
Abstract
Conventional adenomas are initiated by APC gene mutation that activates the WNT signal. Serrated neoplasia is commonly initiated by BRAF or KRAS mutation. WNT pathway activation may also occur, however, to what extent this is owing to APC mutation is unknown. We examined aberrant nuclear β-catenin immunolocalization as a surrogate for WNT pathway activation and analyzed the entire APC gene coding sequence in serrated and conventional pathway polyps and cancers. WNT pathway activation was a common event in conventional pathway lesions with aberrant nuclear immunolocalization of β-catenin and truncating APC mutations in 90% and 89% of conventional adenomas and 82% and 70% of BRAF wild-type cancers, respectively. WNT pathway activation was seen to a lesser extent in serrated pathway lesions. It occurred at the transition to dysplasia in serrated polyps with a significant increase in nuclear β-catenin labeling from sessile serrated adenomas (10%) to sessile serrated adenomas with dysplasia (55%) and traditional serrated adenomas (9%) to traditional serrated adenomas with dysplasia (39%) (P=0.0001). However, unlike the conventional pathway, truncating APC mutations were rare in the serrated pathway lesions especially sessile serrated adenomas even when dysplastic (15%) and in the BRAF mutant cancers with microsatellite instability that arise from them (8%). In contrast, APC missense mutations that were rare in conventional pathway adenomas and cancers (3% in BRAF wild-type cancers) were more frequent in BRAF mutant cancers with microsatellite instability (32%). We conclude that increased WNT signaling is important in the transition to malignancy in the serrated pathway but that APC mutation is less common and the spectrum of mutations is different than in conventional colorectal carcinogenesis. Moderate impact APC mutations and non-APC-related causes of increased WNT signaling may have a more important role in serrated neoplasia than the truncating APC mutations common in conventional adenomas.
Similar content being viewed by others


Main
Two major molecular pathways leading to the development of colorectal cancer are recognized. Approximately 70% of sporadic colorectal carcinomas arise via the conventional adenoma–carcinoma pathway1 with the remaining 30% thought to arise via a different series of precursors and molecular changes known as the serrated neoplasia pathway.2, 3, 4, 5, 6 Both pathways show mutations of genes involved in cellular signaling central to colorectal homeostasis. In the conventional pathway, a relatively consistent sequence of genetic and epigenetic changes in tumor suppressor genes and oncogenes have been found to parallel histological adenoma–carcinoma progression. This includes somatic APC inactivation, usually owing to pathogenic biallelic mutations in ~70% of very early microadenomas, adenomas with low or high-grade dysplasia and carcinomas, consistent with APC being an important initiating event in colorectal tumorigenesis in this pathway.7, 8 The most important consequence of APC mutation is thought to be aberrant activation of the WNT signaling pathway.9 Conversely, in the serrated neoplasia pathway mutations of BRAF or KRAS are thought to initiate development of serrated polyps including sessile serrated adenomas and traditional serrated adenomas via activation of the MAP kinase pathway.3 The role of aberrant WNT signaling in the serrated pathway is less clear than for the conventional adenoma–carcinoma pathway.
Nuclear translocation of the normally membranous β-catenin protein indicates canonical WNT pathway activation. Most conventional adenomas show nuclear β-catenin accumulation, which is a downstream consequence of pathogenic APC mutation.10, 11 Whereas some studies have shown increased nuclear β-catenin staining by immunohistochemistry in serrated precursor polyps with neoplastic progression,12, 13, 14 reports are inconsistent.15 Truncating APC mutations appear to be uncommon in lesions of the serrated pathway, but again the literature shows marked variation of reported rates in serrated pathway lesions including precursor polyps16, 17, 18, 19, 20, 21, 22, 23 and the BRAF mutant cancers that arise from them.24, 25, 26 Many studies show limited mutational analysis focusing only on the mutational cluster region at codons 1281–1556 of the APC gene. Although this is thought to contain ~50–60% of truncating APC mutations,27, 28 it has the potential to miss up to 40% of mutations. Inconsistent and confusing nomenclature of serrated pathway lesions29, 30 are likely contributors for the widely variable rates of nuclear β-catenin and APC mutations found in literature. Fortunately, a standardized diagnostic nomenclature of different serrated polyps was formulated in 2010.31
To address these defects in our knowledge and understanding of the role of APC mutations and WNT pathway activation in the serrated neoplasia pathway, we have undertaken a comprehensive mutational analysis of the entire coding region of the APC gene in combination with immunohistochemical assessment of β-catenin cellular location in a large cohort of colorectal cancers stratified by BRAF mutation and a broad spectrum of currently defined serrated pathway lesions. We find that although truncating APC mutations are rare, missense APC mutations are relatively common in the serrated pathway cancers. We suggest the possibility that missense APC mutations may be contributors to the ‘just-right’ level of WNT/β-catenin signaling32 required for colorectal tumorigenesis via the serrated neoplasia pathway. We also confirm that truncating APC mutation does not account for most of the increased WNT signaling in the serrated pathway.
