
Abstract
Insect odorant receptors (ORs) comprise an enormous protein family that translates environmental chemical signals into neuronal electrical activity. These heptahelical receptors are proposed to function as ligand-gated ion channels and/or to act metabotropically as G protein-coupled receptors (GPCRs). Resolving their signalling mechanism has been hampered by the lack of tertiary structural information and primary sequence similarity to other proteins. We use amino acid evolutionary covariation across these ORs to define restraints on structural proximity of residue pairs, which permit de novo generation of three-dimensional models. The validity of our analysis is supported by the location of functionally important residues in highly constrained regions of the protein. Importantly, insect OR models exhibit a distinct transmembrane domain packing arrangement to that of canonical GPCRs, establishing the structural unrelatedness of these receptor families. The evolutionary couplings and models predict odour binding and ion conduction domains, and provide a template for rationale structure-activity dissection.
Similar content being viewed by others

Introduction
In almost all of the many millions of insect species, repertoires of several dozen to several hundred highly divergent odorant receptors (ORs) are responsible for detecting a myriad of volatile chemical signals in the environment1. By transducing binding of specific odours into sensory neuron activity, insect ORs play critical roles in mediating olfactory behaviours such as mosquito host seeking2, moth mate identification3 and drosophilid pathogen avoidance4.
Surprisingly, the insect OR signalling mechanism has been challenging to define5,6. Like G protein-coupled receptors (GPCRs), insect ORs contain seven putative transmembrane helices (TMHs)7,8. This observation prompted a long-standing supposition that the insect receptors—similar to their mammalian counterparts—act metabotropically8,9,10,11,12. However, insect ORs have the opposite membrane orientation to GPCRs and function as heteromeric complexes (of unknown stoichiometry) of an odour ligand-specific ‘tuning’ OR and a co-receptor, ORCO (formerly called OR83b)13,14,15,16. Moreover, functional analyses in heterologous cells provided evidence that OR/ORCO complexes act as odour-gated cation channels11,17,18,19. However, the lack of similarity of insect ORs to known ion channel classes has made it difficult to dissect the molecular basis of this ionotropic function20.
An important way to help resolve the mechanistic basis of insect OR signalling would be to obtain three-dimensional (3D) structural information of these receptors. Unfortunately, recombinant expression of these polytopic transmembrane proteins is technically very challenging21, which has precluded X-ray crystal structure determination. Furthermore, because insect ORs have no significant similarity to sequences of known 3D structure, homology modelling22,23 is not possible. A complementary approach to predict both protein structure and functionally important sites is to use information contained within sequence variation among the members of large protein families24. Interactions between pairs of amino acids that are important for protein structure and function impose evolutionary constraints on the sets of mutations acceptable at interacting sites25. Reciprocally, identification of such evolutionary couplings (ECs) within primary sequences of isostructural proteins can provide information on distance restraints between amino acid pairs and thereby insight into higher-order structure and functional domains25. Identified coevolving residues can also, depending on the analysis method25, reflect distally coupled residues important for allosteric communication within proteins26,27. Here, we applied this approach to identify ECs across the large insect OR family, which we use to build de novo the first 3D models of these proteins and identify functionally important sites.
Results
We used an extended version of the EVfold-transmembrane method28 (Fig. 1a) to compute ECs for selected insect ORs, starting with multiple sequence alignments containing a set of 5,907 known and newly annotated receptors (Supplementary Table 1 and Supplementary Data 1–4). From these alignments, we extracted patterns of amino acid coevolution to produce a ‘contact map’ of evolutionarily constrained pairs of residues along the OR sequence (Fig. 1b, Supplementary Fig. 1 and Supplementary Data 5–8). These ECs were used in two ways: first, to probe functional sites, as evolutionary co-conservation reveals selection pressures beyond single position conservation (Supplementary Data 9 and 10); second, to produce 3D models by combining ECs with secondary structure predictions to fold the OR polypeptide (Fig. 1c and Supplementary Data 11–14).

