
Abstract
Methylammonium lead halide perovskites are attracting intense interest as promising materials for next-generation solar cells, but serious issues related to long-term stability need to be addressed. Perovskite films based on CH3NH3PbI3 undergo rapid degradation when exposed to oxygen and light. Here, we report mechanistic insights into this oxygen-induced photodegradation from a range of experimental and computational techniques. We find fast oxygen diffusion into CH3NH3PbI3 films is accompanied by photo-induced formation of highly reactive superoxide species. Perovskite films composed of small crystallites show higher yields of superoxide and lower stability. Ab initio simulations indicate that iodide vacancies are the preferred sites in mediating the photo-induced formation of superoxide species from oxygen. Thin-film passivation with iodide salts is shown to enhance film and device stability. The understanding of degradation phenomena gained from this study is important for the future design and optimization of stable perovskite solar cells.
Similar content being viewed by others


Introduction
Methylammonium lead halide perovskites have become one of the most promising classes of materials for the development of low-cost, solution-processed optoelectronics such as solar cells, light-emitting diodes and lasers1,2,3,4,5. Solar cell applications in particular have attracted intense interest in recent years with a rapid rise in power conversion efficiencies of up to 22% for perovskite photovoltaics6,7,8,9,10,11,12,13,14,15,16,17.
However, despite such remarkable progress, serious issues related to the long-term stability of perovskite halides need to be addressed before they can be used successfully in commercial solar cell applications. It has been generally observed that moisture, elevated temperature, oxygen and UV radiation all cause degradation of hybrid perovskite materials and device instability at higher rates than those typically observed in polymer and dye-sensitized photovoltaics18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37. We recently demonstrated38,39 that exposure of CH3NH3PbI3 photoactive layers to light and oxygen leads to the formation of superoxide (O2−) species. This reactive O2− species can deprotonate the methylammonium cation (CH3NH3+) of photo-excited CH3NH3PbI3*, leading to the formation of PbI2, water, methylamine and iodine,38,39 as shown schematically in Fig. 1.

Schematic representation of the reaction steps of O2 with CH3NH3PbI3. (a) Oxygen diffusion and incorporation into the lattice, (b) photoexcitation of CH3NH3PbI3 to create electrons and holes (c) superoxide formation from O2, and (d) reaction and degradation to layered PbI2, H2O, I2 and CH3NH2.
This oxygen-induced degradation pathway has been shown to affect the stability of both CH3NH3PbI3 photoactive layers and solar cell devices40. Transient absorption spectroscopy studies of interfacial charge transfer in CH3NH3PbI3-based films revealed that such oxygen-induced degradation results in a large decrease in the yield of photo-induced charge carriers39. While recent work has demonstrated the importance of electron extraction in reducing the severity of this degradation pathway20,38,39,40, it is highly unlikely that charge extraction alone will completely solve this problem. Hybrid lead halide layers deposited from solution typically produce polycrystalline films with varied microstructure and particle morphologies. The impact of film microstructure on charge carrier transport, photoluminescence, device performance parameters (for example, Jsc, Voc, fill factor) and tolerance to moisture has been reported recently41,42,43,44,45,46,47,48,49,50,51. It is reasonable to suppose that the particle size and defect chemistry of the films may also influence oxygen diffusion into the perovskite layer and its susceptibility to oxidative reactions.
Studies on CH3NH3PbI3 suggest a significant equilibrium defect concentration of I−, Pb2+ and CH3NH3+ vacancies at room temperature, which could provide vacancy-mediated pathways for ion transport52. Our previous work53 and other studies54,55,56,57,58 indicate that these hybrid perovskites are mixed ionic-electronic conductors, and also implicate vacancy-mediated iodide ion diffusion as being responsible for the observed hysteresis effects. Recent studies have also investigated the role of ion migration in perovskite degradation21. Ionic defect and transport phenomena in hybrid perovskites thus have important implications in terms of the long-term stability and performance of perovskite solar cell devices. However, the exact mechanism of oxygen diffusion and the defect species associated with oxygen and light-induced degradation are poorly understood. Additionally, these stability issues raise important questions that have not been fully addressed. Specifically, these questions relate to: (i) the origin of the observed fast rate of oxygen-induced degradation of CH3NH3PbI3 films and (ii) the relationship between the film particle size, oxygen transport, intrinsic vacancies and the mechanism of oxygen-induced degradation.
Here we combine experimental and computational methods to address these important questions at the microscopic level, extending our previous work on hybrid perovskites38,39,40,53. Specifically, we have probed the dynamics of oxygen diffusion in perovskite films and investigated how the film morphology influences reactivity to molecular oxygen. Oxygen diffusion into perovskite films is observed to occur remarkably fast with, for example, a typical film (500 nm thick) reaching complete saturation within 10 min. Density functional theory (DFT) calculations also reveal the importance of iodide vacancies as reaction mediators to form superoxide species. The results provide valuable insights for the design of perovskite devices exhibiting improved environmental stability.
Results
Oxygen diffusion
We first consider the dynamics of oxygen diffusion in hybrid perovskite films. CH3NH3PbI3 and CH3NH3PbI3(Cl) were chosen for a comparative study, since both have been widely tested in photovoltaic devices. (Note that the formula CH3NH3PbI3(Cl) refers to a perovskite material fabricated from a combination of iodide and chloride precursors.) Of the two systems, CH3NH3PbI3(Cl) films are reported to be more stable59,60 but the origin(s) of this superior stability remains unclear. In our study, films were fabricated as described in the Methods section onto clean glass substrates. X-ray diffraction (XRD) (see Supplementary Fig. 1) was used to characterize both materials, and confirmed that in both cases CH3NH3PbI3 is the compound formed. We recognize that the concentration of chlorine within the CH3NH3PbI3(Cl) film may be too low to determine through XRD. The presence of any chlorine may give rise to differences in charge carrier recombination, which could influence stability, but the low Cl levels mean this is unlikely to be the dominant effect. Chlorine substitution is thus unlikely to be responsible for the improved stability of the material.
Isothermal gravimetric analysis (IGA) was used to probe the dynamics of oxygen diffusion as illustrated in Fig. 2a. In a typical experiment, the sample chamber with the perovskite film was first flushed with He gas for 30 min. Next, the weight of the perovskite sample was recorded as a function of exposure to dry air (N2=80%, O2=20%) over the course of 20 min at room temperature (25 °C). As can be seen from the IGA traces in Fig. 2a, oxygen diffusion into both CH3NH3PbI3 and CH3NH3PbI3(Cl) films was rapid, with saturation being achieved within 5–10 min. Further evidence for the fast rate of oxygen diffusion in these two materials was obtained from time-of-flight secondary ion mass spectrometry (ToF-SIMS) measurements, in which the depth profile of oxygen throughout the films was determined. In these experiments, films of both CH3NH3PbI3 and CH3NH3PbI3(Cl) were soaked in dry air in the absence of light and ToF-SIMS data collected. The ToF-SIMS images shown in Fig. 2b,c represent slices through CH3NH3PbI3 and CH3NH3PbI3(Cl) layers at a depth of approximately 200 nm in the films, after soaking for 30 min. 3D profiles (raw data) showing oxygen concentration as a function of film depth are also provided in Supplementary Fig. 2. Taken together, these findings demonstrate that oxygen enters the perovskite samples and is uniformly distributed throughout the films. As the time period for oxygen uptake to reach saturation is in agreement with the fast degradation rate previously observed in CH3NH3PbI3 photoactive layers and devices38,39,40, we conclude that the high sensitivity of CH3NH3PbI3 devices to oxygen is owed, in part, to the rapid rate at which oxygen can diffuse into the films.