Materials and methods
Samples
The WNT pathway was examined in a range of colorectal polyps and cancers by immunohistochemical assessment of β-catenin cellular localization and APC mutational analysis. The BRAFV600E mutation was detected by allele-specific PCR as previously described.33 Lynch syndrome cancers were effectively excluded as BRAF mutant cancers do not arise in the context of Lynch Syndrome and all the BRAF wild-type cancers showed retention of mismatch repair proteins.
This included immunohistochemical assessment of serrated pathway lesions comprising 102 BRAF mutant cancers (stratified by microsatellite instability status including 62 cancers with microsatellite instability and 40 microsatellite stable cancers), 162 traditional serrated adenomas, 38 traditional serrated adenomas with dysplasia, 20 sessile serrated adenomas, and 137 sessile serrated adenomas with dysplasia. β-catenin was scored separately in the dysplastic and non-dysplastic parts of the sessile serrated adenomas with dysplasia to give 157 results for β-catenin in non-dysplastic sessile serrated adenomas. As a control group 354 BRAF wild-type cancers and 96 conventional adenomas (including 46 tubular adenomas and 50 tubulovillous adenomas) were assessed. BRAF mutant carcinomas and BRAF wild-type carcinomas were included in the serrated pathway group and the conventional pathway group respectively based on evidence of the occurrence of BRAF mutations in these pathways.3, 34 Histological assessment of serrated precursor polyps followed the guidelines of the World Health Organization.31 Our traditional serrated adenoma group consists of those traditional serrated adenomas without overt morphological atypia or mitotic activity (sometimes also referred to as ordinary traditional serrated adenomas), and our traditional serrated adenoma with dysplasia group refers to those, which have developed overt cytological dysplasia and/or mitotic activity (also sometimes referred to as advanced traditional serrated adenomas).
The APC-coding region was sequenced in a subset of 189 of these cases including 80 BRAF mutant cancers (50 microsatellite unstable and 30 microsatellite stable), 20 sessile serrated adenoma, 20 sessile serrated adenoma with dysplasia, 14 traditional serrated adenoma, 6 traditional serrated adenoma with dysplasia, 30 BRAF wild-type colorectal cancer, and 19 conventional adenomas (10 tubulovillous adenomas and 9 tubular adenomas).
Traditional serrated adenomas, traditional serrated adenomas with dysplasia and sessile serrated adenomas with dysplasia were collected in a consecutive fashion by a single pathologist and author (NW) between June 2007 and June 2013 and have been included in prior studies.12, 35 The sessile serrated adenomas, tubular adenomas, and tubulovillous adenomas were also sourced from archival specimens at Envoi Specialist Pathologists over the same time period but were not consecutive. The 456 carcinomas were collected in a consecutive fashion by all pathologists at Envoi Specialist Pathologists between January 2011 and June 2012. Cases with insufficient material for complete immunohistochemical and molecular analysis were excluded, including 23 rectal cancers with pre-operative radiotherapy and without pre-operative biopsy. The study was approved by the ethics committee of QIMR Berghofer Medical Research Institute (P1298).
Immunohistochemical Analysis
For all cases, aberrant nuclear immunolocalization of β-catenin was used as a surrogate for WNT pathway activation, compared with the normal membranous staining pattern. Cytoplasmic β-catenin staining found in 98% of cases did not further contribute to analysis. Immunohistochemistry was performed on tissue sections cut from the formalin-fixed, paraffin-embedded blocks. Sections were cut at 4 μm and then dewaxed and rehydrated. Antigen retrieval for β-catenin was performed by incubation in low pH antigen retrieval solution (pH 6.0, Biocare Medical, Concord, CA, USA) at 112 °C for 7 min. Sections were then stained following the manufacturer’s instructions (1:600, Cell Marque, Rocklin, CA, USA). Slides were counterstained with Mayer’s haematoxylin. Cases were assessed for both intensity and the proportion of cells staining positive in the nucleus by JB as previously described.12 Intensity was scored as 0–3 (0=no staining, 1=weak staining, 2=moderate staining, 3=strong staining) and extent 0–4 (0=no staining, 1=1–10% of cells, 2=11–50% of cells, 3=51–90% of cells, 4≥90% of cells). When intensity was variable, an average of the intensity was used. Positive β-catenin required nuclear staining with a score of ≥2 in the intensity and/or extent category. A semiquantitative nuclear score was calculated by multiplying the nuclear intensity and nuclear extent giving three categories of low (1–4), moderate (5–8), and high (9–12) and used to test for possible association between β-catenin expression and the type of APC mutation.