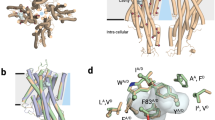
(a) Left: the structural proximity of residues in a protein leaves a visible record of amino acid covariation in the protein family sequence alignment due to the evolutionary pressure to maintain favourable interactions. By analysing these patterns with a statistical model of sequence coevolution, ECs between residue pairs can be inferred and used to predict 3D contacts. Right: schematic of the EVfold-transmembrane prediction workflow (see Methods). A multiple alignment of insect OR protein sequences is used to identify ECs between pairs of coevolving amino acids with EVfold-PLM. After discarding pairs that are inconsistent with the predicted membrane topology and secondary structure, the remaining highest-ranking EC pairs are used as distance restraints on an extended polypeptide to fold a set of 3D OR models. (b) Contact map representation of the top 200 predicted ECs for D. melanogaster OR85b. The axes represent the indices along the OR85b primary sequence, along which predicted TMH segments are annotated as blue bars and predicted helical secondary structure as grey bars. Black dots represent ECs between pairs of residues; the representation is mirror-symmetric along the diagonal. The lines of ECs parallel and anti-parallel to the diagonal of the contact map are characteristic of the helix packing arrangements observed in alpha-helical transmembrane proteins28; the dashed orange lines highlights one of these between TMH1 and TMH2. Three high-density regions of ECs within the N-terminal tail, EL2 and IL3 are highlighted by red dashed circles (see also Fig. 3a). (c) Top-ranked predicted 3D structural models for OR85b (left, model 140_12) and ORCO (right, model 310_2; the ORCO-specific IL2 insertion is not modelled) viewed from within the membrane bilayer (side view) and from the extracellular face (top view). Models are colour coded from N-terminus (blue) to C-terminus (red). As expected for members of the same protein family, the 3D models agree in their overall helical packing arrangement; the observed differences may be caused by actual structural differences of different subfamilies in the OR protein family sequence space and/or by inaccuracies in sequence alignment, statistical inference and 3D modelling.
We used Drosophila melanogaster OR85b (a tuning OR that responds to 2-heptanone and several esters29,30) and ORCO as anchors for these analyses to obtain models for both a ligand-specific OR and the co-receptor. ORs and ORCO have the same transmembrane topology16,17,31 and can—with the exception of an ORCO-specific ~70 amino acid insertion in the intracellular loop 2 (IL2)—be confidently aligned over their whole length (Supplementary Data 2). We therefore expect OR85b and ORCO to share the same overall 3D fold and our predicted ECs and structural models to be similar. However, differences in alignments arising from distinct anchor sequences that cover different parts of the sequence space of the protein family may capture subfamily-specific restraints25.
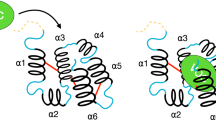
Examination of the predicted contact maps for OR85b and ORCO revealed a number of lines of ECs parallel or perpendicular to the diagonal within predicted TMHs (Fig. 1b, orange dashed box, and Supplementary Fig. 1), which are characteristic of parallel and anti-parallel helix arrangements in alpha-helical membrane proteins28. This observation provides one validation of our approach as the EC analysis is given no prior knowledge that insect ORs are transmembrane proteins. Importantly, the predicted helical contacts bear no resemblance to those of GPCRs or to those of the adiponectin receptor 1, a seven TMH protein that has the same membrane orientation as insect ORs32, but which appears to have a convergent helical arrangement to that of GPCRs (Fig. 2)28. These observations indicate that insect ORs adopt a seven TMH packing arrangement that is distinct from GPCR structures. Thus, the shared heptahelical secondary structure of these receptor families is, in contrast to initial assumptions8,9, likely to be coincidental and not necessarily indicative of a shared signalling mechanism.

Top: comparison of the helical packing arrangements of insect ORs, a GPCR and the adiponectin receptor 1 (AR1); the structures are colour coded from N-terminus (blue) to C-terminus (red). The OR is the top-ranked model of OR85b (see Fig. 1c); the GPCR is the crystal structure of the β2-adrenergic receptor (PDB 2RH1 (ref. 55)); the AR1 structure is the top-ranked model described previously28. As the N-termini of ORs and AR1s are located in the cytosol, but the N-terminus of GPCRs is located extracellularly (or lumenally), the GPCR structure is shown in the opposite orientation so that the packing arrangements are visually comparable. Structures were rotated so that the positioning of TMH1 and TMH2 agrees, as far as possible, between the different molecules. Bottom: corresponding simplified two-dimensional representations of the helical contacts highlighting the difference between the helical packing arrangement of insect ORs when compared to GPCRs and AR1s.