(a) Isothermal gravimetric analysis plot (IGA) of CH3NH3PbI3 (MAPI) and CH3NH3PbI3(Cl) (MAPIC) thin films coated on non-conductive cleaned glass with 30 min of Helium soaking before oxygen exposure, where t=0 corresponds to the time at which oxygen was introduced into the system. (b,c) ToF-SIMS surface images (100 × 100 μm) of CH3NH3PbI3 and CH3NH3PbI3(Cl) respectively after exposure to dry air flow with no illumination, where the maximum counts (MC) and the total number of secondary oxygen ion counts (TC) are shown with the colour scales corresponding to the interval [0, MC].
From the IGA data we can estimate the air (80% N2 and 20% O2) diffusion coefficient, Da, to be in the range 10−7–10−9 cm2 s−1 for the CH3NH3PbI3 perovskite material (calculations provided in SI). This value is comparable to the fast diffusion of gases into polymer thin films where, for example, the diffusion coefficient61 in poly-(3-hexylthiophene-2,5-diyl) (P3HT) thin films is of the order of 10−8 cm2 s−1. It is widely accepted that these fast diffusion kinetics are responsible for the relatively low stability of semiconducting polymer films to molecular oxygen62,63,64. As such, we propose that the fast oxygen diffusion kinetics are critical to the observed oxygen- and light-induced degradation rates seen in perovskite-based optoelectronic devices.
Particle size dependence
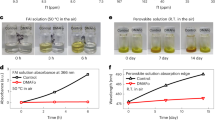
Next we considered the effect of visible light and oxygen on the relative stability of CH3NH3PbI3 and CH3NH3PbI3(Cl) films and devices. Figure 3a,b shows the absorption spectra of CH3NH3PbI3 and CH3NH3PbI3(Cl) films on glass substrates measured as a function of ageing under illumination in dry air. Figure 3a,b reveals that both perovskite materials rapidly degrade under these conditions. However, it is apparent that CH3NH3PbI3(Cl) films degrade at a significantly slower rate than CH3NH3PbI3 films. Figure 3c shows the power conversion efficiency (PCE) versus time profile for CH3NH3PbI3 and CH3NH3PbI3(Cl)-based solar cells, with the devices being continuously aged in dry air under light. The corresponding current–voltage curves are provided as Supplementary Fig. 3 in Supplementary information. For these studies, a solar cell architecture of the type [FTO/compact-TiO2/mesoporous-TiO2/perovskite/spiro-OMeTAD/Au] was employed and all PCE measurements were performed on un-encapsulated devices as in previous work44. The PCEs of the solar cells were determined from current–voltage characteristics ascertained at regular time intervals over the course of 4 h. In Fig. 3c, CH3NH3PbI3(Cl) devices show a relatively small 10% drop in PCE over the 4-h ageing period whereas the CH3NH3PbI3 devices display a more substantial (≈80%) drop in PCE under the same ageing conditions. The better stability of the CH3NH3PbI3(Cl) solar cells (compared to CH3NH3PbI3) is consistent with the absorption data in Fig. 3a,b, as well as other recent reports59. The difference in stability between CH3NH3PbI3 and CH3NH3PbI3(Cl) observed here may be related to the smaller size of the particles.

(a,b) Light absorption spectrum for ageing CH3NH3PbI3 and CH3NH3PbI3(Cl) under dry air and illumination (25 mW cm−2) respectively. (c) Normalized power conversion efficiency loss for photovoltaic devices employing CH3NH3PbI3 (referred to as MAPI) and CH3NH3PbI3(Cl) (referred to as MAPIC) as the light harvesting materials in an [FTO/planar-TiO2/mesoporous-TiO2/perovskite/spiro-OMeTAD/Au] architecture. J-V curves obtained for these studies are shown in Supplementary Fig. 4. (d) Normalized fluorescence intensity increase of the HE probe at 610 nm (excitation at 520 nm). IF(t) is the fluorescence maximum at time t, while IF(t0) is the background fluorescence intensity. IF(t)/IF(t0) ratio corresponds to the yield of superoxide generation for the perovskite films. (e,f) Surface SEM images of CH3NH3PbI3 and CH3NH3PbI3(Cl) films deposited on cleaned glass substrates.
The next question that arises relates to the origin of the difference in stability observed between CH3NH3PbI3 and CH3NH3PbI3(Cl). To identify the cause(s) for this difference we first determined the yield of photo-induced superoxide (O2−) formation for the two perovskites. We have previously established that O2− is the key reactive species responsible for the hybrid perovskite and device degradation38,40. Here the yield of O2− was determined for both CH3NH3PbI3 and CH3NH3PbI3(Cl) films using a hydroethidine (HE) fluorescent probe (as described in the Methods section). As detailed elsewhere38, the degradation effect we observe cannot be ascribed to degradation of the hole transporting material spiro-OMeTAD since we have not used spiro-OMeTAD in these experiments.
Figure 3d shows the rate of increase in HE emission and, accordingly, the O2− generation yield. The data in Fig. 3d show that CH3NH3PbI3 produces a significantly higher yield of superoxide than CH3NH3PbI3(Cl), consistent with the film and device stability data in Fig. 3a–c. It is reasonable to propose that this difference in reactivity stems from differences in the particle sizes within the films. For example, the presence of chloride ions in the precursor mixture is known to slow down the rate of crystal formation, leading to larger crystals28,29. This was confirmed by taking scanning electron microscopy (SEM) images of the CH3NH3PbI3 and CH3NH3PbI3(Cl) films. Figure 3e,f shows that the CH3NH3PbI3(Cl) film consists of crystal domains that are several hundreds of nanometres in diameter, considerably larger than those observed in the CH3NH3PbI3 sample. This strongly suggests that crystal size plays a crucial role in determining the stability.