APC Mutational Analysis
Targeted amplicon sequencing was conducted using a custom enrichment panel including 242 amplicons spanning across the entire coding region of APC (GeneRead DNAseq Targeted Panel, Qiagen). DNA was extracted from the formalin-fixed, paraffin-embedded blocks using the Chelex-100 extraction method (Bio-Rad Laboratories, Hercules, CA, USA). In brief, three 10 μm sections were cut from the FFPE blocks and heated to 90 °C in 200 μl of 0.5% Tween-20 in 1 × TE and then digested with 80 mg of proteinase K at 55 °C for 3 h. After digestion, 200 μl of 5% Chelex-100 was added to the samples and they were heated to 99°, centrifuged and then cooled on ice, and then the paraffin removed. Then, 200 μl of chloroform was added, the samples centrifuged for 15 min, and the final product from the surface phase removed by manual pipette. DNA concentration was established by spectrophotometry (NanoDrop 2000, Thermo Scientific, Fremont, CA, USA). In cases where there was contamination of the formalin-fixed, paraffin-embedded block by non-lesional tissue, manual microdissection was performed using a sterile scalpel blade with a marked haematoxylin and eosin-stained section as a guide. Library preparation was completed using the TruSeq Nano DNA HT Sample Prep Kit (Illumina) as per the manufacturers’ recommendations. Libraries were subsequently sequenced on MiSeq.
For data processing reads were aligned to hg19 using BWA-MEM (version 0.7.10-r879). Variants were called using the QIAGEN low variance CLC Biomedical workbench variant caller. We annotated all genotyped variants with SnpEff 4.2 (build 2015-12-05)36 classifying variants based on their putative effect on the protein product. Truncating mutations categorized as high impact include nonsense, frameshift, and splice site mutations. The significance of moderate impact missense mutations were assessed by pathogenicity prediction tools (CADD, MetaSMV, PolyPhen2 HDIV and HVAR, Mutation Assessor, LRT, MetaLR, SIFT, FATHMM, and Mutation Taster softwares). The websites were simultaneously consulted using dedicated Alamut Interactive Biosoftware.37 In addition, variant allele frequency within the sample was considered since a somatic mutation within one allele would not be expected to be present in >40% of the reads since all samples had at least 20% non-neoplastic cell contamination. Further, the literature was searched for information regarding identified missense variants. Silent and modifier variants were considered to be low impact and were excluded from downstream analysis.
Statistical Analysis
Categorical variables were compared by Fisher’s exact test. A P-value of ≤0.05 was considered significant. GraphPad Prism version 6.02 was used for statistical analyses.
Results
Immunohistochemical assessment of cellular β-catenin location showed nuclear β-catenin staining in most conventional pathway lesions including 86/96 (90%) of conventional adenomas and 290/354 (82%) of BRAF wild-type cancers (Table 1). Lower rates of nuclear β-catenin staining were seen in serrated polyps including sessile serrated adenomas 15/157 (10%) and traditional serrated adenomas 15/162 (9%). A significant increase in nuclear β-catenin staining was seen at the transition to dysplasia in both serrated polyps: sessile serrated adenomas with dysplasia 75/137 (55%) and traditional serrated adenomas with dysplasia 15/38 (39%) (P=0.0001) (Table 1 and Figures 1a and d). This was similar to the prevalence of nuclear β-catenin staining in BRAF mutant colorectal cancer (45/102, 44%) and was approximately half of that seen in BRAF wild-type colorectal cancers (290/354, 82%).

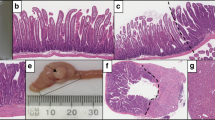
Sessile serrated adenoma (a) and traditional serrated adenoma (c) showing transition to dysplasia, with aberrant nuclear β-catenin in the dysplastic component only (b, d) respectively). Invasive carcinoma (e) with high power view of nuclear β-catenin positivity (f).
The coding region of APC was sequenced in 189 samples. Rates of β-catenin staining were similar between the large immunostained cohort and smaller mutation cohorts for all morphological subgroups except for traditional serrated adenomas, which showed staining in 15/162 (9%) of the overall cohort compared with 4/14 (29%) of cases selected for mutational analysis.