In analyses with both OR85b and ORCO, three regions of the sequences contain patches of residues with multiple, strong ECs (N-terminal tail, extracellular loop 2 (EL2) and intracellular loop 3 (IL3)/TMH7) (Fig. 1b and Supplementary Fig. 1, red dashed circles, and Fig. 3a). These EC clusters suggest that these regions are under strong evolutionary constraint and are therefore predictive of functional importance28. Remarkably, although molecular insights into insect ORs are still in their infancy—and dispersed across several different ‘model’ receptors—experimental validation of the importance of all three regions is available. A single amino acid mutation in the N-terminus of the D. melanogaster pheromone receptor OR67d (C23W) abolishes receptor function in vivo33; mapping of this residue onto the contact map by identifying the homologous position in OR85b (F11) and ORCO (A23) revealed it to lie within the N-terminal cluster of ECs (Fig. 1b, Fig. 3a, Supplementary Fig. 1 and Supplementary Table 2), suggesting that this segment fulfils an important role in all ORs; we test this prediction below. The second patch of EC-enriched residues, in EL2, contains a number of predicted beta-turns separated by conserved proline residues; mutation of these, and adjacent residues, in several different ORs strongly or completely disrupts function34. Although our analysis is unable to predict the precise conformation of this loop, the presence of strongly constrained residues in this region is consistent with the detrimental effect of their mutation. Last, IL3 forms at least part of the molecular interface for assembly of ORCO and ligand-specific ORs into heteromeric complexes16, which may explain the very high frequency of strong ECs within this region (Fig. 3a). We are currently unable to distinguish ECs that mediate intramolecular versus inter-subunit interactions in these receptors. Nevertheless, a systematic mutational analysis of cysteine residues in D. melanogaster ORCO revealed a particularly important functional requirement for those cysteines in IL3 (ref. 35) (Supplementary Table 2). In summary, although these observations do not validate the ECs and structural models per se, the striking coincidence of known functionally important sites with highly coupled residues gives basic confidence in our results.

(a) Positions with above-average EC strength (top 25% of sites, blue spheres) on the top-ranked model of OR85b (140_12; Supplementary Data 13; membrane-integral side view). Strongly coupled residues cluster around three regions of the model: (i) N-terminus, (ii) EL2, (iii) IL3 and along the span of TMH7. Many of the strongly coupled TMH7 residues do not feature in the contact map (Fig. 1b), because they were excluded as structural contacts with our standard transmembrane clash filter. (b) Experimentally characterized residues in different OR and ORCO proteins (spheres coloured by different shades of blue according to functional categorization; Supplementary Table 2) mapped onto the 3D model of OR85b (140_12), based on a sequence alignment of OR and ORCO sequences (Supplementary Data 2). Among many residues whose mutation have general deleterious effects on ion channel function, only a few residues influence ion selectivity (which are strong candidates for pore-lining residues), including two sites in ORCO (double asterisks marking residues at the extracellular end of TMH6 and TMH7) and two in a tuning OR (B. mori OR1) (single asterisks marking residues at the intracellular end of TMH5 and TMH6) (see text and Supplementary Table 2 for details).
To validate the predictive power of our ECs in identifying important residues in insect ORs, we focussed on the N-terminal cytoplasmic region of ORCO, whose role (if any) is unknown. We generated a version of this receptor (ORCO6Mut) bearing mutations in six of the top-ranked constrained residues in the sequence of D. melanogaster ORCO (A23, M24, F30, M31, H32 and N33) (Supplementary Data 10); several of these residues feature as pairs in the top-ranked ORCO ECs (that is, M24-F30, M24-M31 and M24-H32; Supplementary Data 6). We also generated an ORCO mutant bearing a small deletion spanning these residues (ORCOΔ23–33). Wild-type and mutant ORCO proteins were expressed in Xenopus oocytes for functional analysis by two-electrode voltage clamp recording of ligand-evoked current responses20. We first co-expressed ORCO with OR47a or OR85b, whose most potent agonists are pentyl acetate and 2-heptanone, respectively18,30,36. Wild-type ORCO co-expressed with these receptors can reconstitute dose-dependent odour-evoked currents (Fig. 4a,b). By contrast, ORCO6Mut and ORCOΔ23–33 have highly diminished or abolished response to these odours, respectively (Fig. 4a,b). In a second set of experiments, we tested current responses of wild-type and mutant ORCOs to VUAA1, an artificial small-molecule agonist that can activate this co-receptor when expressed alone (without a tuning OR partner)37. Wild-type ORCO produced robust currents on VUAA1 presentation, but neither mutant protein responded to this ligand (Fig. 4c). Taken together, our computational and experimental data support a previously unappreciated function for the ORCO N-terminus; that the tuning receptor OR67d is also sensitive to a mutation in this region33 suggests that this sequence may have a conserved role across the OR family. Determining the precise contribution of the N-terminus to receptor function—for example, in folding, trafficking and/or complex assembly—will nevertheless require further experimentation beyond the scope of this study.