In order to test this hypothesis, we next investigated, in a systematic manner, the stability of CH3NH3PbI3 films composed of different crystal sizes. The controlled growth of CH3NH3PbI3 crystals was achieved following the method previously reported by Grätzel and co-workers50. In this way, we prepared samples with small (100 nm), medium (160 nm) and large (250 nm) perovskite crystals as confirmed by SEM. Representative SEM images are shown in Fig. 4a–c. In Fig. 4d the magnitude of the absorbance at 750 nm is plotted against ageing time, with black, red and blue curves corresponding to films composed of small (sample 1), medium (sample 2) and large (sample 3) CH3NH3PbI3 crystallites, respectively. The corresponding absorption spectra are provided in Supplementary Fig. 4. We stress that these measurements were recorded until the CH3NH3PbI3 film had turned completely yellow, indicating full conversion of the perovskite to the degradation product, PbI2. It is apparent from the data in Fig. 4d that the films with large CH3NH3PbI3 crystals are considerably more stable than the films composed of small CH3NH3PbI3 crystals. We note that all three samples showed similar rates of oxygen uptake, with the films reaching saturation within 10 min, as determined by IGA experiments (Supplementary Fig. 5).

(a–c) Surface SEM images of small (sample 1), medium (sample 2) and large (sample 3) crystal sizes of CH3NH3PbI3. (d) Normalized absorbance decay at 750 nm for methylammonium lead iodide of sample 1 (small crystals, 100 nm), sample 2 (medium crystals, 150 nm) and sample 3 (large crystals, 250 nm) with degradation conditions of illumination (25 mW cm−2) and dry air. (e) Superoxide yield plot for CH3NH3PbI3 (with small (black), medium (red), large (blue) crystal sizes and a toluene dripped prepared sample (green) and CH3NH3PbI3(Cl) (brown).
Next, we consider the possible correlation between CH3NH3PbI3 crystal size, materials stability and superoxide (O2−) generation yield. Figure 4e shows the measured rate of increase of O2− species in these three samples; a correlation is observed between the CH3NH3PbI3 crystallite size, stability (that is, tolerance to visible light and oxygen stress) and O2− yield. It is evident that the CH3NH3PbI3 films with large crystallites show a relatively low yield of superoxide formation and display better stability. Conversely, the CH3NH3PbI3 films composed of smaller crystallites show a higher yield of superoxide generation and rapidly degrade within 2 days. There thus appears to be a direct correlation between yield of superoxide generation (and subsequently degradation rate) and perovskite crystallite size.
Superoxide formation sites
To obtain atomic-scale insights into the energetics and defect mechanisms of the degradation process we have used ab initio simulations based on DFT. We first probed the reaction with oxygen (with no photo-generated electrons in the perovskite lattice), described by the following equation:

The enthalpy was calculated to be +1.60 eV per O2 molecule, indicating that the reaction in the absence of light is unfavourable, which is consistent with observation. We have already shown that in order for the overall degradation reaction to occur, the film must be exposed to both O2 and light in order for superoxide species to form38,39. The implication is that O2 acts as an electron scavenger and absorbs electrons generated by light or an external electrical bias.
The next step in the reaction sequence is for superoxide O2− to react with photo-oxidized CH3NH3PbI3* to produce PbI2, I2, H2O and CH3NH2 according to the following reaction (illustrated in Fig. 1):

In contrast to reaction (1), the calculated enthalpy for reaction (2) is negative (−1.40 eV per O2 molecule), indicating that the degradation reaction is now highly favourable, again in agreement with observation. This indicates that both the photo-oxidized CH3NH3PbI3 and the O2− species are unstable with respect to the reaction products.
A key step in the degradation reaction is the formation of the superoxide species according to O2+e−=O2−. This raises the important question of where the superoxide forms in the crystal lattice. It is plausible that ionic defects such as iodide vacancies play a key role in mediating superoxide formation from O2 and hence the degradation reaction. As noted, recent work52 indicates a significant population of intrinsic Schottky-type defects (a stoichiometric combination of anion and cation vacancies), whose formation can be expressed using Kröger–Vink notation as:

where nil represents the perfect (defect-free) CH3NH3PbI3 lattice, V indicates a vacancy, subscripts indicate the type of lattice site and superscripts the effective charge of the defect (a dot for each positive charge and prime for each negative charge). Such vacancy defects may act as molecular or charge traps which in turn could mediate the electron transfer reaction with oxygen. It is known that the anion vacancy in binary lead halides can trap a photoelectron to form an F centre24.
To investigate this process, we calculated the energetics for superoxide formation from O2 molecules on various lattice and vacancy sites in the CH3NH3PbI3 perovskite structure; the most favourable configurations and the corresponding superoxide formation energies are summarized in Fig. 5. Two main findings emerge. First, superoxide formation by direct electron transfer from the perovskite to oxygen is energetically favourable. Analysis of the electron density and bond lengths shows that the photo-generated electron resides on the O2 molecule; upon adsorption the O2 bond length increases from 1.22 to 1.33 Å and the species becomes spin polarized. Second, superoxide formation energies indicate that vacant iodine sites are the preferred location for the reduction process in the crystal bulk. Interestingly, an iodide ion is of similar size to the superoxide species (as illustrated in Fig. 6), and in occupying an iodide vacancy, the superoxide ion restores the full octahedral coordination of Pb2+.

Schematic representation of possible O2 binding and reduction sites in CH3NH3PbI3 ([001] plane) and corresponding superoxide formation energy: (a) face site neighbouring four iodide ions, (b) neutral iodide vacancy (with a photoelectron on the defect site) and negatively charged lead (c) and methylammonium (d) vacancies (with no photoelectron on them since this was found to be unphysical).

Atomic structure of CH3NH3PbI3 showing (a) superoxide ion, O2−, occupying an iodide vacancy,  (for clarity, a pseudo-cubic sub-region of the structure is shown and not the full tetragonal supercell used in the calculations). (b) Comparison of relative size of iodide and superoxide anions (using ionic radius of I− and, for the superoxide ion, interpolation between covalent radius in O2 and ionic radius of O2−).
(for clarity, a pseudo-cubic sub-region of the structure is shown and not the full tetragonal supercell used in the calculations). (b) Comparison of relative size of iodide and superoxide anions (using ionic radius of I− and, for the superoxide ion, interpolation between covalent radius in O2 and ionic radius of O2−).
Further evidence for the favourable energetics of O2 reduction is given by the calculated band structures for O2 incorporation into CH3NH3PbI3 reported in Fig. 7. In Fig. 7a it can be seen that the unoccupied oxygen π* anti-bonding orbital is located in the middle of the CH3NH3PbI3 band gap where it can readily act as an acceptor state for photo-excited electrons in the conduction band. Moreover, when the O2− superoxide species occupies an iodide vacancy (Fig. 7b), the oxygen states are shifted down into the valence band as a result of changes in bonding interactions, clearly indicating that it is even more energetically favourable for O2 to be reduced by photo-excited electrons in the conduction band.

(a) O2 in defect-free CH3NH3PbI3 and (b) O2 at iodide vacancy in CH3NH3PbI3. Key: blue—CH3NH3PbI3, red—O2. Note the band structure is folded due to the use of a large supercell.