Samples contained an average of 646074.93 reads. A total of 99.96% of bases were covered at × 100 depth. A total of 129 variants were detected. Sixty-one were annotated as high impact variants consisting of truncating mutations including 40 nonsense, 17 frameshift and 4 splice site mutations. Twenty-nine were annotated as moderate impact and consisted of missense mutations. Of the missense variants, eight were excluded based on a high variant allele frequency of 40–60%, suggesting a germline polymorphism rather than somatic mutation and two variants (V1822D and G2502S) had previously been shown to be non-deleterious38 (See Supplementary Table 1). Alamut in silico prediction provided information pertaining to possible pathogenicity of missense variants and is presented in Supplementary table 1, however these predictions were not included in further analyses due to the difficulty in assessing pathogenicity of sporadic mutations in programs primarily designed for analyzing germline variants. The remainder of variants was low impact and modifier variants and were excluded from further analysis. Some samples had two or more truncating APC mutations. This included 11 conventional adenomas, 6 BRAF wild-type colorectal cancer, 1 sessile serrated adenomas with dysplasia, 2 traditional serrated adenomas, 1 traditional serrated adenoma with dysplasia, and 3 BRAF mutant microsatellite stable colorectal cancer. In addition, cases showing a truncating and a missense mutation included one case each from the groups conventional adenoma, BRAF wild-type colorectal cancer and traditional serrated adenoma. One BRAF mutant microsatellite unstable colorectal cancer showed two missense mutations.
As expected, adenomas and cancers of the conventional pathway showed high rates of truncating APC mutations in conventional adenomas 17/19 (89%) and BRAF wild-type cancers 21/30 (70%) and this correlated with the high rates of nuclear β-catenin staining. More intense or diffuse β-catenin staining reflected by moderate or high nuclear scores5, 6, 7, 8, 9, 10, 11, 12 was more frequent in cases with truncating APC mutations 24/57 (42%) than those with missense mutations 1/21 (5%) (P=0.0020) (Table 2). Conversely, truncating APC mutations were rare in sessile serrated adenomas (none) and sessile serrated adenomas with dysplasia 3/20 (15%), as well as in BRAF mutant cancers 9/80 (11%) (Table 1 and Supplementary Table 2). Traditional serrated adenomas 5/14 (36%), and traditional serrated adenomas with dysplasia 2/6 (33%) had intermediate rates although the small numbers limit interpretation in this rare polyp type. Four cases with one or more truncating APC mutation did not show nuclear β-catenin staining. This included two BRAF wild-type colorectal cancers, one BRAF mutant cancer with microsatellite unstable, and one traditional serrated adenoma. Tissue section preparation was good for these cases and lesional tissue showed membranous and areas of cytoplasmic staining, indicative of satisfactory immunohistochemical technique. No explanatory histological features were observed, and three of the four cases had truncating mutations within the mutational cluster region of the APC gene. Furthermore, no specific histological features were identified within the subsets of polyp types and colorectal cancers, to correlate with or predict APC mutation status.
The rate of truncating mutations in the sessile serrated adenomas with dysplasia (15%) and BRAF mutant cancers (11%) was significantly lower than immunohistochemical evidence of WNT signaling activation (P=0.0079 and P=0.0001, respectively)) suggesting there must be other causes of WNT activation. In BRAF mutant microsatellite unstable cancers, there was a significantly higher rate of missense APC mutation compared to BRAF mutant microsatellite stable cancers (P=0.0299) and BRAF wild-type cancers (P=0.0019) (Table 1). It is possible that at least some of these mutations may be contributing to increased WNT signaling in these cancers.
A common pattern of positivity was a score of 1 for nuclear extent and score of 2 for nuclear intensity (Supplementary Table 1). An extent score of 1 (1–10% of cells) would be consistent with staining in a small number of cells. In total, 42/105 (40%) cases showed this pattern (nuclear intensity 2, nuclear extent 1). Of those 42 cases, most were identified in cancers 33/42 (79%), particularly those with BRAF mutation 29/42 (69%), both microsatellite stable 13/30 (43%) and microsatellite unstable 16/50 (32%) cancers. A pattern of strong nuclear β-catenin staining at the invasive front of colorectal cancers with no nuclear staining in the center of the tumor toward the luminal surface was commonly observed.
Evidence of combined truncating and missense mutation was identified in a tubulovillous adenoma (X1486FS and D1484E), a BRAF wild-type cancer (Q1090X and P2158R), a traditional serrated adenoma (S811X and N30Y) and as a ‘third hit’ in a traditional serrated adenoma with dysplasia (X1307FS, splice site mutation, and N30Y).
Of the truncating mutations identified, 62% in the conventional pathway, and 33–37% in the serrated pathway occurred outside of the mutational cluster region. In total, 88% of missense mutations identified occurred outside of the mutational cluster region (see Figure 2). Evidence of retention of 1–2 20 amino-acid repeats was identified in 82% of conventional adenomas and 80% of BRAF wild-type cancers (see Supplementary Table 2 and Figure 2).

