
Abstract
Mitochondria contribute to cellular innate immunity against RNA viruses. Mitochondrial-mediated innate immunity is regulated by signalling molecules that are recruited to the mitochondrial membrane, and depends on the mitochondrial inner membrane potential (Δψm). Here we examine the physiological relevance of Δψm and the mitochondrial-associating influenza A viral protein PB1-F2 in innate immunity. When expressed in host cells, PB1-F2 completely translocates into the mitochondrial inner membrane space via Tom40 channels, and its accumulation accelerates mitochondrial fragmentation due to reduced Δψm. By contrast, PB1-F2 variants lacking a C-terminal polypeptide, which is frequently found in low pathogenic subtypes, do not affect mitochondrial function. PB1-F2-mediated attenuation of Δψm suppresses the RIG-I signalling pathway and activation of NLRP3 inflammasomes. PB1-F2 translocation into mitochondria strongly correlates with impaired cellular innate immunity, making this translocation event a potential therapeutic target.
Similar content being viewed by others

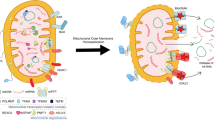

Introduction
Mitochondria, unique organelles that are compartmentalized by outer (MOM) and inner (MIM) membrane bilayers, are the cellular powerhouses of eukaryotes. In addition to their primary function as energy producers via the generation of ATP by aerobic respiration1, mitochondria participate in a wide variety of crucial cellular processes, such as apoptosis2,3, calcium homoeostasis4,5, cell signalling6 and ageing7.
Recent research has revealed that mitochondria act as a central hub for cellular innate immunity in vertebrates, particularly mammals8,9,10. The link between mitochondria and innate immunity was initially discovered based on the activation of the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) signal transduction pathway that leads to the production of type I interferons (IFNs) and proinflammatory cytokines as the first line of defence against RNA viruses. In this signalling pathway, cytoplasmic virus-derived RNA that gains entry into the host cell is recognized by RLRs, and the protein-nucleotide complex translocates to the mitochondrial antiviral signalling protein (MAVS)11, followed by the recruitment of various downstream signalling effectors on the MOM to form a supramolecular assembly essential for immune responses8,9,10. In addition to their involvement with host cell defence via the RLRs pathway, mitochondria are also involved in other types of inflammation, such as the activation of the NOD-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasomes. NLRP3 inflammasomes are activated by sensing mitochondrial reactive oxygen species12,13 that are generated through mitochondrial respiration, or by cytosolic mitochondrial DNA that are released from damaged organelles14, which results in the secretion of inflammasome-dependent cytokines, such as interleukin-1β (IL-1β) and IL-18. Although these mitochondrial-mediated immune responses are tissue/cell type-specific and primed by different stimuli, the convergence of multiprotein complexes on the MOM surface is a key process of each signalling event, thereby promoting the development of immune responses8,9,10,15.
Our previous studies demonstrated that the mitochondrial membrane potential (Δψm), a fundamental mitochondrial phenomenon, is essential to activate both the RLRs pathway and NLRP3 inflammasomes against viral infection16,17. An interesting aspect of these findings is that Δψm across the MIM and the supramolecular signalling assembly on the MOM platform that is topologically flanked by the biological membrane is coupled and activates the immune response in concert. However, the physiological relevance of Δψm in innate immunity has not been elucidated given that previous studies were mostly performed under chemical treatment using the protonophore, carbonyl cyanide m-chlorophenylhydrazone (CCCP)16,17.
PB1-F2 (ref. 18), an influenza A viral protein that is encoded in most influenza A viral strains in the +1 alternate open-reading frame of the RNA polymerase subunit (PB1 gene segment), can be expressed as several different polypeptide lengths with a full size of predominantly 87 or 90 amino acids19 (Supplementary Fig. 1a). An intriguing genomic feature of PB1-F2 is that the highly pathogenic strains (for example, H5N1 subtype) express a longer polypeptide that contains most of the 90 amino acids of PB1-F2, whereas the low pathogenic subtypes (for example, H1N1, with the exception of the 1918 pandemic strain) tend to express a shorter version containing 57 amino acids known as C-terminal truncated form (Supplementary Fig. 1b). Although the PB1-F2 derived from the A/Puerto-Rico/8 (denoted A/PR8 (H1N1)) strain, which has 87 amino acids (longer polypeptide), is known to localize in mitochondria18,20,21,22 and induce apoptosis18,22, the mechanisms by which it translocates into mitochondria and affects the immune response remain unclear. In the present study, we investigated the mechanistic and functional roles of PB1-F2 in mitochondrial-mediated innate immunity. We observed that PB1-F2 translocation into mitochondria leads to Δψm attenuation that strongly correlated with impaired cellular innate immunity.
Results
Distinct subcellular localization of PB1-F2 variants
We first confirmed the specificity of PB1-F2 in associating with mitochondria rather than other organelles, such as the endoplasmic reticulum (ER) and/or peroxisomes. Using a biochemical approach, we fractionated the cell extract from A/PR8-infected cells. The translated PB1-F2 polypeptide in infected HEK293 cells was predominantly observed in the heavy membrane fraction, which contains mitochondrial proteins, and not in the light microsomal membrane or cytosolic fractions (Fig. 1a). Because the infected cells also express other viral proteins that might bypass PB1-F2 targeting to the heavy membrane fraction, we next confirmed whether the protein alone was sufficient to be fractionated into the heavy membrane. As expected, a similar trend was observed in cellular extracts from transiently transfected cells (Supplementary Fig. 2; top panel, PR8). To verify that the observed results were not due to unique characteristics of the viral strain used, we repeated the same experiment using other PB1-F2 variants originating from a different strain (A/California(CA)/04/09 (H1N1) mutants)23 and demonstrated that those variants were also exclusively present in the heavy membrane fraction (Supplementary Fig. 2; 88W and 58W). By contrast, a C-terminal truncated version of PB1-F2 encoding 57 amino acids (denoted 12S), a major population of the low pathogenic subtype, was predominantly observed in the cytosolic fraction. Immunofluorescence microscopy supported these biochemical observations. Consistent with previous reports20,21, the full-length proteins (PR8, 88W and 58W) clearly localized to mitochondria (but not to other organelles, including the ER and peroxisomes; Fig. 1b), and the N66S polymorphism24 in PB1-F2 did not affect its subcellular localization (Supplementary Fig. 3). However, the 12S variant was diffusely localized to the cytoplasm (Fig. 1b). The observed affinity of the full-length PB1-F2 variants for mitochondria was further corroborated using an in vitro import assay (Fig. 1c).

(a) Cellular fractions from A/PR8 (16 HA U ml−1)-infected HEK293 cells were collected by differential centrifugation and analyzed by Western blotting using the indicated organelle markers. C, cytosolic fraction; H, heavy membrane fraction; L, light microsomal membrane fraction. (b) Subcellular localization of PB1-F2 variants. The indicated constructs (PR8, 88W, 58W and 12S) were expressed in HeLa cells, and immunofluorescence with PB1-F2 specific antibodies was used to detect expression (middle panels). Mitochondria in the same cells were identified by stable expression of mitochondrially targeted red fluorescent protein (Mito-RFP, left panels). The bottom four rows depict other organelle markers for the endoplasmic reticulum (ER-green and anti-PDI) and peroxisomes (anti-catalase). Scale bar, 10 μm. (c) Each full-length PB1-F2 variant was synthesized in vitro and incubated with isolated mitochondria (Mt) from HEK293 cells for the indicated times at 25 °C. The reactants were then treated with proteinase K and analyzed by Western blotting. Bottom panel, CoxIV was used as a loading control.