Proposed degradation mechanism
To summarize our four key observations: (i) photo-induced oxygen degradation of CH3NH3PbI3 shows a strong particle size dependence, (ii) structural degradation from the black perovskite phase to the yellow lead PbI2 phase occurs on a timescale of days (see absorbance data, Fig. 3a), whereas the device performance degrades in a matter of hours (see efficiency data, Fig. 3c), (iii) rapid oxygen uptake occurs on a timescale of less than 1 h and results in superoxide generation, and (iv) superoxide formation is always associated with degradation and is facilitated by iodide vacancies.
To rationalize these findings, we propose a mechanism that is summarized schematically in Fig. 8. The CH3NH3PbI3 sample is under illumination throughout this process to provide a constant source of photo-excited electrons in the bulk and surface regions. Oxygen is admitted to the sample and diffuses between the particles and within an hour permeates the inter-particle regions (Fig. 8a). Superoxide species immediately begin to form at the particle surfaces as O2 is reduced while occupying surface iodide vacancies. Over the first few hours there is an initial reaction between these superoxide species and the particle surfaces, leading to their passivation. The degradation of the particle surfaces prevents the extraction of the photocurrent65, causing the device properties such as efficiency (PCE) to decline rapidly. On a timescale of days (Fig. 8b) oxygen diffuses into the interior of the particles where it occupies bulk iodide vacancies while being reduced, leading to full structural degradation of the material.

(a) O2 diffusion in inter-particle regions and initial reaction with particle surfaces over a timescale of hours leading to reduction in device efficiency, open circuit voltage and photocurrent and (b) O2 diffusion into particle bulk regions over a time scale of days leading to a phase change from the photo-absorbing perovskite phase into the non-absorbing lead iodide phase (yellow).
For both the surface and bulk reactions we would expect the particle size and iodide vacancy levels to influence the rate of reaction. It is well known that particle surfaces are normally much more reactive than the bulk; in the halide perovskites it has been shown that different particle facets can display markedly different photovoltaic behaviour66. Furthermore, Haruyama et al.67 have shown that in CH3NH3PbI3 the most prominent surfaces are vacancy rich with defect concentrations of 1012 cm−2, and our data show that oxygen reduction occurs most readily at the iodide vacancies. Thus, the increased initial degradation rate for smaller crystallites may be directly related to the surface vacancy concentration. Figure 3 shows SEM micrographs of large crystallites with typical sizes of ca. 250 nm and small crystallites of ca. 100 nm. Based on these average values, we would expect the number density of surface vacancies to be around 2.4 × 1017 cm−3 in the large crystallites and around 6.0 × 1017 cm−3 in the small crystallites. The small crystallites thus provide more than twice as many surface adsorption/reaction sites per unit volume as the large crystallites.
In addition, bulk adsorption/reaction sites will contribute significantly to the rate of degradation over long time-scales. Iodide vacancy concentrations in the bulk have been estimated52 at 1022 cm−3, which suggests that in a typical particle, bulk vacancies will be five orders of magnitude more numerous than surface vacancies. Since our ToF-SIMS data show that O2 can diffuse into the bulk, the increased degradation rate for smaller crystallites may be due to the shorter O2 diffusion path lengths to the particle interiors. In general, the characteristic time constant, t, for ion diffusion is given by t=L2/D, where L is the diffusion path length and D is the diffusion coefficient. The rate of oxygen permeation into the bulk of CH3NH3PbI3 can be expected to drop by a factor of four for every doubling of the particle size. This model can thus account for our observations. We recognize that other factors such as the rate of reaction between the superoxide species and methylammonium cation may also limit the overall rate of degradation, but such topics are left for future investigations.
Film passivation using salt additives
Finally, a key question that arises relates to whether the stability of CH3NH3PbI3 films can be improved by inhibiting superoxide formation at iodide vacancies, for example, by defect passivation. To investigate this, a solution of either chloride or iodide salts of cations phenylethylammoniun or methylammonium and trimethlsulfonium iodide was spin-coated onto the CH3NH3PbI3 perovskite films. Ionic passivating agents carrying bulky cations were chosen to ensure that substitution with methylammonium cations in the perovskite was unfavourable. The successful preparation of the films was confirmed by XRD analysis (see Supplementary Fig. 6).
The stabilities of the coated films were examined after exposure to light and oxygen. As can be seen in Fig. 9a (raw absorbance data are presented in Supplementary Fig. 7), all the iodide salt-coated samples display an enhanced tolerance to light and oxygen (relative to the uncoated control sample), showing little or no degradation over the ageing time period. In contrast, the chloride salt derivatives showed no stability enhancement, exhibiting a comparable rate of degradation as that of an uncoated perovskite sample. We note that the iodide treated films showed no sign of any degradation over 3 weeks of ageing under light and oxygen. Furthermore, the enhanced stability of the coated samples is consistent with the superoxide generation data shown in Fig. 9b. In particular, it can be seen again that all the iodide salt coated samples show a significantly lower yield of superoxide species than the uncoated sample and the chloride salt-treated samples.

(a) Normalized absorbance decay at 750 nm under illumination (25 mW cm−2) and dry air flow for a pristine CH3NH3PbI3 and CH3NH3PbI3 coated with phenylethylammonium iodide (PhEtNH3I), methylammonium iodide (MeNH3I), trimethylsulfonium iodide (Me3SI), phenylethylammonium chloride (PhEtNH3Cl) and methylammonium chloride (MeNH3Cl) as described in the experimental section. (b) Superoxide yield plot comparing the generation of superoxide for CH3NH3PbI3 treated with the coatings and without. (c) Normalized power conversion efficiency loss for photovoltaic devices employing CH3NH3PbI3 with and without a 10 mM treatment of MeNH3I as the light harvesting material in an [FTO/planar-TiO2/mesoporous-TiO2/perovskite/spiro-OMeTAD/Au] architecture. J-V curves obtained for these studies are shown in the Supplementary Fig. 13.
To confirm that the iodide salt treatments are indeed passivating iodide defects rather than some other effect, we performed time-resolved photoluminescence, IGA, SEM and superoxide yield measurements. IGA data presented in Supplementary Fig. 8 indicate similar fast oxygen diffusion kinetics in both CH3NH3I treated and untreated films. In contrast, a control sample comprising a CH3NH3PbI3 film encapsulated in glass showed little or no weight increase upon exposure to oxygen; in this instance the glass layer functions as an oxygen blocking layer. In addition, time-resolved photoluminescence measurements were performed on perovskite films treated with different concentrations (0.001 M, 0.005 M and 0.01 M) of CH3NH3I. As can be seen from the data provided in Supplementary Fig. 9, increasing the iodide salt concentration leads to an increased photoluminescence lifetime. Moreover, this observation is consistent with the iodide salt treatment reducing the number of defects and therefore the number of trap states for non-radiative recombination, resulting in longer emission lifetimes. Recent studies68,69 have found that alkali metal halide salts introduced at the perovskite interface can decrease halide vacancy levels, resulting in improved device performance.
