, G. T. et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 43, 1450–1455 (2011).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Markowitz, V. M. et al. IMG 4 version of the Integrated Microbial Genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567 (2014).
VanInsberghe, D. et al. Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. 9, 2435–2441 (2015).
Fierer, N., Strickland, M. S., Liptzin, D., Bradford, M. A. & Cleveland, C. C. Global patterns in belowground communities. Ecol. Lett. 12, 1238–1249 (2009).
Dunfield, P. F. et al. Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450, 879–882 (2007).
Everard, A. et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl Acad. Sci. USA 110, 9066–9071 (2013).
Fierer, N. et al. Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342, 621–624 (2013).
Sangwan, P., Chen, X., Hugenholtz, P. & Janssen, P. H. Chthoniobacter flavus gen. nov., sp. nov., the first pure-culture representative of subdivision two, Spartobacteria classis nov., of the phylum Verrucomicrobia. Appl. Environ. Microbiol. 70, 5875–5881 (2004).
Kant, R. et al. Genome sequence of Chthoniobacter flavus Ellin428, an aerobic heterotrophic soil bacterium. J. Bacteriol. 193, 2902–2903 (2011).
Herlemann, D. P. R. et al. Metagenomic de novo assembly of an aquatic representative of the verrucomicrobial class spartobacteria. mBio 4, e00569-12 (2013).
Vandekerckhove, T. T., Willems, A., Gillis, M. & Coomans, A. Occurrence of novel verrucomicrobial species, endosymbiotic and associated with parthenogenesis in Xiphinema americanum-group species (Nematoda, longidoridae). Int. J. Syst. Evol. Microbiol. 50, 2197–2205 (2000).
Felske, A. & Akkermans, A. D. L. Prominent occurrence of ribosomes from an uncultured bacterium of the Verrucomicrobiales cluster in grassland soils. Lett. Appl. Microbiol. 26, 219–223 (1998).
Guo, J., Cole, J. R., Zhang, Q., Brown, C. T. & Tiedje, J. M. Microbial community analysis with ribosomal gene fragments from shotgun metagenomes. Appl. Environ. Microbiol. 82, 157–166 (2016).
Leff, J. W. et al. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl Acad. Sci. USA 112, 10967–10972 (2015).
Fierer, N. et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl Acad. Sci. USA 109, 21390–2139 5 (2012).
Howe, A. C. et al. Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl Acad. Sci. USA 111, 4904–4909 (2014).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Varghese, N. J. et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771 (2015).
Bengtsson-Palme, J. et al. metaxa2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 15, 1403–1414 (2015).
Ciccarelli, F. D. et al. Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287 (2006).
Hou, S. et al. Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia. Biol. Direct 3, 26 (2008).
Sabath, N., Ferrada, E., Barve, A. & Wagner, A. Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol. Evol. 5, 966–977 (2013).
Raes, J., Korbel, J. O., Lercher, M. J., von Mering, C. & Bork, P. Prediction of effective genome size in metagenomic samples. Genome Biol. 8, R10 (2007).
Giovannoni, S. J., Thrash, J. C. & Temperton, B. Implications of streamlining theory for microbial ecology. ISME J. 8, 1553–1565 (2014).
Nayfach, S. & Pollard, K. S. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 16, 51 (2015).
Konstantinidis, K. T. & Tiedje, J. M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl Acad. Sci. USA 101, 3160–3165 (2004).
Barberán, A. et al. Why are some microbes more ubiquitous than others? Predicting the habitat breadth of soil bacteria. Ecol. Lett. 17, 794–802 (2014).
Button, D. K. & Robertson, B. R. Determination of DNA content of aquatic bacteria by flow cytometry. Appl. Environ. Microbiol. 67, 1636–1645 (2001).
Khadem, A. F. et al. Genomic and physiological analysis of carbon storage in the verrucomicrobial methanotroph ‘Ca. methylacidiphilum fumariolicum’ solV. Front. Microbiol. 3, 345 (2012).
Jørgensen, T. S., Kiil, A. S., Hansen, M. A., Sørensen, S. J. & Hansen, L. H. Current strategies for mobilome research. Front. Microbiol. 5, 750 (2014).
Kotak, M. et al. Complete genome sequence of the Opitutaceae bacterium strain TAV5, a potential facultative methylotroph of the wood-feeding termite Reticulitermes flavipes. Genome Announc. 3, e00060-15 (2015).
D'Souza, G. et al. Less is more: selective advantages can explain the prevalent loss of biosynthetic genes in bacteria. Evolution 68, 2559–2570 (2014).
Wook, K. & Levy, S. B. Increased fitness of Pseudomonas fluorescens Pf0-1 leucine auxotrophs in soil. Appl. Environ. Microbiol. 74, 3644–3651 (2008).
Lochhead, A. G. Soil bacteria and growth promoting substances. Bacteriol. Rev. 22, 145–153 (1958).
Akashi, H. & Gojobori, T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl Acad. Sci. USA 99, 3695–3700 (2002).