Full-length PB1-F2 localizes to the inner membrane space
We then analyzed the sub-mitochondrial localization of the PB1-F2 variants. When mitochondria isolated from PB1-F2-expressing cells were treated with a high-salt buffer (1 M KCl), none of the variants were extracted from the pellet (Fig. 2a), suggesting that PB1-F2 did not associate with the MOM via electrostatic interactions. Hypotonic swelling of mitochondria also failed to release PB1-F2 into the supernatant (Fig. 2b), demonstrating that the protein was either in the matrix or integrated and/or tightly associated with the MIM. Sucrose density gradient centrifugation demonstrated that PB1-F2 co-fractionates with CoxIV, an intrinsic MIM protein (Fig. 2c). However, treatment of the mitochondrial pellet with an alkaline solution (pH 11.5) allowed us to extract PB1-F2 (partially) and unanchored mitochondrial proteins (Cyt c, AIF and Hsp60) from the soluble fraction (Fig. 2a; far right lanes). The behaviour of these proteins was distinct from that of the integrated MOM or MIM proteins (Mfn1, Mfn2, Tom20 and CoxIV).

(a) Isolated mitochondria from PB1-F2-expressing HEK293 cells were treated with either 1 M KCl or 0.1 M Na2CO3 (pH 11.5) for 30 min on ice. After centrifugation, the supernatant (S) and pellets (P) were analyzed by immunoblotting with antibodies against PB1-F2 or several mitochondrial proteins as indicated. (b) Mitochondria isolated from A/PR8 (16 HA U ml−1)-infected HEK293 cells were diluted into either regular (−SW) or hypotonic swelling (+SW) buffer and maintained on ice for 30 min. After centrifugation, the supernatant (S) and pellets (P) were analyzed by Western blotting using the indicated sub-mitochondrial markers. HtrA2 was used as a positive control for an unanchored IMS protein. CoxIV and mtHsp70 are MIM and matrix proteins, respectively. (c) Sub-mitochondrial fractionation of PB1-F2 (A/PR8) by sucrose density gradient centrifugation. After centrifugation, fractions (numbered) were analyzed by Western blotting with antibody against PB1-F2 or against indicated sub-mitochondrial markers. (d) The mitochondrial fraction isolated from A/PR8 (16 HA U ml−1)-infected HEK293 cells was treated with proteinase K (100 μg ml−1) in the absence or presence of 500 μg ml−1 digitonin (pre-permeabilized condition) for 15 min on ice. The reactants were developed by immunoblotting with antibodies against PB1-F2 or against several mitochondrial membrane markers as indicated. MOM proteins: MAVS, Mfn1, Mfn2 and Tom20. IMS protein: HtrA2. MIM proteins: AIF, OPA-1 and CoxIV. Matrix proteins: Tfam and mtHsp70. (e) HEK293 cells were co-transfected with 5 ng of NLuc-PB1-F2 (88W) expression plasmid with increasing amounts (0–200 ng) of either Venus(N)-PB1-F2 (88W) (open triangle), Venus(C)-PB1-F2 (88W) (open circle), or equal mass pairs of Venus(N)- and Venus(C)-PB1-F2 (88W) (filled square) expression plasmids and were analyzed 21 h later using a biomolecular fluorescence complementation BRET saturation assay. Inset figure depicts the dose-dependent complementation (Venus(N+C)-PB1-F2 (88W)) of Venus fluorescence signal. All data represent the mean values±s.d. (n=3 experiments).
We confirmed that PB1-F2 was resistant to proteinase K digestion in the absence of digitonin, whereas it was completely digested in the presence of a low digitonin concentration that specifically permeabilizes the MOM, excluding the possibility that PB1-F2 was localized to the matrix side (Fig. 2d). The viral protein topology was further confirmed using semi-permeabilized cells25 to monitor the accessibility of PB1-F2 antibodies. PB1-F2 was visible when the MOM was specifically permeabilized by a low digitonin concentration (compare Supplementary Fig. 4a,b), which was consistent with the proteolysis results. We then analyzed the structural characteristics of the conformation of mitochondrially targeted PB1-F2 using a bioluminescence resonance energy transfer (BRET)-based assay26 in live cells. The interaction between Venus- and NanoLuc luciferase (NLuc)-tagged PB1-F2 but not other Venus-tagged proteins was successfully monitored (Supplementary Fig. 5). This homotypic interaction combined with a complementation assay (biomolecular fluorescence complementation)26 revealed that the PB1-F2 complex is composed of more than three molecules (Fig. 2e). Taken together, these biochemical lines of evidence indicate that PB1-F2 constitutively localizes to the mitochondrial inner membrane space (IMS), is tightly associated with the MIM, and assembles into a highly ordered oligomer.
Tom40-dependent import of PB1-F2 into mitochondria
Having identified that PB1-F2 is imported into the mitochondrial IMS, we next investigated whether this protein could be transported via the translocase of the outer membrane (TOM) machinery of the host import system27. Using an RNA interference approach, we treated HeLa cells with representative TOM-specific (TOM22-, TOM40-, or TOM70-) small interference RNAs (siRNAs) and confirmed that the target Tom proteins were efficiently suppressed by >80% (Fig. 3a).

(a) HeLa cells were treated with either control siRNA or siRNA directed against representative TOM components (TOM22, TOM40 and TOM70) to evaluate the effect of PB1-F2 import into mitochondria. The silencing efficiency for each Tom protein was confirmed by immunoblotting with specific antibodies, and β-actin was used as a loading control. (b,c) Subcellular localization of (b) mitochondrially targeted enhanced GFP (Su9-eGFP) and (c) PB1-F2 (PR8) in the siRNA-treated HeLa cells (TOM40 and TOM70). In each in vivo import experiment, at least 150 cells were scored, and the green and red bars represent mitochondrial and cytosolic localization, respectively. All data represent the mean values±s.d. (n=3 experiments). Scale bar, 10 μm. N.D., not detected, **P<0.01 and ***P<0.001 (by unpaired t-test). (d) Immunofluorescence images of the subcellular localization of Su9-eGFP or PB1-F2 (PR8) in HeLa cells treated with either TOM22 or TOM20 siRNAs. Scale bar, 10 μm.
As expected, the knockdown of Tom40, which is a major MOM import channel, clearly inhibited the import of mitochondrial matrix-targeted enhanced green fluorescent protein (Su9-eGFP; Fig. 3b; middle images); however, the integration of MOM proteins (Omp-25, MARCH-V/MITOL and Mfn2) was unaffected by Tom40 depletion (Supplementary Fig. 6a–c) as reported previously28. We observed that the mitochondrial import of PB1-F2 was also greatly suppressed by Tom40 knockdown, resulting in a clear diffuse cytosolic distribution (Fig. 3c, middle images). To eliminate the possibility of off-target effects, we tested secondary siRNAs against TOM40, and both pairs of oligos (TOM40_#B and TOM40_#C) caused similar impairment of PB1-F2 incorporation into mitochondria as observed with the Su9-eGFP construct (Supplementary Fig. 7a). By contrast, knockdown of Tom70, which is an essential component of the translocation of MOM proteins with a multi-spanning transmembrane domain28, did not affect mitochondrial import of either Su9-eGFP or PB1-F2 (Fig. 3b,c, bottom images and Supplementary Fig. 7a), although the integration of the polytopic MOM proteins MARCH-V/MITOL and Mfn2, which were used as controls, was significantly inhibited by Tom70 depletion (Supplementary Fig. 6b,c).