Next, the relative stability and superoxide yield of CH3NH3PbI3 films treated with different concentrations of CH3NH3I were investigated. The relationship between CH3NH3I concentration, stability and superoxide yield is shown in Supplementary Fig. 10, which shows that increasing the concentration of CH3NH3I salt leads to lower yields of superoxide generation and consequently better stability. Finally, surface SEM images were taken of a treated and an untreated CH3NH3PbI3 film to show that the treatment does not induce any significant morphological changes to the film’s surface (provided in Supplementary Fig. 11). Taken together, the results in Supplementary Figs 8–11 strongly suggest that salt treatments passivate the crystal defects rather than providing a physical barrier layer to oxygen diffusion.
Passivation of crystal defects using iodide salts reduces superoxide yields and enhances film stabilities, but it is not known whether they improve device stability. To address this question, cells were fabricated and characterized under different conditions (as described in the experimental section). The results, shown in Fig. 9c and Supplementary Figs 12,13 and Supplementary Table 1, indicate that solar cells that use CH3NH3I-coated CH3NH3PbI3 films exhibit better stability than those that use uncoated perovskite layers. More specifically, exposure of devices using uncoated perovskite layers to light and dry air for just 2.5 h leads to a 50% drop in the PCE. In contrast, devices containing CH3NH3I coated layers exhibit a significantly smaller drop in efficiency (10%) over the same ageing period. The relationships between concentration of the CH3NH3I coating in solution and the device stability are shown in Supplementary Figs 12,13 and Supplementary Table 1. It is clear from these results that increasing the concentration of CH3NH3I salt leads to progressively higher stability.
These results further confirm that the iodide anion from the salt passivates the iodide vacancies in the crystal, and by occupying the otherwise vacant iodide sites leads to increased stability by suppressing superoxide formation. We note that degradation of the coated devices is not halted completely, which suggests that other factors also influence overall device stability. For example, it is possible that iodide vacancies in the bulk are not filled by iodide ions from the coating and these can still act as sites for superoxide generation. Nevertheless, it is clear from the present findings that the iodide salt coatings lead to significant improvements in device stability.
Discussion
The present study suggests that iodide defects in the CH3NH3PbI3 structure are key sites for superoxide formation. In addition, the results demonstrate that iodide salt treatment can be employed to reduce the number of problematic iodide vacancies, thereby hindering the electron transfer reaction that generates superoxide species. Although this study is not exhaustive it does highlight a critical issue for further work, and could include probing the combined effect of oxygen and water on CH3NH3PbI3 as well as large-scale atomistic simulations of oxygen diffusion and iodine interstitials.
In conclusion, a combination of IGA, photoluminescence, SIMS and ab initio simulation techniques has provided mechanistic insights into oxygen- and light-induced degradation of perovskite solar cells. We found that fast oxygen diffusion into CH3NH3PbI3 films is accompanied by formation of superoxide species, which are critical to oxygen-induced degradation. The yield of superoxide (O2−) species and thus the degradation rate are dependent on crystallite size when exposed to light and oxygen: perovskite films composed of small crystallites show high yields of photo-induced superoxide formation and therefore low stability. Ab initio simulations indicate that iodide vacancies are the preferred sites in mediating the photo-induced formation of superoxide species from O2. We also demonstrated that thin-film passivation with iodide salts leads to reduced superoxide formation, and consequently enhanced film and device stabilities. These combined results improve our fundamental understanding of degradation phenomena in perovskite solar cells, and provide a strategy for greatly improving their long-term stability.
Methods
Materials and synthesis
All chemicals were purchased from Sigma-Aldrich and used as received, except TiO2 nanoparticles from Dyesol and methylammonium iodide (MAI), which were synthesized in the lab. Methylamine 33% wt solution in ethanol (6.2 ml, 0.046 mol) was cooled down in an ice bath. Hydroiodic acid 55% wt solution in water (10 ml, 0.073 mol) was then added dropwise under vigorous stirring. The reaction was stirred for 1 h at 0 °C. The product precipitated from the solution as a white-yellowish solid. Ethanol (5 ml) was added to ensure full precipitation of the solid, which was filtered and washed with cold ethanol. Recrystallization of the product in ethanol/diethyl ether afforded the pure compound as white crystalline solid (6.4 g, 87%). Synthesis of phenyl-ethylammonium iodide (PEAI): Phenylethylamine 33% wt solution in ethanol (6.2 ml, 0.046 mol) was cooled down in an ice bath. Hydroiodic acid 55% wt solution in water (10 ml, 0.073 mol) was then added dropwise under vigorous stirring. The reaction was stirred for 1 h at 0 °C. The product precipitated from the solution as a white-yellowish solid. Ethanol (5 ml) was added to ensure full precipitation of the solid, which was filtered and washed with cold ethanol. Recrystallization of the product in ethanol/diethyl ether afforded the pure compound as white crystalline solid.
Film fabrication
All films were deposited onto clean glass substrates of ca. 1 cm by 1 cm in size. The glass substrates were washed sequentially in acetone, water and isopropylalcohol (IPA) under sonication for 10 min during each washing cycle. A Laurell Technologies WS-650MZ-23NPP Spin Coater was used to fabricate the films. (a) CH3NH3PbI3: A 1 M solution of CH3NH3PbI3 was formed by adding PbI2 in a 1:1 molar ratio with MAI in a solvent mixture of 7:3 γ-butyrolactone to DMSO. This solution was then spin-coated onto the substrates using a consecutive two-step spin program under a nitrogen atmosphere in a glove box. The first spinning cycle was performed at 1,000 r.p.m. for 10 s followed by 5,000 r.p.m. for 20 s, as reported by Jeon et al.70. During the second phase, the substrate was treated with toluene (ca. 350 ml) drop-casting. The films were then annealed at 100 °C for 10 min. (b) Crystal size variation: Films were prepared with controlled crystal size in accordance with the two-step deposition method described by Grätzel et al.50. Twenty microlitres of a 1 M solution of PbI2 in DMF was spin-coated onto glass substrates at 3,000 r.p.m. for 5 s and then at 6,000 r.p.m. for another 5 s. The films were then annealed at 40 °C for 3 min followed by heating at 100 °C for 5 min. Once the films had cooled to room temperature, 200 μl of 0.038 M (sample 3), 0.050 M (sample 2) and 0.063 M (sample 1) MAI in IPA solution were loaded on top of them for 20 s, before spinning at 4,000 r.p.m. for 20 s. Films were then annealed at 100 °C for 5 min. (c) Chlorine treated CH3NH3PbI3: A 1 M solution was created by dissolving PbCl2 and MAI in a 1:3 ratio in DMF. The solution was then spin-coated onto substrates at 1,000 r.p.m. for 10 s followed by 5,000 r.p.m. for 20 s. Annealing was carried out by leaving the films at room temperature for 30 min followed by heating at 100 °C for an hour.