(a–c) Left: representative whole-cell current traces to the indicated stimuli with two-electrode voltage-clamp in Xenopus oocytes injected with cRNAs for the indicated combinations of wild-type or mutant D. melanogaster ORs. ORCO6Mut contains amino acid substitutions in the six top-ranked N-terminal residues (A23S, M24A, F30A, M31A, H32A and N33A); ORCOΔ23–33 bears a deletion of this region (A23–N33). Ligand solutions were applied for 3 s (arrowheads). Right: quantification of current amplitudes (mean±s.e.m.; n=5 oocytes for wild-type ORCO and ORCO6Mut; n=4 for ORCOΔ23–33).
Finally, we asked whether our analysis could provide insight into the proposed function of insect ORs as ligand-gated ion channels. Four studies have experimentally defined mutations that affect (directly or indirectly) ligand recognition properties of different receptors30,38,39,40 (Supplementary Table 2). Notably, when mapped onto the OR85b model, these cluster in the external half of the TM2-4 region (Fig. 3b), suggesting that this region comprises a part of a ligand-binding pocket. The proposition that the relatively long EL2 loop might form a ‘lid’ that could cover/uncover this pocket34 is inline with the location of these residues.
Although ORs do not bear strong similarity to known ion channels, previous characterization of a sequence suggested to bear some resemblance to a K+ channel selectivity filter in D. melanogaster ORCO TMH6 (T393VVGYLG399) demonstrated a role in controlling K+ permeability11 (Supplementary Table 2). Independently, a systematic screen of residues in Bombyx mori ORCO showed mutation of Y464 (in TMH7)—and the equivalent Y478 in D. melanogaster ORCO—affected K+ selectivity20. Strikingly, this residue lies directly opposite the TVVGYLG sequence in our model, suggesting that this region forms a key part of the pore (Fig. 3b, double asterisks). Channel properties depend on both ORCO and the tuning OR, indicating that both subunits contribute to formation of the ion-conducting passage18,20,41,42. We therefore mapped the position of additional residues that have direct or indirect effects on channel function20 (Supplementary Table 2). The majority of these are in TMH5-7 region (Fig. 3b), consistent with this region forming a central part of the ion-conducting channel; notably, the only two of these residues that have effects on K+ ion selectivity (D299 and E356 in B. mori OR1; ref. 20)—as opposed to more general deleterious effects on ion channel function—are also located directly opposite to each other at the cystosolic end of TMH5 and TMH6 (Fig. 3b, single asterisks). Future determination of the stoichiometry of the heteromeric complex and identification of inter-subunit ECs may permit modelling of its quaternary structure to visualize how this C-terminal region forms the ion-conducting channel. The function of these receptors in odour-evoked signalling clearly depends on other regions too, such as the N-terminus and EL2.
Discussion
Our analysis of ECs in insect ORs has allowed us to build the first 3D models of this receptor family. These proteins appear to define a novel fold, as searches with DALI43 did not reveal highly significant similarity of our models to experimentally determined membrane protein structures (data not shown). It is, however, important to keep in mind that our current models are coarse grained, and the accuracy of the folded 3D structures is limited by multiple factors, including: (i) the availability of OR sequences, (ii) accurate secondary structure predictions and (iii) the confounding of monomer folding by the presence of multimer contacts, conformational plasticity and functional couplings, which are not easily disambiguated. As additional receptor sequences become available and as the EVfold method itself is further developed to fold multimers, future OR models—which we plan to update on the EVfold website ( http://evfold.org)—will be more accurate. It will be of particular interest to determine whether ORCO and ligand-specific ORs have distinct structural features. For ORCO-specific predictions, many hundreds of additional ORCO sequences from other insects are required. Similarly, increased sequence availability and improvement of alignments will aid analysis of the divergent tuning ORs. Nevertheless, the current models are both consistent with available and new experimental data and offer a number of predictions—notably, through revelation of highly constrained residues (Supplementary Data 9 and 10)—to guide and visualize structure–function analyses of these unusual sensory receptors. Moreover, these predictions may be the first step to assist in the design of novel pharmacological reagents to manipulate OR function, and thereby control olfactory behaviours of pest insects.
Methods
Annotation of OR sequences
New OR sequences from nine additional drosophilid genomes (Supplementary Table 1 and Supplementary Data 1) were identified by TBLASTN44 using D. melanogaster OR protein sequences as queries.
Multiple sequence alignment
All identified OR sequences were compiled into a FASTA database (Supplementary Data 1) and aligned iteratively with either D. melanogaster OR85b or D. melanogaster ORCO as reference sequences using jackhmmer with default parameters45. In the ORCO-anchored alignment, homologues that split into two separate aligned domains (due to the presence of the ORCO-specific insertion in IL2) were re-merged using custom Python scripts. In the OR85b-anchored alignment, the inclusion E-value (1E-40) was manually chosen to maximize EC contrast over residual background couplings. In both cases, alignment columns with >50% gaps were excluded from EC analysis as described28.