Intriguingly, we demonstrated that PB1-F2 translocation into mitochondria did not rely on the canonical TOM machinery because the attenuation of endogenous Tom20 or Tom22 production, which are central import receptors that recognize the positively charged N-terminal presequence of proproteins, did not affect PB1-F2 localization in mitochondria (Fig. 3d and Supplementary Fig. 7b). Indeed, in silico analysis revealed the absence of the mitochondrial targeting sequence (MTS) in PB1-F2 proteins (Supplementary Fig. 8a), and substitutions of basic residues predicted to be essential for mitochondrial targeting21 did not alter its translocation (Supplementary Fig. 8b). Specifically, the omission of several basic residues from the C-terminal region of PB1-F2 potentially destabilized the mutant proteins in vivo (Supplementary Fig. 8c). Taken together, these findings indicate that the translocation mechanism of the viral protein into mitochondria is distinct from the canonical host-protein import system that is generally mediated by Tom20/Tom22 receptors (and Tom70 for precursors with a hydrophobic presequence), which might account for the unique structural properties of the MTS in PB1-F2.
Defective mitochondrial dynamics in PB1-F2-expressing cells
In the course of examining PB1-F2 import into mitochondria, we found that the morphological balance of mitochondrial networks was extensively shifted towards fragmented structures in the A/PR8-infected cells compared with uninfected cells (Fig. 4a and Supplementary Fig. 9). When the cells were challenged against the A/CA/04/09 strain, which naturally lacks the PB1-F2 gene (Supplementary Fig. 1a; encoding only 11 amino acids), the vast majority of mitochondria exhibited morphologies similar to uninfected cells (Fig. 4a, bottom panels and Supplementary Fig. 9). We therefore quantified mitochondrial morphology in PB1-F2-expressing cells, and confirmed that >50% of the population exhibit fragmented mitochondria (Fig. 4b; red bar). This phenomenon was also observed in cells that transiently expressed full-length PB1-F2 variants, whereas Mock- and 12S variant-transfected cells exhibited no obvious abnormal phenotypes (Fig. 4c). Interestingly, the accumulation of punctate mitochondria in PB1-F2-expressing cells was unrelated to the expression level of mitochondrial GTPases (Drp-1, Mfn1 and Mfn2) involved in mitochondrial dynamics, with the exception of a slight reduction in long OPA-1 isoforms (L-OPA-1), which regulate mitochondrial fusion (Fig. 4d). These findings suggest that the accumulation of PB1-F2 in mitochondria causes defects in mitochondrial dynamics and raises the possibility that the physiological function of PB1-F2 is to modulate mitochondrial-mediated cellular functions.

(a) A549 cells were infected with either the A/PR8 (16 HA U ml−1) or A/CA/04/09 (multiplicity of infection (MOI) of 10) strains for 22 h, and the mitochondrial morphology of infected cells was monitored by immunofluorescence microscopy (left panels). Mitochondria in the same cells were stained with an anti-HtrA2 polyclonal antibody (middle panels). Images on the right depict a magnified version of the boxed areas in each middle image. To detect A/CA/04/09-infection, an anti-M2 monoclonal antibody was used. (b) Quantification of mitochondrial morphology in a. Cells were scored as one of the four morphological categories as depicted in the inset. In each infection experiment, at least 100 cells were scored. (c) HeLa cells were transfected with each PB1-F2 variant plasmid, and their mitochondrial morphologies were classified. In each transfection experiment, at least 100 cells were scored. (d) Protein expression levels (Drp-1, Mfn1, Mfn2 and OPA-1) in the A/PR8-infected (I) and uninfected (U) cells were analyzed by immunoblotting. CoxIV was used as a loading control. (e) A549 cells were infected with A/PR8 (16 HA U ml−1) or uninfected, and the cells were stained with MitoTracker Red CMXRos, which detects mitochondrial membrane potential (Δψm). The two right panels are magnified images of the boxed area in the infected cells (left panel), and the circles in the images are highlighted examples of mitochondria loss of Δψm. All data represent the mean values±s.d. (n=3 experiments). Scale bar, 10 μm. **P<0.01 and ***P<0.001 (by unpaired t-test), respectively.
Mitochondrially translocated PB1-F2 attenuates Δψm
We investigated Δψm in PB1-F2-expressing cells because reduction of L-OPA-1 by proteolytic processing is a Δψm-dependent process29. Although mitochondria in uninfected cells were uniformly stained with the Δψm-sensitive dye MitoTracker Red (Fig. 4e; top image), mitochondria in the A/PR8-infected cells displayed non-uniform staining, and these less intensely stained mitochondria likely correlated with PB1-F2 co-localization (bottom figures, circled areas). In line with these observations, HeLa cells transfected with full-length PB1-F2 variant plasmids exhibited similar patterns of widespread loss of Δψm, but these dimly stained mitochondria were not observed in cells expressing the 12S variant or in A/CA/04/09-infected cells (Supplementary Fig. 10).
Moreover fluorescence-activated cell sorting (FACS) analysis verified the relationship between Δψm and PB1-F2 expression because the fluorescence intensity of another Δψm-sensitive dye, tetramethylrhodamine methyl ester (TMRM), was significantly decreased in A/PR8-infected cells (Fig. 5a; red line in the right panel) compared with uninfected cells (black line). We hypothesized that this effect was due to the accumulation of PB1-F2 in mitochondria because cells transfected with plasmids encoding full-length PB1-F2 variants also retained the Δψm-decreased phenotype (Fig. 5b; PR8 and 88W), although other influenza A viral M2 proteins, a proton-selective ion channel, and 12S failed to demonstrate this effect (Fig. 5b and Supplementary Fig. 11a).

(a) HEK293 cells were infected with A/PR8 (16 HA U ml−1) for about 15 h, and the cells were stained with the cationic fluorescent dye TMRM and analyzed by flow cytometry (right panel; red trace). As a control, CCCP-treated cells (40 μM) were also analyzed (left panel). Grey histograms in both panels represent a profile of unstained cells. (b) Similar to a, except that HEK293 cells were transfected with the expression plasmid indicated inside the panel. Cells were analyzed by flow cytometry at 24 h post transfection. (c) The kinetics of Δψm disruption in A/PR8-infected J774A.1 macrophages. Cells infected with A/PR8 were collected at the indicated time points (0, 3, 6, 9, 15, 18 and 21 h), stained with JC-1 for 30 min, and analyzed by flow cytometry. The percentage of JC-1 reduction (y axis) is presented. The immunoblot on the right represents a profile of PB1-F2 expression at each time point as well as the loading controls β-actin and OPA-1. Five bands (a–e) of OPA-1 isoforms were detected by immunoblotting with the antibody against OPA-1, and bands a and b are a mixture of L-OPA-1 isoforms. (d) Δψm is dispensable for PB1-F2 translocation into mitochondria. HeLa cells transfected with either mitochondrial-targeted dihydrofolate reductase (mtDHFR) or PB1-F2 were treated with (+) or without (−) CCCP (40 μM), and their translocation into mitochondria was monitored by immunofluorescence microscopy (left images). Quantification is listed in the right score panel. In each transfection experiment, at least 100 cells were scored, and the green and red bars represent mitochondrial and cytosolic localization, respectively. All data represent the mean values±s.d. (n=3 experiments). Scale bar, 10 μm. N.D., not detected, **P<0.01 and ***P<0.001 (by unpaired t-test).
We then monitored the kinetic profile of PB1-F2 expression and its Δψm reduction in J774A.1 macrophages infected with A/PR8. The Δψm of the infected cells as detected by Mitoprobe JC-1 started to decrease at ~9 h post infection (Fig. 5c; left panel), and this profile matched the expression pattern of PB1-F2 (right blots) as well as L-OPA-1 processing within the cells (Fig. 5c, bands a and b; and Supplementary Fig. 11b). The translocation of PB1-F2 into mitochondria was not affected by CCCP-treatment (Fig. 5d) perhaps because the protein could not cross the MIM (Fig. 2). Taken together, these observations indicate that mitochondrially targeted PB1-F2 leads to the attenuation of Δψm.