Device fabrication
FTO-coated glass substrates (100 × 25 mm, 2.3 mm thick TEC15, Pilkington) were first etched with Zn power and aqueous hydrochloric acid (37%), and then cut into 25 × 25 mm pieces followed by cleaning sequentially in acetone, distilled water and IPA under sonication for 10 min during each washing cycle. A compact TiO2 layer was prepared by spin coating a solution comprised of 350 μl titanium isopropoxide (Aldrich), 35 μl hydrochloric acid (37%) and 5 ml anhydrous ethanol at 5,000 r.p.m. for 30 s. The TiO2 films were sintered at 160 °C for 5 min and then at 500 °C for 45 min. Next, a mesoporous-TiO2 film was deposited onto this using a solution of 20 nm particle transparent titania paste (18NR-T, Dyesol). The solution was spin-coated onto the substrates at 5,000 r.p.m. for 30 s. Once spun, the films were dried on a hotplate at 80 °C for 5 min, and then sintered at 500 °C for 45 min. The desired perovskite layer was prepared using a consecutive five-step spin program inside glovebox as previously described40. In instances where iodine salt layer were used, 1, 2, 6 and 10 mM solution of MAI with IPA were prepared respectively. The solution was then spin-coated onto the perovskite layer. The Spiro-OMeTAD hole conductor layer was spin-coated onto the perovskite films from a solution of 72.3 mg ml−1 2,2′,7,7′-tetrakis-(N,N-di-p-methoxyphenylamine)9,9′-spirobifluorene (spiro-OMeTAD) powder in 1 ml anhydrous chlorobenzene. The spiro-OMeTAD solution contained additives including 17.5 μl lithium bis(trifluoromethane) sulfonimide lithium salt (Li-TFSl) and 28.8 μl 4-tert-butylpyridine (tBP). Finally, a 100 nm-thick gold contact was evaporated under vacuum (approximately 10−6 T at a rate of 0.2 nm s−1) with an active pixel area of 0.12 cm2.
Current–voltage (JV) measurements
The JV characteristics were carried out using an AM1.5 simulated solar illumination (Oriel Instruments) and a Keithley 2400 source meter. Calibration was performed with a silicon photodiode before measurement. The scans were performed at a rate of 0.125 V s−1 for both forward scan (from short circuit open circuit) and backward scan (in the opposite direction). Cells were placed unmasked under continuous 1 sun illumination during aging and were masked during each scan to ensure the active area (0.12 cm2) is same for all measured devices.
Superoxide measurements
A stock 31.7 μM solution of the HE probe was prepared by dissolving 10 mg in 10 ml of dry toluene; sonication was used to facilitate miscibility. Films were then added to 10 ml of 0.317 μM solution created from the stock solution. Photoluminescence spectra were recorded using an excitation wavelength of 520 nm and slit widths of 10 mm on a Horiba Yobin-Ybon Fluorolog-3 spectrofluorometer.
Film coatings
A 0.01 M solution was prepared by dissolving the iodide salt (phenylethylammonium iodide, MAI or trimethylsulfonium iodide) in a 1:4 solvent mixture of IPA to chlorobenzene. One hundered microlitres of this solution was then dripped onto pre-deposited perovskite films with a 20 s loading time before spinning at 4,000 r.p.m. and annealing at 100 °C for 5 min. The chloride salt derivatives of the cations phenylethylammonium and methylammonium were prepared using the same protocol.
Ageing conditions
Films were sealed in a controlled environment, where dry air was gassed through for the duration of the degradation and illumination was provided by a tungsten lamp equipped with a UV-blocking filter as previously reported38,40.
Spectroscopy and microscopy
1H NMR was recorded on a 400 MHz Bruker spectrometer running and analysed using TopSpin software. Deuterated acetone was employed as the reference. UV–Vis was performed on a PerkinElmer UV/VIS Spectrometer Lambda 25. XRD patterns were measured on a PANalytical X’Pert Pro MRD diffractometer using Ni filtered Cu Kα radiation at 40 kV and 40 mA. SEM-EDX measurements were carried out on a JEOL 6400 scanning electron microscope operated at 20 kV. IGA measurements were conducted on a Mettler Toledo TGA spectrometer. For ToF-SIMS samples were fabricated onto clean glass substrates. The samples were then soaked under dry flux in the dark for an hour before the ToF-SIMS measurements were recorded. Data were obtained using an IONTOF ToF.SIMS-Qtac LEIS spectrometer employing an Argon sputter gun for oxygen ion detection.
Ab initio calculations
DFT calculations were performed using the numeric atom-centred basis set all-electron code FHI-AIMS71,72. Tight basis sets were used with tier 2 basis functions for all species. Electronic exchange and correlation were modelled with the semi-local PBE exchange-correlation functional73. For the treatment of spin orbit coupling we used an atomic zeroth-order regular approximation (ZORA)71. Van der Waals forces were accounted for by applying a Tkatchenko–Sheffler electrodynamic screening scheme74. O2 absorption in the bulk was calculated using a 2 × 2 × 1 supercell (giving a tetragonal cell of 192 atoms). A gamma point offset grid at a density of 0.04 Å−1 was used for k-point sampling. Structures were relaxed with convergence criteria of 10−4 eV Å−1 for forces, 10−5 electrons for the electron density and 10−7 eV for the total energy. These settings ensured highly converged energies and equilibrium distances.
Data availability
The data that support the findings of this study are available from the authors on reasonable request.
Additional information
How to cite this article: Aristidou, N. et al. Fast oxygen diffusion and iodide defects mediate oxygen-induced degradation of perovskite solar cells. Nat. Commun. 8, 15218 doi: 10.1038/ncomms15218 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Tan, Z.-K. et al. Bright light-emitting diodes based on organometal halide perovskite. Nat. Nanotechnol. 9, 687–692 (2014).
Lin, Q., Armin, A., Nagiri, R. C. R., Burn, P. L. & Meredith, P. Electro-optics of perovskite solar cells. Nat. Photonics 9, 106–112 (2015).
Hoye, R. L. Z. et al. Enhanced performance in fluorene-free organometal halide perovskite light-emitting diodes using tunable, low electron affinity oxide electron injectors. Adv. Mater. 27, 1414–1419 (2015).
Xing, G. et al. Low-temperature solution-processed wavelength-tunable perovskites for lasing. Nat. Mater. 13, 476–480 (2014).
Chin, X. Y., Cortecchia, D., Yin, J., Bruno, A. & Soci, C. Lead iodide perovskite light-emitting field-effect transistor. Nat. Commun. 6, 7383 (2015).
Grätzel, M. The light and shade of perovskite solar cells. Nat. Mater. 13, 838–842 (2014).
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Lee, M. M., Teuscher, J., Miyasaka, T., Murakami, T. N. & Snaith, H. J. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 338, 643–647 (2012).