ECs inference from sequence variation
ECs were calculated with an updated version of our protocol (EVfold-PLM) based on a pseudolikelihood maximization statistical inference procedure46,47,48 instead of the original mean-field approximation24. Sequences in the alignment were downweighted at a 90% identity threshold to reduce the influence of sampling bias on statistical inference. Webserver and code for EC calculations are available on evfold.org.
De novo folding from protein sequences
The folding protocol is described in more detail elsewhere24,28. In brief, ECs were filtered using a combination of predicted transmembrane topology and predicted secondary structure. Transmembrane topology was predicted using PolyPhobius49 and validated by consensus prediction using TOPCONS50 and MEMSAT-SVM51. Secondary structure was predicted using PSIPRED52 and low confidence predictions of helix or beta-strand in ORCO (reliability index <3) were set to coil. Increasing numbers of top-ranked ECs were then used to fold D. melanogaster OR85b and ORCO from an extended polypeptide with CNS distance geometry and simulated annealing53. Local secondary structure was constrained based on the predicted topology and secondary structure. Due to the lack of alignment coverage on the ORCO-specific insertion in IL2 (residues 247–329), this region was not modelled to avoid interference with the folding of the membrane integral domain.
Clustering and ranking of predicted models
All generated protein models were clustered and ranked using the default quality assessment protocol of EVfold-transmembrane24,28. The ranking procedure uses a score composed of the agreement of each model with predicted lipid exposure, predicted secondary structure, and ECs. Models with structural knots are excluded from consideration as top-ranked models. Additional structure-based clustering ensures that high-scoring outlier structures can be excluded from consideration as confident predictions.
Model visualization and annotation
Protein models were visualized and annotated using PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC. To annotate functionally analysed residues of different ORs (Supplementary Table 2) on the common model, the equivalent positions in OR85b and ORCO were determined using the jackhmmer multiple sequence alignment anchored around OR85b (Supplementary Data 2).
Functional analysis of ORs in Xenopus oocytes
Mutant orco plasmid constructs were generated by standard cloning procedures; primer sequences are as follows: ORCO6Mut (forward: 5′-GCGCGAATTCGCCACCACCATGACAACCTCGATG-3′, reverse: 5′-CAGGCCGGAGTACTTCGCCGACCGGATGTTGGGCAT-3′; and forward: 5′-AAGTACTCCGGCCTGGCCGCGGCCGCCTTCACGGGCGGCAGT-3′, reverse: 5′-GCGCCTCGAGTTACTTGAGCTGCAC-3′) and ORCOΔ23–33 (forward: 5′-TTCACGGGCGGCAGTGCCTTC-3′, reverse: 5′-CCGGATGTTGGGCATCAGGTC-3′). cRNAs were synthesized from linearized modified pSPUTK vector54 containing wild-type or mutant versions of D. melanogaster ORCO, OR47a or OR85b. Stage V–VII oocytes were treated with 2 mg ml−1 of collagenase B (Roche Diagnostics, Tokyo, Japan) in Ca2+-free saline solution (82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2 and 5 mM HEPES, pH 7.5) for 1–2 h at 18 °C. Oocytes were microinjected either with 6.25 ng cRNA encoding a ligand tuning OR (OR47a or OR85b) and 6.25 ng cRNA encoding ORCO (Fig. 4a,b) or 12.5 ng of ORCO cRNA (Fig. 4c). Injected oocytes were incubated for 3–4 days at 18 °C in Barth’s solution supplemented with 84.7 mg l−1 of gentamycin. Whole-cell currents were recorded using the two-electrode voltage-clamp technique as previously described3. Intracellular glass electrodes were filled with 3 M KCl. Signals were amplified with an OC-725C amplifier (Warner Instruments, Hamden, CT, USA), low-pass filtered at 50 Hz and digitized at 1 kHz. Ligand solution was delivered via a silicon tube connected to a computer-driven solenoid. Before experiments, pentyl acetate (CAS: 628-63-7; Wako, Osaka, Japan) and 2-heptanone (CAS: 110-43-0; TCI, Tokyo, Japan) were directly diluted into the bath solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM BaCl2 and 10 mM HEPES, pH 7.2). VUAA1 (CAS: 525582-84-7; Vitas-M Laboratory, Moscow, Russian Federation) was prepared in dimethylsulphoxide as 20 mM, 66.7 mM or 200 mM stock solutions, which were then diluted into the bath solution at 0.15% to give the final desired concentration.