PB1-F2 functions upstream in mitochondrial fission pathway
Given that the accumulation of PB1-F2 in mitochondria leads to remarkable mitochondrial fragmentation, we attempted to elucidate whether certain molecules involved in mitochondrial dynamics correlate with this phenomenon. PB1-F2-induced mitochondrial fragmentation was not observed in cells treated with Drp-1-specific siRNA, rather extensively elongated tubules were noted (Fig. 6a; middle row; PR8, 88W and 58W). These phenotypes indicate that the mitochondrial fission observed in wild-type cells occurs via a Drp-1-dependent pathway, and PB1-F2 acts upstream of Drp-1 in the cascade. We next tested the effect of PB1-F2 in cells with attenuated endogenous OMA-1, a mitochondrial zinc metallopeptidase, because the protease mediates L-OPA-1 degradation and negatively regulates mitochondrial fusion under stress conditions30. In these cells, mitochondria remain to be elongated structures, even in the presence of PB1-F2 variants, with no obvious punctate mitochondria (Fig. 6a,b). These results demonstrate that the viral protein acts upstream of OMA-1. Collectively, these findings highlight the mechanism for PB1-F2 involvement in the early pathway of mitochondrial fission (Fig. 6c) and potentially link this process to OMA-1 activation.

(a) Mitochondrial morphology in HeLa cells treated with control siRNA, Drp-1 siRNA or OMA-1 siRNA. Each siRNA-treated cell was transfected with PB1-F2 expression plasmids as indicated or treated with 20 μM CCCP (DMSO as a control). PB1-F2-expressing cells were identified by immunofluorescence, and mitochondria were visualized using an antibody against CoxIV. Insets depict the magnified images of each boxed area. Scale bar, 10 μm. (b) Quantification of mitochondrial morphology in a. Cells were scored as one of three morphological categories as shown in the inset. In each transfection experiment, at least 100 cells were scored. All data represent the mean values±s.d. (n=3 experiments). N.D., not detected. (c) Model of mitochondria fission induced by PB1-F2. We propose that the observed mitochondrial fragmentation occurs via a Drp-1-dependent pathway.
PB1-F2 inhibits the RIG-I signalling pathway
The aforementioned experiments indicated that the influenza A viral protein PB1-F2 has the potential to interfere with fundamental mitochondrial function. To evaluate the physiological relevance of this protein in host cells, we sought to determine the effect of PB1-F2 on a mitochondrial-mediated immune response, the RIG-I signalling pathway. We examined whether the mitochondrial translocation of PB1-F2 modulates the RIG-I-mediated activation of IFN-β and nuclear factor κB (NF-κB) reporter constructs. The overexpression of Myc-tagged MAVS, the downstream adaptor molecule of RIG-I, potently activated both IFN-β and NF-κB luciferase reporters (Fig. 7a,b; black bars) as described previously31. However, the co-expression of full-length PB1-F2 variants, but not 12S, with MAVS was sufficient to inhibit the MAVS-dependent activation of the RIG-I signalling pathway in a dose-dependent manner (Fig. 7a,b; PR8, 88W and 58W), even with equal amounts of MAVS expression (inset blots) or its mitochondrial localization (Supplementary Fig. 12). The inhibitory activities of PB1-F2 variants were also confirmed by stimulating cells transfected with a plasmid that expresses RIG-I, which is the upstream molecule of MAVS (Supplementary Fig. 13a). In addition, the delivery of poly(I:C) into HEK293 cells by transient transfection stimulated the phosphorylation of endogenous IFN regulatory factor 3 (IRF-3), a hallmark of IRF-3 activation, which was suppressed by full-length PB1-F2 variants (Supplementary Fig. 13b). Consistent with these findings, the production of the endogenous proinflammatory cytokine IL-6 in response to A/PR8 infection was substantially downregulated compared with the A/Beijing/262/1995 (H1N1) strain that encodes a 57 amino acid PB1-F2 (Supplementary Fig. 13c). Furthermore, the cells decreased Δψm due to the expression of full-length PB1-F2 variants were more susceptible to infection by a recombinant vesicular stomatitis virus than cells expressing 12S due to defective mitochondrial-mediated immunity (Supplementary Fig. 13d). Verifying that the observed inhibitory effects of PB1-F2 were not due to the induction of apoptosis under our experimental conditions, we confirmed that cells expressing full-length variants remained viable by examining cytochrome c release (Supplementary Fig. 14a) or activation of caspase-3 or poly (ADP-ribose) polymerase (PARP; Supplementary Fig. 14b).

(a,b) HEK293 cells were co-transfected with 75 ng of plasmid encoding Myc-tagged MAVS, increasing amounts (20, 50 and 100 ng) of plasmids encoding the PB1-F2 variants, and either the (a) IFN-β or the (b) NF-κB reporter plasmids. Transfected cells were analyzed 24 h later for reporter gene-dependent luciferase activities. In these assays, we used Su9-eGFP plasmids as the control. Western blots reveal the abundance of Myc-MAVS proteins. (c) HEK293 cells were co-transfected with 75 ng of plasmid encoding Myc-tagged RIG-I (1-250), 100 ng of PB1-F2 chimeric mutants, and IFN-β reporter plasmids. (d) HEK293 cells were co-transfected with 30 ng of plasmid encoding NLRP3, ASC (5 ng), procaspase-1 (5 ng), pro-IL-1β (150 ng) and each PB1-F2 variant (300 ng). Cell-free supernatants were collected 24 h post transfection, and secreted IL-1β was measured by ELISA. In these assays, we used the eGFP plasmid as the control. Inset: the functional role of the MOM-localized PB1-F2 chimeric mutant in NLRP3 inflammasomes. (e) The reconstituted level of secreted IL-1β in d was confirmed by immunoblotting with antibodies against human IL-1β and pro-IL-1β, and β-actin was used as a loading control in whole cell lysates. (f) The inhibition of ASC oligomerization by mitochondrial-targeted PB1-F2 variants. HEK293 cells transfected with plasmids encoding NLRP3, FLAG-tagged ASC, and each PB1-F2 variant were lysed and their lysates were cross-linked with BS3. The samples were analyzed by immunoblotting with antibody against FLAG. The bottom blots confirm the expression levels of NLRP3 and ASC proteins in their lysates. All data represent the mean values±s.d. (n=3 experiments). N.S., not significant, *P<0.05, **P<0.01, and ***P<0.001 (by unpaired t-test), respectively.
To test whether the topology of PB1-F2 in mitochondria is essential for its inhibitory effect on signalling events, we designed chimeric proteins expected to incorrectly localize to the MOM. We fused a C-terminal transmembrane segment from Omp-25, which is a MOM protein, with PB1-F2 variants and confirmed that these mutants (PR8-Omp25 and 88W-Omp25) were integrated into the MOM without attenuating Δψm (Supplementary Fig. 15a,b). Strikingly, neither of the chimeric proteins exhibited any functional effect on the RIG-I-mediated activation of IFN-β compared with intact proteins (Fig. 7c). We believe that PB1-F2-induced inhibition of the RIG-I pathway was not attributable to a jamming of MAVS oligomerization, because the presence of PB1-F2 variants did not interfere structurally with the MAVS–MAVS interaction (Supplementary Fig. 16). Together, these findings demonstrate that PB1-F2 acts downstream of MAVS activation and impairs RLRs-dependent antiviral signalling, further highlighting the importance of Δψm in upregulating MAVS-mediated signal transduction16,32.
