
Abstract
Inflammatory leiomyosarcoma is a soft-tissue tumor resembling conventional leiomyosarcoma, but with a prominent intrinsic inflammatory component. Previous studies have suggested that inflammatory leiomyosarcoma differs genetically from leiomyosarcoma, but in-depth analyses are lacking. Here we provide a comprehensive picture of the genome and transcriptome of inflammatory leiomyosarcoma by combining cytogenetic, single-nucleotide polymorphism array, mRNA-sequencing, and whole-exome sequencing data. The results show that inflammatory leiomyosarcoma has a specific genetic profile characterized by near-haploidization with or without subsequent whole-genome doubling. Consistently, both parental copies of chromosomes 5 and 22 are preserved. Apart from recurrent mutation of the NF1 gene, additional somatic events that could serve as driver mutations were not found at either the nucleotide or the genome level. Furthermore, no fusion transcripts were identified. Global gene expression profiling revealed particularly prominent differential expression of genes, including ITGA7, MYF5, MYF6, MYOD1, MYOG, and PAX7, involved in muscle development and function, providing strong argument for grouping inflammatory leiomyosarcoma with myogenic sarcomas, rather than with myofibroblastic lesions. Combined with previously published data, there are now 10 cases of inflammatory leiomyosarcoma with confirmed near-haploid genotype. These patients differ from leiomyosarcoma patients in being younger (median 41 years), showing a male predominance (9:1), and few relapses (1 of 8 informative patients). Thus, the clinical, morphological, and genetic data provide compelling support for inflammatory leiomyosarcoma being a distinct subtype of myogenic tumors.
Similar content being viewed by others


Main
Inflammatory leiomyosarcoma, first described by Merchant et al.,1 is a spindle cell tumor of the deep soft tissues that shares morphological features with conventional leiomyosarcoma but that also has a prominent, intrinsic inflammatory component.
Although genetic information is restricted to chromosome banding and/or single-nucleotide polymorphism array analyses on only seven cases, these preliminary data indicate that inflammatory leiomyosarcoma has a specific and highly unusual genetic profile. In six of the cases, massive reduction of the chromosome number to a hyperhaploid level (25–28 chromosomes), with or without subsequent polyploidization, was found to constitute an early step in tumorigenesis.2, 3, 4 Furthermore, the copy number distribution of individual chromosomes in inflammatory leiomyosarcoma is clearly nonrandom with all six cases with a hyperhaploid origin showing retained biparental disomy for chromosomes 5 and 22, and often also for chromosomes 18, 20, and 21.2 Nord et al.2 also performed global gene expression analysis on 3 inflammatory leiomyosarcomas and 15 leiomyosarcomas and found that these tumors have distinct transcriptional profiles, and that genes located on the disomic chromosomes are expressed at higher levels than genes on the monosomic chromosomes.
Thus, currently available information suggests that near-haploidization is an important pathogenetic mechanism in inflammatory leiomyosarcoma, a phenomenon that is very rare in other tumor types.5 Here we present single-nucleotide polymorphism array data on four new cases of inflammatory leiomyosarcoma, providing further support for the distinct, near-haploid origin of inflammatory leiomyosarcoma. Furthermore, in an attempt to understand better the pathogenesis of inflammatory leiomyosarcoma and to obtain clues about the mechanisms behind haploidization, we also performed whole-exome and mRNA sequencing on inflammatory leiomyosarcoma cases.
Materials and methods
Patients
In all cases preoperative diagnostics were performed with fine-needle aspirations and core-needle biopsies.
Cases 1–3 correspond to cases 1–3 in Nord et al.2
Case 4 was a 23-year-old man with a tumor in his left thigh. At surgery, a 3 cm tumor in the sartorial muscle was removed with wide margins. Microscopy showed a spindle cell neoplasm with fascicular pattern and focally infiltrative growth. The tumor showed no necrosis or vascular invasion. Spindled tumor cells, partly with distinct nuclear pleomorphism, were mixed with inflammatory cells, predominantly lymphocytes. Tumor cells were positive for desmin, α-smooth muscle actin, and caldesmon, and negative for CKAE1/AE3, S100, and CD34. There were 3 mitoses per 10 HPF. The diagnosis was inflammatory leiomyosarcoma, grade 1. The patient is disease-free 26 months after surgery.