Elharar, Y. et al. Survival of mycobacteria depends on proteasome-mediated amino acid recycling under nutrient limitation. EMBO J. 33, 1802–1814 (2014).
Batut, B., Knibbe, C., Marais, G. & Daubin, V. Reductive genome evolution at both ends of the bacterial population size spectrum. Nat. Rev. Microbiol. 12, 841–850 (2014).
Kantor, R. S. et al. Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio 4, e00708-13 (2013).
Vos, M., Wolf, A. B., Jennings, S. J. & Kowalchuk, G. A. Micro-scale determinants of bacterial diversity in soil. FEMS Microbiol. Rev. 37, 936–954 (2013).
Fierer, N. & Lennon, J. T. The generation and maintenance of diversity in microbial communities. Am. J. Bot. 98, 439–448 (2011).
Giovannoni, S. J. & Vergin, K. L. Seasonality in ocean microbial communities. Science 335, 671–676 (2012).
Friedel, J. K. & Scheller, E. Composition of hydrolysable amino acids in soil organic matter and soil microbial biomass. Soil Biol. Biochem. 34, 315–325 (2002).
Farrell, M. et al. Oligopeptides represent a preferred source of organic N uptake: a global phenomenon? Ecosystems 16, 133–145 (2013).
Mee, M. T., Collins, J. J., Church, G. M. & Wang, H. H. Syntrophic exchange in synthetic microbial communities. Proc. Natl Acad. Sci. USA 111, E2149–E2156 (2014).
Morris, J. J., Lenski, R. E. & Zinser, E. R. The black queen hypothesis: evolution of dependencies through adaptive gene loss. mBio 3, e00036-12 (2012).
Portillo, M. C., Leff, J. W., Lauber, C. L. & Fierer, N. Cell size distributions of soil bacterial and archaeal taxa. Appl. Environ. Microbiol. 79, 7610–7617 (2013).
Carini, P., Steindler, L., Beszteri, S. & Giovannoni S. J. Nutrient requirments for growth of the extreme oligotroph ‘Candidatus Pelagibacter ubique’ HTCC1062 on a defined medium. ISME J. 7, 592–602 (2013).
Crowther, T. W. et al. Predicting the responsiveness of soil biodiversity to deforestation: a cross-biome study. Glob. Change Biol. 20, 2983–2994 (2014).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R. C. UPARSE: highly accurate phylotype sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Miller, C. S. et al. Short-read assembly of full-length 16S amplicons reveals bacterial diversity in subsurface sediments. PLoS ONE 8, e56018 (2013).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
Asnicar, F., Weingart, G., Tickle, C. H., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029 (2015).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R. & Wu, C. H. Uniref: comprehensive and non-redundant uniProt reference clusters. Bioinformatics 23, 1282–1288 (2007).
Dick, G. J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009).
Anantharaman, K., Breier, J. A. & Dick, G. J. Metagenomic resolution of microbial functions in deep-sea hydrothermal plumes across the Eastern Lau Spreading Center. ISME J. 10, 225–239 (2016).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Baker, B. J. et al. Genomic inference of the metabolism of cosmopolitan subsurface Archaea, Hadesarchaea. Nat. Microbiol. 1, 16002 (2016).
Garcia, S. L. et al. Auxotrophy and intrapopulation complementary in the ‘interactome’ of a cultivated freshwater model community. Mol. Ecol. 24, 4449–4459 (2015).
Acknowledgements
Funding to support this work was provided by grants from the National Science Foundation to N.F. (DEB0953331, EAR1331828), a Visiting Postdoctoral Fellowship award to P.C. from the Cooperative Institute for Research in Environmental Sciences and an Alfred P Sloan Foundation grant to J.G. The authors acknowledge infrastructural support provided by the University of Chicago Research Computing Center and University of Colorado Next Generation Sequencing Facility.
Author information
Authors and Affiliations
Contributions
T.E.B., N.F., K.M.H. and J.A.G. conceived and designed the research studies. All authors contributed to the generation and analyses of the data. T.E.B., P.C. and N.F. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
Supplementary Figures 1–5, Supplementary Tables 1–4 and Supplementary References (PDF 803 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Brewer, T., Handley, K., Carini, P. et al. Genome reduction in an abundant and ubiquitous soil bacterium ‘Candidatus Udaeobacter copiosus’. Nat Microbiol 2, 16198 (2017). https://doi.org/10.1038/nmicrobiol.2016.198
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nmicrobiol.2016.198
This article is cited by
-
Long-term push–pull cropping system shifts soil and maize-root microbiome diversity paving way to resilient farming system
BMC Microbiology (2024)
-
A social niche breadth score reveals niche range strategies of generalists and specialists
Nature Ecology & Evolution (2023)
-
Ecological insights into soil health according to the genomic traits and environment-wide associations of bacteria in agricultural soils
ISME Communications (2023)
-
DNA sequencing, microbial indicators, and the discovery of buried kimberlites
Communications Earth & Environment (2023)
-
Taxonomic and environmental distribution of bacterial amino acid auxotrophies
Nature Communications (2023)