PB1-F2 inhibits the activation of NLRP3 inflammasomes
Apart from the RIG-I signalling pathway, mitochondria also act as a platform for activating NLRP3 inflammasomes, which are involved in early stages of the inflammatory response by sensing cellular damage or stress, including viral infection12,13,14,17,33. The activation of NLRP3 inflammasomes is initiated via the formation of a multi-complex with an adaptor protein, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and procaspase-1. This association is followed by the processing of procaspase-1 to its mature form, which ultimately leads to IL-1β maturation34. We reconstituted NLRP3 activation by co-transfecting cells with NLRP3, ASC, procaspase-1 and pro-IL-1β expression plasmids and evaluated the secretion of IL-1β in vitro (Fig. 7d; control) as described previously35,36. In addition to the RIG-I pathway, the expression of full-length PB1-F2 variants (PR8, 88W, and 58W) significantly suppressed the secretion of IL-1β, similar to the uncoupling protein 2 ( used as a positive control17), whereas 12S abolished the effect (Fig. 7d,e) despite similar cytosolic pro-IL-1β expression level (Fig. 7e; middle blot). Again, MOM-localized PB1-F2 mutants were unaffected by the inactivation of NLRP3 inflammasomes (Fig. 7d inset figure, and Supplementary Fig. 15c). Interestingly, when cells were treated with TOM40 siRNA, the suppression of IL-1β induced by the full-length PB1-F2 variants was also less complete, resulting in the recovery of NLRP3 activation (Supplementary Fig. 15d). These findings demonstrate that mitochondrially targeted PB1-F2 acts as a negative regulator of NLRP3 inflammasomes.
We also examined ASC oligomerization, the hallmark assembly of activated NLRP3 inflammasomes, in the presence of PB1-F2 variants. Reduced accumulation of higher-order oligomers was exclusively observed when the mitochondrially targeted version of the variants was present (Fig. 7f). In addition, we confirmed that NLRP3 recruitment to mitochondria, an upstream event of NLRP3 inflammasome formation33, was prohibited by the presence of PB1-F2 (Supplementary Fig. 17).
Taken together, these data demonstrate that Δψm dissipation introduced by translocation of the influenza A virus PB1-F2 into mitochondria correlates with the severity of defective mitochondrial-mediated innate immunity, including the RIG-I signalling pathway and activation of NLRP3 inflammasomes.
Discussion
Some viruses encode genes that have the potential to suppress host cell immunity due to a viral evasion strategy. An example of such a gene product is the hepatitis C virus serine protease NS3/4A, which proteolytically cleaves MAVS and ultimately abolishes mitochondrial-mediated antiviral signalling37,38. The influenza A virus also encodes several gene products that aid in escaping immune responses, for example, nonstructural protein 1, an inhibitor of the RIG-I pathway through its binding to RIG-I39, and PB1-F2, which downregulates mitochondrial-mediated immune responses by attenuating Δψm as demonstrated in the present study.
The gene product PB1-F2, which is encoded by a +1 reading frame shift of PB1, is a unique polypeptide with naturally existing variants of various lengths ranging from 11 to 87 or 90 amino acids. It is curious that a highly pathogenic (H5N1) subtype and pandemic (1918, 1957 and 1968) strains of influenza A virus tends to encode longer polypeptides; thus, we presumed that this molecular signature might affect pathogenicity (Supplementary Fig. 1). In this regard, previous studies demonstrated that PB1-F2 with 87 amino acids colocalizes with mitochondria18,20,21,22, and its targeting is proposed to be caused by the C-terminal region of the polypeptide that acts as an MTS20,21 and is frequently missing in the low pathogenic subtypes (Supplementary Fig. 1). Consistent with these assumptions, we demonstrated that full-length versions of PB1-F2 from different viral strains specifically translocate into mitochondria, and we further revealed mechanistic insight into how the viral protein is incorporated into the organelle.
We first demonstrated that PB1-F2 localizes to the mitochondrial IMS and tightly associates with the MIM (Fig. 2). A sequence profile of PB1-F2, which has an extremely basic isoelectric point (~pKa>10), supports the notion that PB1-F2 has an electrostatic interaction with its negatively charged membrane. We next observed that the membrane-associated PB1-F2 forms a stable homotypic complex at the membrane and that the complex is a highly ordered assembly that contains more than three molecules (Fig. 2e). Given that the accumulation of PB1-F2 in mitochondria causes defects in mitochondrial function as described in the following section, this complex would presumably interact with other mitochondrial proteins. Finally, we clarified that PB1-F2 translocation into the mitochondria relied on a specific TOM component, the Tom40 import channel (Fig. 3). Interestingly, our in vivo import analysis clearly demonstrated that PB1-F2 bypasses the general import receptors Tom20/Tom22 and is targeted directly to the Tom40 channel likely by evading host cell surveillance during import. Collectively, our findings provide a mechanism for PB1-F2 import into mitochondria and localization in mitochondrial sub-compartments. Further studies are needed to identify other viral proteins (includes another influenza A viral protein) that have an affinity for mitochondria and to elucidate whether these proteins highjack the host import machinery.
Our results from influenza A-infected cells clearly demonstrate that PB1-F2 translocates into mitochondria, leading to a loss of Δψm, which results in abnormal fragmented mitochondria (Fig. 4; Supplementary Figs 9 and 10). The most likely explanation for the dissipation of Δψm by PB1-F2 is that the formation of positively charged PB1-F2 oligomers on the MIM leads to some functional defects in the respiratory chain complexes and ultimately to widespread heterogeneity of Δψm. Importantly, PB1-F2-induced mitochondrial fragmentation was not observed in cells in which either Drp-1 or OMA-1 were individually depleted by siRNA treatment, suggesting that mitochondrial fission occurs via a Drp-1-dependent pathway (Fig. 6). In particular, OMA-1-mediated processing of L-OPA-1 is a stress-related cellular response30, and thus it we speculate that mitochondrially targeted PB1-F2 might trigger OMA-1 activation and induce such punctate mitochondria.
We also revealed that cells with abnormal mitochondria exhibited defective mitochondrial-mediated innate immune responses, including activation of the RIG-I signalling pathway and NLRP3 inflammasomes (Fig. 7). These observations are interesting in light of our previous findings that cells completely lacking mitochondrial fusion due to targeted deletion of both Mfn1 and Mfn2 also have severely impaired immune responses against RNA viruses16. Arnoult and colleagues40 reported that OPA-1 knockdown negatively regulates virus-induced activation of the transcriptional factor NF-κB and depletes the fission factor Drp-1, thereby upregulating the RIG-I signalling pathway. Cellular stress, including viral infection, does not always induce mitochondrial fragmentation40,41, and thus our results highlight the physiological impact of influenza A viral evasion of host immunity via Δψm regulation in connection with mitochondrial dynamics. Although we believe that Δψm manipulation by PB1-F2 is important in relation to innate immunity, we cannot rule out the possibility that mitochondrially targeted PB1-F2 affects innate immunity via another unknown route. In addition, PB1-F2-induced mitochondrial dysfunction also potentially impacts the pathogenicity of secondary bacterial infections. McCullers and colleagues reported that A/PR8-challenged mice exhibit significant weight loss and have greater susceptibility to pneumonia caused by Streptococcus pneumoniae, a Gram-positive bacteria that causes secondary infection42. It is notable that the pathogenic phenotypes are diminished when using a truncated version of the PB1-F2 polypeptide42,43, which corresponded to the lack of the C-terminal region that is important for mitochondrial translocation. In general, secondary bacterial infections are associated with increased severity and lethality, and future studies aimed at evaluating the role of PB1-F2 in the differential susceptibility to pathogen infection may prove interesting.
Mitochondria are involved in various cellular processes and also appear to act as a platform for first-line innate immunity against RNA viruses. The findings of the present study reveal a role for mitochondrial targeting of PB1-F2 in immune evasion, and the inhibition of PB1-F2 translocation into the organelle might serve as a potential therapeutic target.