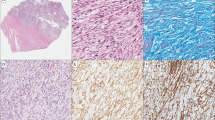
Case 5 was a 41-year-old man who for almost a year had noticed a lump in his right thigh. A 7 cm tumor in the hamstring muscle was removed with free surgical margins. Morphological analysis showed a partly pseudocystic lesion with extensive areas of infarction and domination of chronic inflammation with lymphocytes, histiocytes, and cholesterol clefts (Figure 1a). Within the inflammatory background a population of plump spindled, ovoid, and epithelioid cells was seen with immunohistochemically strong and diffuse positivity for desmin, caldesmon, and multifocally for α-smooth muscle actin (Figures 1c and e). Initially a reactive lesion was suggested, but the final diagnosis was low-grade inflammatory leiomyosarcoma after external consultation. The patient received 50 Gy postoperative radiation and remains free of disease 36 months after surgery.

Representative images of inflammatory leiomyosarcoma case 5 (a, c, e) and case 7 (b, d, f). Hematoxylin–eosin stain (a, b), α-smooth muscle actin immunohistochemistry (c, d), and desmin immunohistochemistry (e, f). Case 5 shows tumor cells with partly epithelioid, partly spindled morphology, intermingled with lymphocytes. The tumor cells in case 7 show a rather plump ovoid morphology with some lymphocytes and macrophages in the background. Both tumors show a strong and diffuse expression of desmin. Note the variable expression of α-smooth muscle actin in inflammatory leiomyosarcoma.
Case 6 was an 18-year-old man who presented with a sports-related knee injury. Incidentally, a tumor was found in the medial aspect of his right thigh at MRI. An 8 cm tumor in m. vastus intermedius was resected with free surgical margins. The tumor showed a fascicular growth pattern of atypical spindle cells with infiltration of the surrounding muscle, but there was no necrosis or vascular invasion. The tumor cells that were positive for α-smooth muscle actin and desmin were embedded among inflammatory cells. There were 2 mitoses per 10 HPF. The diagnosis was inflammatory leiomyosarcoma, grade 1. The patient remains free of disease 11 months after surgery.
Case 7 was a 42-year-old man who for 6 months had noticed a lump in his right deltoid muscle. The tumor, 4.5 cm, was excised with wide margins. Microscopy revealed an encapsulated tumor growing with pushing borders without areas of necrosis. The slightly atypical ovoid to spindled tumor cells were exceptionally arranged in fascicles among abundant inflammatory cells and macrophages (Figure 1b). One mitosis per HPF was counted. Immunohistochemistry showed positivity for desmin, α-smooth muscle actin, and caldesmon in the tumor cell population (Figure 1d and f). The diagnosis was inflammatory leiomyosarcoma, grade 1. Three months after surgery, there is no sign of relapse.
All samples were obtained after informed consent and the study was approved by the local review board.
DNA and RNA Extractions
DNA was extracted from fresh frozen tumor biopsies using the DNeasy blood and tissue kit, including the optional RNaseH treatment according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Quality and concentration of the extracted material were measured with a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and a NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA, USA).
RNA was extracted from fresh frozen tumor biopsies using the RNeasy lipid tissue mini kit according to the manufacturer’s instructions (Qiagen). Quality and concentration of the extracted material were measured with a 2100 Bioanalyzer (Agilent Technologies) and a Qubit 3.0 fluorometer (Thermo Fisher Scientific).
Cytogenetic Analyses
Cytogenetic analyses were performed according to standard procedures, and karyotypes were written following the recommendations of the International System for Human Cytogenetic Nomenclature.6 Fluorescence in situ hybridization (FISH) with centromere-specific probes (Vysis, Abbott park, IL, USA) for chromosomes X, 17, 18, and 20 was used to establish the ploidy level in case 4.