Chen, W. et al. Efficient and stable large-area perovskite solar cells with inorganic charge extraction layers. Science 350, 944–948 (2015).
Yang, W. S. et al. High-performance photovoltaic perovskite layers fabricated through intramolecular exchange. Science 348, 1234–1237 (2015).
Berry, J. et al. Hybrid organic–inorganic perovskites (HOIPs): opportunities and challenges. Adv. Mater. 27, 5102–5112 (2015).
Kaltenbrunner, M. et al. Flexible high power-per-weight perovskite solar cells with chromium oxide-metal contacts for improved stability in air. Nat. Mater. 14, 1032–1039 (2015).
Xiao, Z. et al. Giant switchable photovoltaic effect in organometal trihalide perovskite devices. Nat. Mater. 14, 193–198 (2015).
McMeekin, D. P. et al. A mixed-cation lead mixed-halide perovskite absorber for tandem solar cells. Science 351, 151–155 (2016).
Babayigit, A., Ethirajan, A., Muller, M. & Conings, B. Toxicity of organometal halide perovskite solar cells. Nat. Mater. 15, 247–251 (2016).
Eperon, G. E. et al. Perovskite-perovskite tandem photovoltaics with optimized band gaps. Science 354, 861–865 (2016).
Saliba, M. et al. Incorporation of rubidium cations into perovskite solar cells improves photovoltaic performance. Science 354, 206–209 (2016).
Soon, Y. W. et al. Correlating triplet yield, singlet oxygen generation and photochemical stability in polymer/fullerene blend films. Chem. Commun. 49, 1291–1293 (2013).
Berhe, T. A. et al. Organometal halide perovskite solar cells: degradation and stability. Energy Env. Sci. 9, 323–356 (2016).
Pearson, A. J. et al. Oxygen degradation in mesoporous Al2O3/CH3NH3PbI3-xClx perovskite solar cells: kinetics and mechanisms. Adv. Energy Mater. 6, 1600014 (2016).
Yuan, H. et al. Degradation of methylammonium lead iodide perovskite structures through light and electron beam driven ion migration. J. Phys. Chem. Lett. 7, 561–566 (2016).
Christians, J. A., Miranda Herrera, P. A. & Kamat, P. V. Transformation of the excited state and photovoltaic efficiency of CH3NH3PbI3 perovskite upon controlled exposure to humidified air. J. Am. Chem. Soc. 137, 1530–1538 (2015).
Yang, J., Siempelkamp, B. D., Liu, D. & Kelly, T. L. Investigation of CH3NH3PbI3 degradation rates and mechanisms in controlled humidity environments using in situ techniques. ACS Nano 9, 1955–1963 (2015).
Schoonman, J. Organic–inorganic lead halide perovskite solar cell materials: a possible stability problem. Chem. Phys. Lett. 619, 193–195 (2015).
Tiep, N. H., Ku, Z. & Fan, H. J. Recent advances in improving the stability of perovskite solar cells. Adv. Energy Mater. 6, 1501420 (2016).
Tian, Y. et al. Mechanistic insights into perovskite photoluminescence enhancement: light curing with oxygen can boost yield thousandfold. Phys. Chem. Chem. Phys. 17, 24978–24987 (2015).
Leguy, A. M. A. et al. Reversible hydration of CH3NH3PbI3 in films, single crystals, and solar cells. Chem. Mater. 27, 3397–3407 (2015).
Zhang, L. & Sit, P. H.-L. Ab initio study of interaction of water, hydroxyl radicals, and hydroxide ions with CH3NH3PbI3 and CH3NH3PbBr3 surfaces. J. Phys. Chem. C 119, 22370–22378 (2015).
Misra, R. K. et al. Temperature- and component-dependent degradation of perovskite photovoltaic materials under concentrated sunlight. J. Phys. Chem. Lett. 6, 326–330 (2015).
Deretzis, I. et al. Atomistic origins of CH3NH3PbI3 degradation to PbI2 in vacuum. Appl. Phys. Lett. 106, 131904 (2015).
Han, Y. et al. Degradation observations of encapsulated planar CH3NH3PbI3 perovskite solar cells at high temperatures and humidity. J. Mater. Chem. A 3, 8139–8147 (2015).
Guerrero, A. et al. Interfacial degradation of planar lead halide perovskite solar cells. ACS Nano 10, 218–224 (2016).
Song, D. et al. Degradation of organometallic perovskite solar cells induced by trap states. Appl. Phys. Lett. 108, 093901 (2016).
Nie, W. et al. Light-activated photocurrent degradation and self-healing in perovskite solar cells. Nat. Commun. 7, 11574 (2016).
Ahn, N. et al. Trapped charge-driven degradation of perovskite solar cells. Nat. Commun. 7, 13422 (2016).
Li, B., Li, Y., Zheng, C., Gao, D. & Huang, W. Advancements in the stability of perovskite solar cells: degradation mechanisms and improvement approaches. RSC Adv. 6, 38079–38091 (2016).
You, J. et al. Improved air stability of perovskite solar cells via solution-processed metal oxide transport layers. Nat. Nanotechnol. 11, 75–81 (2016).
Aristidou, N. et al. The role of oxygen in the degradation of methylammonium lead trihalide perovskite photoactive layers. Angew. Chem. Int. Ed. 54, 8208–8212 (2015).
O’Mahony, F. T. F. et al. Improved environmental stability of organic lead trihalide perovskite-based photoactive-layers in the presence of mesoporous TiO2. J. Mater. Chem. A 3, 7219–7223 (2015).
Bryant, D. et al. Light and oxygen induced degradation limits the operational stability of methylammonium lead triiodide perovskite solar cells. Energy Environ. Sci. 9, 1655–1660 (2016).
Ball, J. M., Lee, M. M., Hey, A. & Snaith, H. J. Low-temperature processed meso-superstructured to thin-film perovskite solar cells. Energy Environ. Sci. 6, 1739–1743 (2013).
Bastiani, M. D., D’Innocenzo, V., Stranks, S. D., Snaith, H. J. & Petrozza, A. Role of the crystallization substrate on the photoluminescence properties of organo-lead mixed halides perovskites. APL Mater. 2, 081509 (2014).
Grancini, G. et al. The impact of the crystallization processes on the structural and optical properties of hybrid perovskite films for photovoltaics. J. Phys. Chem. Lett. 5, 3836–3842 (2014).
Nie, W. et al. High-efficiency solution-processed perovskite solar cells with millimeter-scale grains. Science 347, 522–525 (2015).
de Quilettes, D. W. et al. Impact of microstructure on local carrier lifetime in perovskite solar cells. Science 348, 683–686 (2015).
Srimath Kandada, A. R. & Petrozza, A. Photophysics of hybrid lead halide perovskites: the role of microstructure. Acc. Chem. Res. 49, 536–544 (2016).