APC mutations in colorectal carcinomas (a) and colorectal polyps (b). Selected regions of the APC gene are shown including OD=oligomerization domain, Armadillo repeats, 15AA repeats, MCR=mutational cluster region, 20AA repeats, EB1-binding site and hDLG region. In colorectal carcinomas in (a) red circles represent BRAF wild-type colorectal cancer, blue circles BRAF mutant microsatellite unstable colorectal cancer, and green circles BRAF mutant microsatellite stable colorectal cancer. In colorectal polyps in (b) red circles represent conventional adenomas (tubular adenoma, tubulovillous adenoma), blue circles sessile serrated adenoma and sessile serrated adenoma with dysplasia, and green circles traditional serrated adenomas and traditional serrated adenomas with dysplasia.
Discussion
Mutations in APC occur in familial adenomatous polyposis,39 and occur frequently in sporadic colorectal cancers.40, 41, 42 Our findings are consistent with the initiating event of truncating APC mutations and subsequent increased WNT signaling in the conventional pathway. Most of our conventional adenomas and BRAF wild-type cancers showed nuclear β-catenin localization (90 and 82%, respectively) and truncating APC mutation (89 and 70%, respectively). The lower rate of APC mutation in BRAF wild-type carcinomas is not unexpected as this group will also contain some serrated pathway cancers with KRAS mutation or BRAF mutations other than V600E.
The evolution of the serrated neoplasia pathway through which 30% of colon cancers progress is less well understood.3, 4, 43 Mutation of BRAF, virtually never observed in conventional adenomas, is thought to be an early event in the initiation of sessile serrated adenomas. Progression to sessile serrated adenomas with dysplasia and subsequent BRAF mutant colorectal cancers is associated with DNA CpG island methylation, which can silence important tumor suppressor genes.44, 45, 46, 47, 48, 49 CpG island methylation of the promoter region of MLH-1 is a common event in sessile serrated adenomas with dysplasia and this causes mismatch repair deficiency leading to microsatellite unstable, which profoundly influences subsequent tumorigenesis. Mismatch repair deficiency greatly increases the rate of mutation and some of these mutations are functionally important. Traditional serrated adenomas are rare, and the least understood of the precursor polyps. These develop via sessile serrated adenomas with BRAF mutation, or alternatively by initiating KRAS mutation. Traditional serrated adenomas progress to microsatellite stable cancers, which can definitely be traced to the serrated pathway when they have a BRAF mutation.50
The role of aberrant WNT signaling in the serrated pathway however is less clear. The present study is consistent with previous evidence of WNT pathway activation at the transition to dysplasia in both sessile serrated adenomas and traditional serrated adenomas with an increase in nuclear β-catenin from 10% in sessile serrated adenomas to 55% in sessile serrated adenomas with dysplasia, and 9% in traditional serrated adenomas to 39% in traditional serrated adenomas with dysplasia. No further increase of WNT activation is seen in BRAF mutant cancers (44%), indicating that altered WNT signaling facilitates progression of serrated polyps. Although high rates of truncating APC mutation correlated with WNT pathway activation in conventional pathway lesions, this correlation was not evident in serrated pathway lesions. In total, 55% of sessile serrated adenomas with dysplasia showed immunohistochemical evidence of WNT pathway activation, but only 15% had truncating APC mutations. Similarly, 44% of BRAF mutant cancers showed evidence of WNT pathway activation, but only 11% had truncating APC mutations, suggesting other mechanisms of activating WNT are common in serrated neoplasia.
A number of alternative mechanisms of WNT activation have been identified and these generally do not result in such marked upregulation of WNT signaling as do truncating APC mutations. Levels of β-catenin in the cytoplasm are regulated via WNT receptors (FZD, LRP5/6),51 RNF43,52 secreted proteins (SFRPS, WIF, DKK, WISE/SOST, Norrin, R-spondins)53, 54, 55, 56, 57 and by a destruction complex including APC, AXIN, GSK-3beta, CK1alpha, which allows phosphorylation, ubiquitination, and proteasomal degradation of β-catenin in the absence of WNT ligand. Concentrations of β-catenin are normally kept low in the cytoplasm to prevent translocation to the nucleus. When WNT signaling is active translocation of β-catenin into the nucleus recruits TCF/LEF family cofactors activating target gene transcription.58 Mutations and methylation events have been identified involving several of WNT pathway components other than APC. Mutations in the gene encoding β-catenin (CTNNB1) are found in 5% of colorectal cancers; however, these predominantly occur in Lynch syndrome cancers.59 Inactivating mutations in the transmembrane ligase antagonists RNF43 and its functional homolog ZNRF3 have been reported in colorectal cancers particularly BRAF mutant microsatellite unstable cancers52, 60, 61, 62, 63, 64 and traditional serrated adenomas.18 Rearrangements involving R-spondin genes RSPO2 and RSPO3 have been reported in 10% of colorectal cancers,65, 66 and PTPRK-RSPO3 fusions were reported in traditional serrated adenomas.18 Methylation induced silencing of upstream WNT suppressors such as the SFRPs, and mutated in colorectal cancer gene (MCC) are reported to be common in sessile serrated adenomas.67, 68, 69 Our results suggest that truncating APC mutations are very uncommon in the serrated pathway and it is likely that WNT signaling is predominantly altered by the mechanisms described above. It is possible that these alternate mechanisms activate the WNT signal to a lesser extent and these synergize better with the existing BRAF mutation and are thus selected in these polyps.