Additional information
How to cite this article: Hopf, T. A. et al. Amino acid coevolution reveals three-dimensional structure and functional domains of insect odorant receptors. Nat. Commun. 6:6077 doi: 10.1038/ncomms7077 (2015).
References
Su, C. Y., Menuz, K. & Carlson, J. R. Olfactory perception: receptors, cells, and circuits. Cell 139, 45–59 (2009).
Degennaro, M. et al. orco mutant mosquitoes lose strong preference for humans and are not repelled by volatile DEET. Nature 498, 487–491 (2013).
Nakagawa, T., Sakurai, T., Nishioka, T. & Touhara, K. Insect sex-pheromone signals mediated by specific combinations of olfactory receptors. Science 307, 1638–1642 (2005).
Stensmyr, M. C. et al. A conserved dedicated olfactory circuit for detecting harmful microbes in Drosophila. Cell 151, 1345–1357 (2012).
Benton, R. Chemical sensing in Drosophila. Curr. Opin. Neurobiol. 18, 357–363 (2008).
Nakagawa, T. & Vosshall, L. B. Controversy and consensus: noncanonical signaling mechanisms in the insect olfactory system. Curr. Opin. Neurobiol. 19, 284–292 (2009).
Vosshall, L. B., Amrein, H., Morozov, P. S., Rzhetsky, A. & Axel, R. A spatial map of olfactory receptor expression in the Drosophila antenna. Cell 96, 725–736 (1999).
Clyne, P. J. et al. A novel family of divergent seven-transmembrane proteins: candidate odorant receptors in Drosophila. Neuron 22, 327–338 (1999).
Hill, C. A. et al. G protein-coupled receptors in Anopheles gambiae. Science 298, 176–178 (2002).
Sakurai, T. et al. Identification and functional characterization of a sex pheromone receptor in the silkmoth Bombyx mori. Proc. Natl Acad. Sci. USA 101, 16653–16658 (2004).
Wicher, D. et al. Drosophila odorant receptors are both ligand-gated and cyclic-nucleotide-activated cation channels. Nature 452, 1007–1011 (2008).
Stengl, M. & Funk, N. W. The role of the coreceptor Orco in insect olfactory transduction. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 199, 897–909 (2013).
Lundin, C. et al. Membrane topology of the Drosophila OR83b odorant receptor. FEBS Lett. 581, 5601–5604 (2007).
Neuhaus, E. M. et al. Odorant receptor heterodimerization in the olfactory system of Drosophila melanogaster. Nat. Neurosci. 8, 15–17 (2005).
Larsson, M. C. et al. Or83b encodes a broadly expressed odorant receptor essential for Drosophila olfaction. Neuron 43, 703–714 (2004).
Benton, R., Sachse, S., Michnick, S. W. & Vosshall, L. B. Atypical membrane topology and heteromeric function of Drosophila odorant receptors in vivo. PLoS Biol. 4, e20 (2006).
Smart, R. et al. Drosophila odorant receptors are novel seven transmembrane domain proteins that can signal independently of heterotrimeric G proteins. Insect Biochem. Mol. Biol. 38, 770–780 (2008).
Sato, K. et al. Insect olfactory receptors are heteromeric ligand-gated ion channels. Nature 452, 1002–1006 (2008).
Silbering, A. F. & Benton, R. Ionotropic and metabotropic mechanisms in chemoreception: ‘chance or design'? EMBO Rep. 11, 173–179 (2010).
Nakagawa, T., Pellegrino, M., Sato, K., Vosshall, L. B. & Touhara, K. Amino acid residues contributing to function of the heteromeric insect olfactory receptor complex. PLoS ONE 7, e32372 (2012).
Carraher, C., Nazmi, A. R., Newcomb, R. D. & Kralicek, A. V. Recombinant expression, detergent solubilisation and purification of insect odorant receptor subunits. Protein Expr. Purif. 90, 160–169 (2013).
Sander, C. & Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 9, 56–68 (1991).
Zhang, Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 18, 342–348 (2008).
Marks, D. S. et al. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 6, e28766 (2011).
Marks, D. S., Hopf, T. A. & Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 30, 1072–1080 (2012).
Lockless, S. W. & Ranganathan, R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–299 (1999).
Suel, G. M., Lockless, S. W., Wall, M. A. & Ranganathan, R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10, 59–69 (2003).
Hopf, T. A. et al. Three-dimensional structures of membrane proteins from genomic sequencing. Cell 149, 1607–1621 (2012).
Hallem, E. A., Ho, M. G. & Carlson, J. R. The molecular basis of odor coding in the Drosophila antenna. Cell 117, 965–979 (2004).