Methods
Reagents
CCCP was purchased from Sigma-Aldrich (St Louis, MO, USA). MitoTracker Red CMXRos, the MitoProbe JC-1 assay kit and TMRM were purchased from Molecular Probes/Invitrogen (Carlsbad, CA, USA), and bis(sulfosuccinimidyl) suberate (BS3) was obtained from Thermo Scientific (Rockford, IL, USA). Furimazine was supplied by Promega (Madison, WI, USA) and poly(I:C) was purchased from InvivoGen (San Diego, CA, USA). All other reagents were biochemical research grade.
Cell lines, viruses and animals
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO BRL) supplemented with 1% GlutaMAX, penicillin (100 U ml−1)–streptomycin (100 μg ml−1), and 10% bovine calf serum at 5% CO2 and 37 °C. HeLa, A549 and J774A.1 cell lines were maintained in DMEM medium supplemented with 1% GlutaMAX, penicillin (100 U ml−1)–streptomycin (100 μg ml−1), and 10% foetal bovine serum. Bone marrow-derived macrophages were prepared from the tibia and femur of mice, and the cells were cultured in DMEM supplemented with 10% foetal bovine serum, 1% L-glutamine and 30% L929 cell supernatant containing the macrophage colony-stimulating factor at 37 °C for 5 days as described previously44. The influenza virus A/PR8 strain (H1N1) was grown in allantoic cavities of 10-day-old fertile chicken eggs45 for 2 days at 35 °C, and the produced virus was stored at −80 °C until infection experiments. The influenza virus A/CA/04/09 strain (H1N1) was a generous gift from Professor Yoshihiro Kawaoka (the University of Tokyo). The animal experiments (six- to eight-week old female C57BL/6J mice, from The Jackson Laboratory) were approved by the Animal Committees of the Institute of Medical Science, The University of Tokyo.
Antibodies
The anti-PB1-F2 (A/CA/04/09 mutants; 88W, 58W and 12S; 1:500) antibody was generated by immunizing a rabbit with the N-terminal region of PB1-F2, which was supplied by peptide synthesis (MEQEQDTPWTQSTEHTNTQKRESGRQT). A monoclonal antibody against PB1-F2 (A/PR8 strain; 1:10) was kindly gifted by Viktor Wixler46 (the Münster University Hospital Medical School). Anti-Mfn1 (H-65, 1:1,000), anti-Tom40 (H-300, 1:1,000), anti-cytochrome c (H-104, 1:500), anti-calnexin (C-20, 1:1,000), anti-IRF-3 (FL-425, 1:1,000) and anti-Myc (A-14, 1:1,000) polyclonal antibodies and anti-AIF (E-1, 1:1,000), anti-β-actin (C4, 1:2,000), anti-Mfn2 (XX-1, 1:1,000) and anti-Tom20 (F-10, 1:500) monoclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-CoxIV (3E11, 1:300), anti-PDI (C81H6, 1:200) and phospho-IRF-3 (Ser396; 4D4G, 1:1,000) rabbit monoclonal antibodies and anti-Hsp60 (D307, 1:1,000), anti-Tfam (1:1,000), anti-PARP (1:1,000), anti-caspase-3 (1:1,000) and anti-cleaved caspase-3 (Asp175; 1:1,000) polyclonal antibodies were purchased from Cell Signalling Technology (Danvers, MA, USA). Anti-Tom22 (1C9-2, 1:1,000) and anti-FLAG (M2, 1:1,000) monoclonal antibodies were supplied by Sigma-Aldrich. Monoclonal antibodies against haemagglutinin (HA; HA.11, 1:1,000) and Myc (9E10, 1:1,000) were obtained from Covance (Princeton, NJ, USA), and a polyclonal antibody against HtrA2 (Omi; 1:200) was purchased from R&D Systems (Minneapolis, MN, USA). Anti-OPA-1 (1:1,000), anti-cytochrome c (1:500) and anti-Drp-1 (1:1,000) monoclonal antibodies were supplied by BD Biosciences (San Jose, CA, USA), and an anti-mitochondrial heat shock protein 70 (mtHsp70; 1:2,000) monoclonal antibody was obtained from Affinity BioReagents (Golden, CO, USA). Anti-influenza A virus M2 protein (1:1,000) monoclonal and human MAVS (1:1,000) and IL-1β (1:1,000) polyclonal antibodies were purchased from Abcam (Cambridge, MA, USA), and anti-human IL-1β (1:500) and anti-mouse IL-6 (1:500) antibodies used for ELISA were obtained from eBioscience (San Diego, CA, USA). The Alexa Fluor 488 (1:500) anti-mouse monoclonal and the Alexa Fluor 488 (1:500) and Alexa Fluor 568 (1:500) anti-rabbit polyclonal antibodies were obtained from Molecular Probes/Invitrogen, and the Cy3-conjugated sheep anti-mouse (1:1,000) monoclonal antibody was from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Plasmids
The PB1-F2 (PR8) encoding gene was obtained from A/PR8-infected HEK293 cells, and the complementary DNA (cDNA) was subcloned into a pcDNA3.1(−) (Invitrogen) vector. Plasmids expressing PB1-F2 variants (88W, 58W and 12S) were constructed by ligating the PB1-F2 amplimers, which were generated using pPolI/CA04-PB1-F2-stop88W23 (a gift from Professor Yoshihiro Kawaoka) as a template for PCR, into the pcDNA3.1(−). To construct the PB1-F2 (12S) plasmid with a N-terminal FLAG epitope tag, the PCR-amplified fragment was subcloned into the p3 × FLAG-CMV-10 (Sigma-Aldrich) vector. The PR8- and 88W-Omp25 expression plasmids were constructed by fusing a segment encoding the last 37 amino acids of murine Omp25 in-frame with each 3′ region of PB1-F2 cDNA. Fusion-tagged PB1-F2 (88W) plasmids used for the BRET saturation assay were constructed by ligating the PCR amplimer into vectors that encoded either an N-terminal Venus or NLuc (Promega) tag as described previously26. All PCR experiments were performed using PrimeSTAR DNA polymerase (Takara, Tokyo, Japan). The following primers (Supplementary Table 1) were used to amplify the PB1-F2 variants: PR8, TK797/TK798; 88W, TK754/TK755; 58W, TK754/TK780 and 12S, TK754/TK779. Plasmids encoding human NLRP3, ASC, caspase-1, pro-IL-1β, MAVS (Myc- and Venus tagged), uncoupling protein 2, RIG-I(1-250), Omp25 and matrix-targeted dihydrofolate reductase were described previously16,17,26,31,36. All constructs used in the present study were confirmed by DNA sequencing (ABI 3100).
Mitochondrial fractionation and proteolysis
Mitochondrial fractionation and proteolysis assays were performed as previously described26 with slight modification. Briefly, either virally infected (A/PR8) or plasmid-transfected HEK293 cells (22 h post infection/transfection) were washed once with cold 1 × phosphate-buffered saline (PBS; pH 7.2), scraped off the culture plate, and lysed in 800 μl of homogenization buffer (20 mM HEPES (pH 7.5), 70 mM sucrose and 220 mM mannitol) by 30 strokes in a Dounce homogenizer. The homogenate was centrifuged at 800 g for 5 min to precipitate the nuclei, and the resulting supernatant was further centrifuged at 10,000 g for 10 min (4 °C) to precipitate the crude mitochondrial fraction. The resulting supernatant was further centrifuged at 100,000 g for 30 min (4 °C) to precipitate light membrane organelles, and the final supernatant was used as the cytosolic fraction.
For the proteinase K resistance assay, the isolated mitochondria pellet was washed once with homogenization buffer. Then, the pellet was resuspended in buffer and treated with proteinase K (100 μg ml−1) in the absence or presence of 0.05% digitonin (weight per volume). The proteolytic reaction was performed on ice for 15 min, and the reactants were subjected to Western blot analysis with the indicated antibodies.