There was a markedly higher incidence of missense APC mutations in serrated pathway cancers (24%), particularly, BRAF mutant cancers with microsatellite unstable (32%) compared with BRAF wild-type cancers (3%). Contrary to their prior lack of association with truncating mutations or loss of heterozygosity we showed missense variants concurrent with truncating mutations in four of our cases. This raises the possibility that they may not be non-pathogenic bystanders.70 These mutations may arise as a consequence of microsatellite instability but may be selected for because they moderately increase WNT signaling, thus providing a growth advantage to the mutated cells.
A particular pattern of β-catenin staining was observed frequently in BRAF mutant cancers (69%). These cases showed moderate nuclear staining in a small percentage of cells at the invasive front, but lack of nuclear localization in the center of the tumor, or toward the luminal surface. This phenomenon has been previously described71, 72, 73 in colorectal cancer and discussion has pertained to its significance in relation to molecular changes related to epithelial–mesenchymal transition. Furthermore, one study showed an association between this pattern of β-catenin staining and increased frequency of liver metastasis.74 This pattern of staining occurred predominantly in BRAF mutant cancers and may contribute to the epithelial–mesenchymal transition in this tumor subgroup.75
There were some limitation in this study related to lack of availability of non-neoplastic tissue for validation of excluded germline missense variants based on variant allele frequency (see methodology), small polyp numbers for the traditional serrated adenoma category, and lack of loss of heterozygosity analysis at the APC locus.
In summary, we have examined WNT activation immunohistochemically and APC mutation by sequencing the entire coding region in a comprehensive cohort of serrated polyp precursors and cancers. Our study confirms the timing WNT pathway activation at the transition to dysplasia in serrated precursor polyps. Interestingly, although truncating APC mutations were infrequent in serrated pathway lesions, we found missense APC mutations to be overrepresented in this group, particularly BRAF mutant cancers with microsatellite instability. Missense APC mutations require further investigation as potential activators or co-activators of WNT signaling in the serrated neoplasia pathway.
References
Fearon ER, Vogelstein B . A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767.
Snover DC . Update on the serrated pathway to colorectal carcinoma. Hum Pathol 2011;42:1–10.
Leggett B, Whitehall V . Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010;138:2088–2100.
Bettington M, Walker N, Clouston A et al. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology 2013;62:367–386.
Rex DK, Ahnen DJ, Baron JA et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 2012;107:1315–1329.
O'Brien MJ, Zhao Q, Yang S . Colorectal serrated pathway cancers and precursors. Histopathology 2015;66:49–65.
Vogelstein B, Fearon ER, Hamilton SR et al. Genetic alterations during colorectal-tumour development. N Engl J Med 1988;319:525–532.
Jen J, Powell SM, Papadopoulos N et al. Molecular determinants of dysplasia in colorectal lesions. Cancer Res 1994;54:5523–5526.
Nathke IS . The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol 2004;20:337–366.
Morin PJ, Sparks AB, Korinek V et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275:1787–1790.
Joo M, Shahsafaei A, Odze RD . Paneth cell differentiation in colonic epithelial neoplasms: evidence for the role of the Apc/beta-catenin/Tcf pathway. Hum Pathol 2009;40:872–880.
Bettington ML, Walker NI, Rosty C et al. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod Pathol 2015;28:414–427.
Yachida S, Mudali S, Martin SA et al. Beta-catenin nuclear labeling is a common feature of sessile serrated adenomas and correlates with early neoplastic progression after BRAF activation. Am J Surg Pathol 2009;33:1823–1832.
Fujita K, Yamamoto H, Matsumoto T et al. Sessile serrated adenoma with early neoplastic progression: a clinicopathologic and molecular study. Am J Surg Pathol 2011;35:295–304.
Fu X, Li L, Peng Y . Wnt signalling pathway in the serrated neoplastic pathway of the colorectum: possible roles and epigenetic regulatory mechanisms. J Clin Pathol 2012;65:675–679.
Boparai KS, Dekker E, Van Eeden S et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology 2008;135:2014–2018.
Boparai KS, Dekker E, Polak MM et al. A serrated colorectal cancer pathway predominates over the classic WNT pathway in patients with hyperplastic polyposis syndrome. Am J Pathol 2011;178:2700–2707.