Nichols, A. S. & Luetje, C. W. Transmembrane segment 3 of Drosophila melanogaster odorant receptor subunit 85B contributes to ligand-receptor interactions. J. Biol. Chem. 285, 11854–11862 (2010).
Tsitoura, P. et al. Expression and membrane topology of Anopheles gambiae odorant receptors in lepidopteran insect cells. PLoS ONE 5, e15428 (2010).
Yamauchi, T. et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769 (2003).
Jin, X., Ha, T. S. & Smith, D. P. SNMP is a signaling component required for pheromone sensitivity in Drosophila. Proc. Natl Acad. Sci. USA 105, 10996–11001 (2008).
Xu, P. & Leal, W. S. Probing insect odorant receptors with their cognate ligands: insights into structural features. Biochem. Biophys. Res. Commun. 435, 477–482 (2013).
Turner, R. M. et al. Mutational analysis of cysteine residues of the insect odorant co-receptor (Orco) from Drosophila melanogaster reveals differential effects on agonist- and odorant/tuning receptor-dependent activation. J. Biol. Chem. 289, 31837–31845 (2014).
Dobritsa, A. A., van der Goes van Naters, W., Warr, C. G., Steinbrecht, R. A. & Carlson, J. R. Integrating the molecular and cellular basis of odor coding in the Drosophila antenna. Neuron 37, 827–841 (2003).
Jones, P. L., Pask, G. M., Rinker, D. C. & Zwiebel, L. J. Functional agonism of insect odorant receptor ion channels. Proc. Natl Acad. Sci. USA 108, 8821–8825 (2011).
Pellegrino, M., Steinbach, N., Stensmyr, M. C., Hansson, B. S. & Vosshall, L. B. A natural polymorphism alters odour and DEET sensitivity in an insect odorant receptor. Nature 478, 511–514 (2011).
Leary, G. P. et al. Single mutation to a sex pheromone receptor provides adaptive specificity between closely related moth species. Proc. Natl Acad. Sci. USA 109, 14081–14086 (2012).
Hughes, D. T., Wang, G., Zwiebel, L. J. & Luetje, C. W. A determinant of odorant specificity is located at the extracellular loop 2-transmembrane domain 4 interface of an Anopheles gambiae odorant receptor subunit. Chem. Senses 39, 761–769 (2014).
Pask, G. M., Jones, P. L., Rutzler, M., Rinker, D. C. & Zwiebel, L. J. Heteromeric Anopheline odorant receptors exhibit distinct channel properties. PLoS ONE 6, e28774 (2011).
Nichols, A. S., Chen, S. & Luetje, C. W. Subunit Contributions to Insect Olfactory Receptor Function: Channel Block and Odorant Recognition. Chem. Senses. 36, 781–790 (2011).
Holm, L. & Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010).
Gertz, E. M., Yu, Y. K., Agarwala, R., Schaffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4, 41 (2006).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 7, e1002195 (2011).
Ekeberg, M., Lovkvist, C., Lan, Y., Weigt, M. & Aurell, E. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 87, 012707 (2013).
Balakrishnan, S., Kamisetty, H., Carbonell, J. G., Lee, S. I. & Langmead, C. J. Learning generative models for protein fold families. Proteins 79, 1061–1078 (2011).
Kamisetty, H., Ovchinnikov, S. & Baker, D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl Acad. Sci. USA 110, 15674–15679 (2013).
Kall, L., Krogh, A. & Sonnhammer, E. L. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21, (Suppl 1): i251–i257 (2005).
Bernsel, A., Viklund, H., Hennerdal, A. & Elofsson, A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 37, W465–W468 (2009).
Nugent, T. & Jones, D. T. Transmembrane protein topology prediction using support vector machines. BMC Bioinform 10, 159 (2009).
Jones, D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999).
Brunger, A. T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 (2007).
Katada, S., Nakagawa, T., Kataoka, H. & Touhara, K. Odorant response assays for a heterologously expressed olfactory receptor. Biochem. Biophys. Res. Commun. 305, 964–969 (2003).
Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007).
Acknowledgements
We are grateful to our colleagues for supplying OR protein sequence files, and thank Vanessa Ruta and members of the Benton lab for discussions and comments on the manuscript. T.A.H. and D.S.M. are supported by the National Institutes of Health (NIH R01 award GM106303). Research in K.T.’s laboratory is supported by the ERATO Touhara Chemosensory Signal Project from JST, Japan. Research in R.B.’s laboratory is supported by the University of Lausanne, European Research Council Starting Independent Researcher and Consolidator Grants (205202 and 615094) and the Swiss National Science Foundation (31003A_140869).