In vitro mitochondrial import assay
Cell-free protein synthesis of each PB1-F2 variant (PR8, 88W and 58W) was performed using TNT Coupled Wheat Germ Extract System (Promega) according to the manufacturer's protocol. The synthesized proteins were incubated with mitochondria isolated from HEK293 cells in import buffer (20 mM HEPES-KOH (pH 7.4), 250 mM sucrose, 2.5 mM magnesium-acetate, 25 mM KCl and 2.5 mM EGTA) at 25 °C. The reactant from each time point was then treated with proteinase K (50 μg ml−1) to avoid non-specific binding of MOM, and the reactant was subjected to Western blot analysis.
Sub-mitochondrial fractionation
Isolated mitochondria (suspended in 100 μl of homogenization buffer) were diluted in 900 μl of hypotonic buffer (10 mM HEPES-KOH buffer (pH 7.5) containing 0.5 mM EDTA and protease inhibitor cocktail (Roche)) and maintained on ice for 30 min to cause swelling. The mixture was then sonicated five times for 30 s each followed by centrifugation at 5,000g for 10 min. The resulting supernatant was further spun at 200,000 g for 45 min at 4 °C, and the pellet was resuspended in 100 μl of 5 mM HEPES-KOH buffer (pH 7.5), followed by layering over a linear gradient (0.8 to 1.6 M) of sucrose in hypotonic buffer and finally centrifuged at 100,000 g for 15 h at 4 °C in an Optima TL Ultracentrifuge (Beckman Coulter). After centrifugation, 200-μl fractions were collected and subjected to Western blot analysis.
BRET saturation assay
All BRET assays were performed as previously described26 with slight modifications. In brief, HEK293 cells (2.5 × 105 cells per well) were co-transfected with a constant amount (5 ng) of NLuc-tagged PB1-F2 (88W) plasmid and increasing amounts of Venus-tagged constructs using Lipofectamine 2000 reagent (Invitrogen). Cells were harvested 21 h post transfection and transferred to each well of white 96-well microplates. NLuc substrate (furimazine; 5 μM) was added, and the plates were analyzed via a BRET saturation assay using a Flexstation 3 Microplate Reader (Molecular Devices) at 37 °C.
Confocal microscopy
Cells were plated on coverslips in 12-well plates (5 × 104 cells per well), and the next day, the cells were infected with influenza A virus (or transfected with expression plasmids using X-tremeGENE HP reagent (Roche)). At 22 h post infection (24 h post transfection), the cells were fixed with 3.7% formaldehyde for 10 min, permeabilized with 1% Triton X-100 in 1 × PBS (pH 7.2), and blocked with 5% bovine calf serum. PB1-F2 was detected either using a monoclonal (PR8) or polyclonal (88W, 58W and 12S) primary antibody and Alexa Fluor 488-conjugated secondary antibodies, and mitochondria were visualized using mitochondrial-targeted red fluorescent protein (Mito-RFP). The ΔΨm was monitored by MitoTracker Red CMXRos as described previously16, and all cells were imaged by confocal microscopy (Carl Zeiss LSM510). An anti-influenza A virus M2 protein monoclonal antibody was used to detect A/CA/04/09-infected cells.
A semi-intact cell assay was performed as previously described25. Briefly, PB1-F2-expressing HeLa cells were fixed with 3.7% formaldehyde for 10 min, permeabilized with either 400 μg ml−1 (pre-permeabilized) or 2,000 μg ml−1 (normal condition) digitonin in PBS, and then processed for immunofluorescence microscopy.
siRNA treatment and mitochondrial protein import assay
For the RNA interference knockdown experiments, 21-nucleotide siRNA (sense strand-only shown) against human TOM20 (5′-aguuaccugaccuuaaagatt-3′), hTOM22 (5′-gcacauugaucuaucuaaatt-3′), hTOM40 (5′-caacugguuggcaacgguatt-3′), hTOM40_#B (5′-cgguauaaaucauguuuautt-3′), hTOM40_#C (5′-gguauaaaucauguuuauatt-3′), hTOM70 (5′-gacaauaagaaggaauguutt-3′), hTOM70_#B (5′-gaaugaccucugacuuuaatt-3′), hDrp-1 (5′-gcagaagaaugggguaaautt-3′) and hOMA-1 (5′-gaucaauuggguuauucautt-3′) were purchased from QIAGEN. HeLa cells were transfected with 50 nM siRNA (final concentration) twice within 24 h using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocols. At 72 h after the initial treatment, the siRNA-treated cells were transfected with each of the indicated expression plasmids using X-tremeGENE HP reagent followed by an in vivo import assay. AllStars Negative Control siRNA (QIAGEN) was used as the control.
Flow cytometry
The ΔΨm was analyzed by BD FACSCalibur (BD Biosciences) with the cationic fluorescent dye TMRM (Molecular Probes/Invitrogen). Cells (~1 × 106 cells ml−1) that were infected with A/PR8 or plasmid transfected were washed once with 1 × PBS (pH 7.2) and harvested into a centrifuge tube. The cells were then resuspended in 1 ml of 1 × PBS (pH 7.2) containing 2 μM TMRM and incubated at 37 °C for 30 min. After washing three times with 1 × PBS (pH 7.2), flow cytometry analysis was performed. Data analyses and graphs were prepared using FlowJo software (Tree Star, Inc.).
Dual-luciferase reporter assays
HEK293 cells were plated in 24-well plates (2 × 105 cells per well). On the following day, the cells were co-transfected with 100 ng of a luciferase reporter plasmid (p125luc or pELAM), 2.5 ng of the Renilla luciferase internal control vector phRL-TK (Promega), and each of the indicated expression plasmids using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's protocols. Empty vector (pcDNA3.1(−)) was used to maintain equivalent amounts of DNA in each well. Cells were harvested 24 h post transfection and analyzed using a dual-luciferase reporter assay on the GloMax 20/20n luminometer (Promega). Each experiment was replicated at least three times.
Reconstitution assay of NLRP3 inflammasomes
HEK293 cells were plated in 24-well plates (4 × 105 cells per well). On the following day, the cells were co-transfected with 30 ng of pCA7-NLRP3, 5 ng of pCA7-ASC-FLAG, 5 ng of pCA7-procaspase-1 and 150 ng of pCA-pro-IL-1β and each of the indicated expression plasmids using Lipofectamine 2000. Empty vectors (pcDNA3.1(−)) were used to maintain equivalent amounts of DNA in each well. Twenty-four hours after transfection, clarified medium supernatants were collected, and IL-1β secretion was analyzed by ELISA. Each experiment was replicated at least three times.
To detect ASC oligomerization, the above cells were washed once with 1 × PBS (pH 7.2), harvested in a centrifuge tube, and then resuspended in 150 mM NaCl solution followed by lysis with 15 strokes through a syringe with a 21-gauge needle. After centrifugation (12,000 g for 15 min), the clarified supernatant was cross-linked with 4 mM BS3 for 30 min at room temperature followed by incubation with 50 mM Tris-HCl buffer for 15 min at room temperature to completely quench the cross-linking reaction. The resulting sample was resolved on 10% SDS–PAGE and immunoblotted with anti-FLAG monoclonal antibody.
Statistical analysis
The analysis of variance test (GraphPad QuickCalcs) was used for the statistical analyses. A P-value<0.05 was considered statistically significant.
Additional information
How to cite this article: Yoshizumi, T. et al. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 5:4713 doi: 10.1038/ncomms5713 (2014).
References
Attardi, G. & Schatz, G. Biogenesis of mitochondria. Annu. Rev. Cell Biol. 4, 289–333 (1988).