Sekine S, Yamashita S, Tanabe T et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J Pathol 2016;239:133–138.
Qiu Y, Fu X, Zhang W et al. Prevalence and molecular characterisation of the sessile serrated adenoma in a subset of the Chinese population. J Clin Pathol 2014;67:491–498.
Hiyama T, Yokozaki H, Shimamoto F et al. Frequent p53 gene mutations in serrated adenomas of the colorectum. J Pathol 1998;186:131–139.
Fu X, Li J, Li K et al. Hypermethylation of APC promoter 1A is associated with moderate activation of Wnt signalling pathway in a subset of colorectal serrated adenomas. Histopathology 2009;55:554–563.
Shi Y, Li J, Wu SY et al. BRAF mutation is associated with the unique morphology of traditional serrated adenoma of the colorectum. Int J Surg Pathol 2013;21:442–448.
Uchida H, Ando H, Maruyama K et al. Genetic alterations of mixed hyperplastic adenomatous polyps in the colon and rectum. Jpn J Cancer Res 1998;89:299–306.
Suehiro Y, Wong CW, Chirieac LR et al. Epigenetic-genetic interactions in the APC/WNT, RAS/RAF, and P53 pathways in colorectal carcinoma. Clin Cancer Res 2008;14:2560–2569.
TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337.
Scholtka B, Schneider M, Melcher R et al. A gene marker panel covering the Wnt and the Ras-Raf-MEK-MAPK signalling pathways allows to detect gene mutations in 80% of early (UICC I) colon cancer stages in humans. Cancer Epidemiol 2009;33:123–129.
Rowan AJ, Lamlum H, Ilyas M et al. APC mtuations in sporadic colorectal tumors: a mutational "hotspot' and interdependence of the "two hits". Proc Natl Acad Sci USA 2000;97:3352–3357.
Cheadle JP, Krawczak M, Thomas MW et al. Different combinations of biallelic APC mutation confer different growth advantages in colorectal tumours. Cancer Res 2002;62:363–366.
Torlakovic EE, Gomez JD, Driman DK et al. Sessile serrated adenoma (SSA) vs. traditional serrated adenoma (TSA). Am J Surg Pathol 2008;32:21–29.
Aust DE, Baretton GB . Serrated polyps of the colon and rectum (hyperplastic polyps, sessile serrated adenomas, traditional serrated adenomas, and mixed polyps)-proposal for diagnostic criteria. Virchows Arch 2010;457:291–297.
Bosman FT . WHO, IARC. WHO classification of tumours of the digestive system. IARC Press: Lyon, France, 2010.
Groves C, Lamlum H, Crabtree M et al. Mutation cluster region, association between germline and somatic mutations and genotype-phenotype correlation in upper gastrointestinal familial adenomatous polyposis. Am J Pathol 2002;160:2055–2061.
Young J, Barker MA, Simms LA et al. Evidence for BRAF mutation and variable levels of microsatellite instability in a syndrome of familial colorectal cancer. Clin Gastroenterol Hepatol 2005;3:254–263.
Pino M, Chung DC . The chromosomal instability pathway in colon cancer. Gastroenterology 2010;138:2059–2072.
Bettington M, Walker N, Rosty C et al. Clinicopathological and molecular features of sessile serrated adenomas with dysplasia or carcinoma. Gut 2017;66:97–106.
Cingolani P, Platts A, Wang le L et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92.
Alamut Visual version 2.6 (Interactive Biosoftware, Rouen, France).
Tranah GJ, Giovannucci E, Ma J et al. APC Asp1822Val and Gly2502Ser polymorphisms and risk of colorectal cancer and adenoma. Cancer Epidemiol Biomarkers Prev 2005;14:863–870.
Groden J, Thliveris A, Samowitz W et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589–600.
Miyoshi Y, Nagase H, Ando H et al. Somatic mutation sin the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992;1:229–233.
Powell S, Zilz N, Beazer-Barclay Y et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235–237.
Samowitz WS, Slattery ML, Sweeney C et al. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res 2007;5:165–170.
Jass JR . Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007;50:113–130.
Yang S, Farraye FA, Mack C et al. BRAF and KRAS mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol 2004;28:1452–1459.
Kambara T, Simms LA, Whitehall VL et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004;53:1137–1144.
Rosenberg DW, Yang S, Pleau DC et al. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer Res 2007;67:3551–3554.
Sheridan TB, Fenton H, Lewin MR et al. Sessile serrated adenomas with low- and high-grade dysplasia and early carcinomas: an immunohistochemical study of serrated lesions "caught in the act". Am J Clin Pathol 2006;126:564–571.