Author information
Authors and Affiliations
Contributions
T.A.H. developed analysis methods, performed multiple sequence alignments, EC calculations and model building, and contributed to the writing of the manuscript. D.S.M. developed analysis methods, analysed data and contributed to the writing of the manuscript. S.M. designed and performed the OR experimental analysis, and S.I. and K.T contributed to experimental design and interpretation. R.B. conceived the project, annotated gene sequences and models, analysed data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Figure, Tables, and References
Supplementary Figure 1, Supplementary Tables 1-2, and Supplementary References. (PDF 274 kb)
Supplementary Data 1
Most sequences were compiled from published sources (Supplementary Table 1). Additional drosophilid sequences were manually annotated as described in the Methods. Note the vast majority of OR sequences are computational predictions, and many are not full-length. Nevertheless, all alignable sequence fragments can be used to calculate ECs. (TXT 2278 kb)
Supplementary Data 2
Sequence alignment of modelled and functionally-annotated ORs. This alignment file was used to map functionally important residues defined in different ORs onto the common model illustrated in Fig. 3b. (TXT 3 kb)
Supplementary Data 3
D. melanogaster OR85b-anchored multiple sequence alignment of OR protein sequences. (TXT 1854 kb)
Supplementary Data 4
D. melanogaster ORCO-anchored multiple sequence alignment of OR protein sequences. (TXT 1826 kb)
Supplementary Data 5
Ranked list of ECs derived from the OR85b-anchored multiple sequence alignment. (XLSX 224 kb)
Supplementary Data 6
Ranked list of ECs derived from the ORCO-anchored multiple sequence alignment. (XLS 512 kb)
Supplementary Data 7
Predicted contact maps displaying increasing numbers of ECs (indicated in the file name; black dots in files with suffix "all" are EC pairs filtered as topology/secondary structure clashes); Fig. 1b shows the contact map with 200 ECs. The maps locate ECs relative to the OR85b sequence, but reflect ECs identified across all aligned insect ORs. (ZIP 1551 kb)
Supplementary Data 8
Contact maps of ORCO. Predicted contact maps displaying increasing numbers of ECs (indicated in the file name; black dots in files with suffix "all" are EC pairs filtered as topology/secondary structure clashes); Supplementary Fig. 1 shows the contact map with 200 ECs. (ZIP 2188 kb)
Supplementary Data 9
Highly constrained residues in insect ORs (OR85b-based). Residue positions with above-average coupling strengths were calculated based on ECs derived from the OR85b-anchored sequence alignment (Supplementary Data 5). ECs clashing with secondary structure were included in this calculation. (XLSX 42 kb)
Supplementary Data 10
Residue positions with above-average coupling strengths were calculated based on ECs derived from the ORCO-anchored sequence alignment (Supplementary Data 6). ECs clashing with secondary structure were excluded in this calculation. (XLS 60 kb)
Supplementary Data 11
Ranking of OR85b structure models. List of 600 OR85b 3D models ranked by EVfold-transmembrane quality assessment score (see Methods). To sample conformational search space sufficiently, 20 models were generated for each set of different numbers of ECs. (XLSX 93 kb)
Supplementary Data 12
Ranking of ORCO structure models. List of 700 ORCO 3D models ranked by EVfold-transmembrane quality assessment score (see Methods). To sample conformational search space sufficiently, 20 models were generated for each set of different numbers of ECs. (XLS 154 kb)
Supplementary Data 13
Structural coordinates in PDB format for the 600 OR85b models listed in Supplementary Data 11. (ZIP 37790 kb)
Supplementary Data 14
Structural coordinates in PDB format for the 700 ORCO models listed in Supplementary Data 12. (ZIP 43194 kb)
Rights and permissions
About this article

Cite this article
Hopf, T., Morinaga, S., Ihara, S. et al. Amino acid coevolution reveals three-dimensional structure and functional domains of insect odorant receptors. Nat Commun 6, 6077 (2015). https://doi.org/10.1038/ncomms7077
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms7077
This article is cited by
-
Nucleotide-based genetic networks: Methods and applications
Journal of Biosciences (2022)
-
Putative ligand binding sites of two functionally characterized bark beetle odorant receptors
BMC Biology (2021)
-
Altered functional properties of the codling moth Orco mutagenized in the intracellular loop-3
Scientific Reports (2021)
-
The structural basis of odorant recognition in insect olfactory receptors
Nature (2021)
-
Functional properties of insect olfactory receptors: ionotropic receptors and odorant receptors
Cell and Tissue Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