Ow, Y. P., Green, D. R., Hao, Z. & Mak, T. W. Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542 (2008).
Wang, C. & Youle, R. J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118 (2009).
Gunter, T. E., Yule, D. I., Gunter, K. K., Eliseev, R. A. & Salter, J. D. Calcium and mitochondria. FEBS Lett. 567, 96–102 (2004).
Leo, S., Bianchi, K., Brini, M. & Rizzuto, R. Mitochondrial calcium signalling in cell death. FEBS J. 272, 4013–4022 (2005).
McBride, H. M., Neuspiel, M. & Wasiak, S. Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560 (2006).
Seo, A. Y. et al. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 123, 2533–2542 (2010).
Arnoult, D., Soares, F., Tattoli, I. & Girardin, S. E. Mitochondria in innate immunity. EMBO Rep. 12, 901–910 (2011).
West, A. P., Shadel, G. S. & Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402 (2011).
Koshiba, T. Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta 1833, 225–232 (2013).
Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and characterization of MAVS, a mitochondrial antiviral signalling protein that activates NF-κB and IRF3. Cell 122, 669–682 (2005).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Tal, M. C. & Iwasaki, A. Mitoxosome: a mitochondrial platform for cross-talk between cellular stress and antiviral signalling. Immunol. Rev. 243, 215–234 (2011).
Koshiba, T., Yasukawa, K., Yanagi, Y. & Kawabata, S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signalling. Sci. Signal. 4, ra7 (2011).
Ichinohe, T., Yamazaki, T., Koshiba, T. & Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl Acad. Sci. USA 110, 17963–17968 (2013).
Chen, W. et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7, 1306–1312 (2001).
Chakrabarti, A. K. & Pasricha, G. An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology 440, 97–104 (2013).
Gibbs, J. S., Malide, D., Hornung, F., Bennink, J. R. & Yewdell, J. W. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 77, 7214–7224 (2003).
Yamada, H., Chounan, R., Higashi, Y., Kurihara, N. & Kido, H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 578, 331–336 (2004).
Zamarin, D., García-Sastre, A., Xiao, X., Wang, R. & Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1, e4 (2005).
Ozawa, M. et al. Impact of amino acid mutations in PB2, PB1-F2, and NS1 on the replication and pathogenicity of pandemic (H1N1) 2009 influenza viruses. J. Virol. 85, 4596–4601 (2011).
Conenello, G. M., Zamarin, D., Perrone, L. A., Tumpey, T. & Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3, 1414–1421 (2007).
Otera, H., Ohsakaya, S., Nagaura, Z., Ishihara, N. & Mihara, K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 24, 1375–1386 (2005).
Sasaki, O. et al. A structural perspective of the MAVS-regulatory mechanism on the mitochondrial outer membrane using bioluminescence resonance energy transfer. Biochim. Biophys. Acta 1833, 1017–1027 (2013).
Neupert, W. & Brunner, M. The protein import motor of mitochondria. Nat. Rev. Mol. Cell Biol. 3, 555–565 (2002).
Otera, H. et al. A novel insertion pathway of mitochondrial outer membrane proteins with multiple transmembrane segments. J. Cell Biol. 179, 1355–1363 (2007).
Ishihara, N., Fujita, Y., Oka, T. & Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977 (2006).
Baker, M. J. et al. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 33, 578–593 (2014).
Yasukawa, K. et al. Mitofusin 2 inhibits mitochondrial antiviral signalling. Sci. Signal. 2, ra47 (2009).
Varga, Z. T., Grant, A., Manicassamy, B. & Palese, P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 86, 8359–8366 (2012).
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z. & Germain, R. N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 (2013).
Stehlik, C. et al. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol. 171, 6154–6163 (2003).
Komune, N., Ichinohe, T., Ito, M. & Yanagi, Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1β secretion. J. Virol. 85, 13019–13026 (2011).
Li, X. D., Sun, L., Seth, R. B., Pineda, G. & Chen, Z. J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signalling protein off the mitochondria to evade innate immunity. Proc. Natl Acad. Sci. USA 102, 17717–17722 (2005).
Meylan, E. et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 (2005).
Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 (2006).
Castanier, C., Garcin, D., Vazquez, A. & Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 11, 133–138 (2010).
Tondera, D. et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600 (2009).
McAuley, J. L. et al. Expression of the 1918 Influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2, 240–249 (2007).
Weeks-Gorospe, J. N. et al. Naturally occurring swine influenza A virus PB1-F2 phenotypes that contribute to superinfection with Gram-positive respiratory pathogens. J. Virol. 86, 9035–9043 (2012).
Ichinohe, T., Lee, H. K., Ogura, Y., Flavell, R. & Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87 (2009).
Ito, M., Yanagi, Y. & Ichinohe, T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 8, e1002857 (2012).
Nordmann, A., Wixler, L., Ludwig, S. & Wixler, V. Monoclonal antibodies against the PB1-F2 protein of H1N1 influenza A virus. Hybridoma (Larchmt) 29, 321–326 (2010).
Acknowledgements
We are grateful to Koji Okamoto (the Osaka University, Japan) and Thomas Langer (the University of Cologne) for their valuable comments on the study. We appreciate technical support from the Research Support Centre, the Graduate School of Medical Sciences, the Kyushu University for the fluorescence measurements and Yuko Fuchigami for her technical assistance with DNA cloning and sequencing. The anti-PB1-F2 (A/PR8 strain) monoclonal antibody was a kind gift from Viktor Wixler (the Münster University Hospital Medical School). We are also grateful to Yoshihiro Kawaoka (the University of Tokyo) for kindly providing the pPolI/CA04-PB1-F2-stop88W plasmid and the A/CA/04/09 (H1N1) virus, Hideki Hasegawa (the National Institute of Infectious Diseases, Japan) for the A/Beijing/262/95 (H1N1) virus, Michael Whitt (the University of Tennessee) for the recombinant vesicular stomatitis virus and Victoria Allan (the University of Manchester) for the ER-green plasmid. This work was supported by the JSPS KAKENHI Grants (No. 25115515, 26291032, and 26620135 to T.K.), the Kyushu University Interdisciplinary Programs in Education and Projects in Research Development (T.K.), the Uehara Memorial Foundation (T.K.), the Takeda Science Foundation (T.K.), the Kao Foundation for Arts and Sciences (T.K. and T.I.), the Tokyo Biochemical Research Foundation (T.I.), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (T.I.), and the Sasakawa Scientific Research Grant from the Japan Science Society (T.Y.). This study was also partly supported by the Grant for Joint Research Project of the Institute of Medical Science, the University of Tokyo (T.K.) and Research Fellow of the Japan Society for the Promotion of Science (O.S.).
Author information
Authors and Affiliations
Contributions
T.Y., T.I. and O.S. performed most of the experiments. H.O. established the knockdown cell lines and performed the semi-intact cell assay, and S.-i.K. provided reagents. T.Y., T.I., K.M. and T.K. Analyzed data and interpreted all experimental results. T.K. designed the overall direction and contributed to writing the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-21, Supplementary Table 1 and Supplementary References (PDF 2684 kb)
Rights and permissions
About this article

Cite this article
Yoshizumi, T., Ichinohe, T., Sasaki, O. et al. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun 5, 4713 (2014). https://doi.org/10.1038/ncomms5713
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5713
This article is cited by
-
The role of autophagy in viral infections
Journal of Biomedical Science (2023)
-
The mitophagy pathway and its implications in human diseases
Signal Transduction and Targeted Therapy (2023)
-
The influenza virus PB2 protein evades antiviral innate immunity by inhibiting JAK1/STAT signalling
Nature Communications (2022)
-
Restructured membrane contacts rewire organelles for human cytomegalovirus infection
Nature Communications (2022)
-
An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates
Virus Genes (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