O'Brien MJ, Yang S, Clebanoff JL et al. Hyperplastic (serrated) polyps of the colorectum: relationship of CpG island methylator phenotype and K-ras mutation to location and histologic subtype. Am J Surg Pathol 2004;28:423–434.
Spring KJ, Zhao ZZ, Karamatic R et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology 2006;131:1400–1407.
Bettington M, Walker N, Rosty C et al. Serrated tubulovillous adenoma of the large intestine. Histopathology 2016;68:578–587.
He X, Semenov M, Tamai K et al. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 2004;131:1663–1677.
Koo BK, Spit M, Jordens I et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012;488:665–669.
Bovolenta P, Esteve P, Ruiz JM et al. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci 2008;121:737–746.
Semenov M, Tamai K, He X . SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 2005;280:26770–26775.
Xu Q, Wang Y, Dabdoub A et al. Vascular development in teh retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 2004;116:883–895.
de Lau W, Barker N, Low TY et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011;476:293–297.
Semenov MV, Tamai K, Brott BK et al. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol 2001;11:951–961.
Barker N, Clevers H . Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov 2006;5:997–1014.
Johnson V, Volikos E, Halford SE et al. Exon 3 beta-catenin mutations are specifically associated with colorectal carcinomas in hereditary non-polyposis colorectal cancer syndrome. Gut 2005;54:264–267.
Giannakis M, Hodis E, Jasmine Mu X et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014;46:1264–1266.
Bond CE, McKeone DM, Kalimutho M et al. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016;7:70589–70600.
Hao HX, Xie Y, Zhang Y et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012;485:195–200.
de Lau W, Peng WC, Gros P et al. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev 2014;28:305–316.
Yan HH, Lai JC, Ho SL et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2016;66:1645–1656.
Seshagiri S, Stawiski EW, Durinck S et al. Recurrent R-spondin fusions in colon cancer. Nature 2012;488:660–664.
Shinmura K, Kahyo T, Kato H et al. RSPO fusion transcripts in colorectal cancer in Japanese population. Mol Biol Rep 2014;41:5375–5384.
Dhir M, Yachida S, Van Neste L et al. Sessile serrated adenomas and classical adenomas: an epigenetic perspective on premalignant neopalstic lesions of the gastrointestinal tract. Int J Cancer 2010;129 1889 1898.
Suzuki H, Watkins DN, Jair KW et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004;36:417–422.
Kohonen-Corish MR, Sigglekow ND, Susanto J et al. Promoter methylation of the mutated in colorectal cancer gene is a frequent early event in colorectal cancer. Oncogene 2007;26:4435–4441.
Christie M, Jorissen RN, Mouradov D et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta-catenin signalling thresholds for tumourigenesis. Oncogene 2013;32:4675–4682.
Gavert N, Ben-Shmuel A, Lemmon V et al. Nuclear factor-kappaB signaling and ezrin are essential for L1-mediated metastasis of colon cancer cells. J Cell Sci 2010;123:2135–2143.
Brabletz T, Jung A, Dag S et al. β-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol 1999;155:1033–1038.
Brabletz T, Jung A, Reu S et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 2001;98:10356–10361.
Suzuki H, Masuda N, Shimura T et al. Nuclear beta-catenin expression at the invasive front and in the vessels predicts liver metastasis in colorectal carcinoma. Anticancer Res 2008;28:1821–1830.
Barras D . BRAF mutation in colorectal cancer: an update. Biomark Cancer 2015;7:9–12.
Acknowledgements
This study was funded by a National Health and Medical Research Council grant (NHMRC: 1050455) and by Pathology Queensland. Vicki Whitehall is supported by a Gastroenterological Society of Australia Senior Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Borowsky, J., Dumenil, T., Bettington, M. et al. The role of APC in WNT pathway activation in serrated neoplasia. Mod Pathol 31, 495–504 (2018). https://doi.org/10.1038/modpathol.2017.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.150
This article is cited by
-
BRAFV600E-mutated serrated colorectal neoplasia drives transcriptional activation of cholesterol metabolism
Communications Biology (2023)
-
Molecular profiling of appendiceal serrated lesions, polyps and mucinous neoplasms: a single-centre experience
Journal of Cancer Research and Clinical Oncology (2021)
-
Oncogenic BRAF, unrestrained by TGFβ-receptor signalling, drives right-sided colonic tumorigenesis
Nature Communications (2021)
-
Clinicopathological and molecular correlations in traditional serrated adenoma
Journal of Gastroenterology (2020)
-
Lnc SMAD5-AS1 as ceRNA inhibit proliferation of diffuse large B cell lymphoma via Wnt/β-catenin pathway by sponging miR-135b-5p to elevate expression of APC
Cell Death & Disease (2019)