Sharenko, A. & Toney, M. F. Relationships between lead halide perovskite thin-film fabrication, morphology, and performance in solar cells. J. Am. Chem. Soc. 138, 463–470 (2016).
Zhang, W. et al. Enhanced optoelectronic quality of perovskite thin films with hypophosphorous acid for planar heterojunction solar cells. Nat. Commun. 6, 10030 (2015).
Salim, T. et al. Perovskite-based solar cells: impact of morphology and device architecture on device performance. J. Mater. Chem. A 3, 8943–8969 (2015).
Im, J.-H., Jang, I.-H., Pellet, N., Grätzel, M. & Park, N.-G. Growth of CH3NH3PbI3 cuboids with controlled size for high-efficiency perovskite solar cells. Nat. Nanotechnol. 9, 927–932 (2014).
Li, X. et al. Improved performance and stability of perovskite solar cells by crystal crosslinking with alkylphosphonic acid ω-ammonium chlorides. Nat. Chem. 7, 703–711 (2015).
Walsh, A., Scanlon, D. O., Chen, S., Gong, X. G. & Wei, S.-H. Self-regulation mechanism for charged point defects in hybrid halide perovskites. Angew. Chem. Int. Ed. 54, 1791–1794 (2015).
Eames, C. et al. Ionic transport in hybrid lead iodide perovskite solar cells. Nat. Commun. 6, 7497 (2015).
Azpiroz, J. M., Mosconi, E., Bisquert, J. & De Angelis, F. Defect migration in methylammonium lead iodide and its role in perovskite solar cell operation. Energy Environ. Sci. 8, 2118–2127 (2015).
Yang, T.-Y., Gregori, G., Pellet, N., Grätzel, M. & Maier, J. The significance of ion conduction in a hybrid organic–inorganic lead-iodide-based perovskite photosensitizer. Angew. Chem. Int. Ed. 54, 7905–7910 (2015).
Richardson, G. et al. Can slow-moving ions explain hysteresis in the current–voltage curves of perovskite solar cells? Energy Environ. Sci. 9, 1476–1485 (2016).
Egger, D. A., Rappe, A. M. & Kronik, L. Hybrid organic–inorganic perovskites on the move. Acc. Chem. Res. 49, 573–581 (2016).
de Quilettes, D. W. et al. Photo-induced halide redistribution in organic-inorganic perovskite films. Nat. Commun. 7, 11683 (2016).
Colella, S. et al. MAPbI3-xClx mixed halide perovskite for hybrid solar cells: the role of chloride as dopant on the transport and structural properties. Chem. Mater. 25, 4613–4618 (2013).
Aldibaja, F. K. et al. Effect of different lead precursors on perovskite solar cell performance and stability. J. Mater. Chem. A 3, 9194–9200 (2015).
Hintz, H. et al. Photodegradation of P3HT—a systematic study of environmental factors. Chem. Mater. 23, 145–154 (2011).
Lüer, L. et al. Oxygen-induced quenching of photoexcited states in polythiophene films. Org. Electron. 5, 83–89 (2004).
Jørgensen, M. et al. Stability of polymer solar cells. Adv. Mater. 24, 580–612 (2012).
Beaupré, S. & Leclerc, M. PCDTBT: en route for low cost plastic solar cells. J. Mater. Chem. A 1, 11097 (2013).
Calloni, A. et al. Stability of organic cations in solution-processed CH3NH3PbI3 perovskites: formation of modified surface layers. J. Phys. Chem. C 119, 21329–21335 (2015).
Leblebici, S. Y. et al. Facet-dependent photovoltaic efficiency variations in single grains of hybrid halide perovskite. Nat. Energy 1, 16093 (2016).
Haruyama, J., Sodeyama, K., Han, L. & Tateyama, Y. Termination dependence of tetragonal CH3NH3PbI3 surfaces for perovskite solar cells. J. Phys. Chem. Lett. 5, 2903–2909 (2014).
Yen, H.-J. et al. Large grained perovskite solar cells derived from single-crystal perovskite powders with enhanced ambient stability. ACS Appl. Mater. Interfaces 8, 14513–14520 (2016).
Boopathi, K. M. et al. Synergistic improvements in stability and performance of lead iodide perovskite solar cells incorporating salt additives. J. Mater. Chem. A 4, 1591–1597 (2016).
Jeon, N. J. et al. Solvent engineering for high-performance inorganic–organic hybrid perovskite solar cells. Nat. Mater. 13, 897–903 (2014).
Blum, V. et al. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 180, 2175–2196 (2009).
Ren, X. et al. Resolution-of-identity approach to Hartree–Fock, hybrid density functionals, RPA, MP2 and GW with numeric atom-centered orbital basis functions. New J. Phys. 14, 053020 (2012).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Tkatchenko, A., Ambrosetti, A. & DiStasi, R. A. Jr. Interatomic methods for the dispersion energy derived from the adiabatic connection fluctuation-dissipation theorem. J. Chem. Phys. 138, 074106 (2013).
Acknowledgements
M.S.I. acknowledges support from the EPSRC for the Energy Materials Programme grant (EP/K016288) and Archer HPC facilities through the Materials Chemistry Consortium (EP/L000202). S.A.H. acknowledges financial support from EPRSC via EP/M023532/1, EP/K010298/1 and EP/K030671/1 grants. We thank L.M. Peter (Bath) and C.A.J. Fisher (JFCC, Nagoya) for useful discussions.
Author information
Authors and Affiliations
Contributions
S.A.H. conceived and supervised the experimental project. M.S.I. and C.E. conceived and performed the computational work. N.A. performed optical spectroscopy, superoxide yield, salt coating experiments and microscopy studies, I.S.-M. conducted optical spectroscopy experiments, J.K. conducted the salt coating experiments and X.B. fabricated and characterized the solar cell devices. S.A.H., N.A., M.S.I. and C.E. drafted the initial versions of the paper. All authors contributed to the analysis, discussion and writing of the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures, Supplementary Table and Supplementary Notes (PDF 1158 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Aristidou, N., Eames, C., Sanchez-Molina, I. et al. Fast oxygen diffusion and iodide defects mediate oxygen-induced degradation of perovskite solar cells. Nat Commun 8, 15218 (2017). https://doi.org/10.1038/ncomms15218
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms15218
This article is cited by
-
Phase stabilization of cesium lead iodide perovskites for use in efficient optoelectronic devices
NPG Asia Materials (2024)
-
Mobile iodides capture for highly photolysis- and reverse-bias-stable perovskite solar cells
Nature Materials (2024)
-
Chemical Aspects of Halide Perovskite Nanocrystals
Topics in Current Chemistry (2024)
-
Ultrafast photo-induced carrier dynamics of perovskite films being degraded by atmospheric exposure
Journal of the Korean Physical Society (2024)
-
Chloride incorporation for the stability improvement of the MAPI hybrid perovskite
Journal of Materials Science: Materials in Electronics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