Genomic Copy Number and Loss of Heterozygosity Analyses
Single-nucleotide polymorphism array analysis was used for combined DNA copy number and loss of heterozygosity investigation. It was performed according to the manufacturer’s instructions on extracted DNA from cases 4–7 using the Affymetrix Cytoscan HD platform (Affymetrix, Santa Clara, CA, USA). The position of the single-nucleotide polymorphisms was based on the UCSC hg19/NCBI Build 37 sequence assembly. Data analysis by visual inspection was performed using the Affymetrix Chromosome Analysis Suite 3.1 software.
Whole-Exome Sequencing
DNA was extracted from fresh frozen tumor tissue from cases 1–5 with matching peripheral blood samples, as described.7 Whole-exome capture was performed with the Ion Ampliseq Exome RDY-IC kit, according to the manufacturer’s instructions (Thermo Fisher Scientific), followed by template preparation and sequencing on the Ion Chef and Ion Proton systems, using the Ion PI IC 200 kit (Thermo Fisher Scientific). Variant calling was performed with the AmpliSeq Exome tumor-normal pair workflow in Ion Reporter (Thermo Fisher Scientific), as well as with the Torrent Variant Caller plugin in the Torrent Suite Software (Thermo Fisher Scientific), using standard parameters for somatic variant detection. Annotation of detected variants was performed in Ion Reporter, using the GRCh37/hg19 assembly. The results were further filtered, keeping only non-synonymous exonic mutations that were supported by at least five reads.
To verify NF1 mutations detected at whole-exome sequencing, a TruSeq Custom Amplicon panel (Illumina, San Diego, CA, USA) was designed. The TruSeq Custom Amplicon panel covered the regions of NF1 harboring mutations detected at whole-exome sequencing. DNA was processed and analyzed as described for whole-exome sequencing analysis.
mRNA Sequencing
mRNA libraries for cases 2–6 were prepared for sequencing using the Truseq RNA sample preparation kit v2 (Illumina). Paired-end 151 bp reads were generated from the mRNA libraries on a NextSeq 500 (Illumina). Library preparation, sequencing, and bioinformatic analysis were performed as described.8 ChimeraScan and FusionCatcher, using default settings, were used to identify candidate fusion transcripts from the sequence data.9, 10 The GRCh37/hg19 build was used as the human reference genome.
For gene expression studies, data were normalized using Cufflinks with default settings.11 Correlation-based principal component analysis and hierarchical clustering analysis were performed using the Qlucore Omics Explorer version 3.2 (Qlucore AB, Lund, Sweden). Differences between tumor types in log2-transformed expression data were calculated using a t-test, and corrections for multiple testing were based on the Benjamini–Hochberg method (Qlucore AB). Genes with P<0.05 and a false discovery rate <0.3 were considered significantly altered.
Results
Copy Number and Heterozygosity States
Case 4 was not analyzed by G-banding. However, single-nucleotide polymorphism array showed relative gain and retained heterozygosity of chromosomes 5, 18, 20, 21, and 22, while the rest of the autosomes displayed complete loss of heterozygosity. One structural aberration was observed: hemizygous deletion in 13q at position 85 618 281–86 939 204, containing the genes LINC00351 and SLITRK6. Interphase FISH analysis confirmed the hyperdiploid state suggested by the single-nucleotide polymorphism profile.
Case 5 showed gains of chromosomes 5 and 22 in hyperhaploid and hyperdiploid clones: 24-25,X,+5,+22(cp8)/43-49,idemx2,?del(5)(p11),+mar,inc(cp15). This karyotype was in accordance with the single-nucleotide polymorphism array profile, which showed relative gain and retained heterozygosity of these two chromosomes, as compared to the rest of the autosomes, which all displayed complete loss of heterozygosity. By G-banding analysis, some clones with a near-diploid state displayed a marker chromosome of unknown origin as well as potential rearrangement of 5p.
Case 6 showed relative gain of chromosomes 5, 18, 20, 21, and 22 in hyperhaploid and hyperdiploid clones: 29,XY,+5,+18,+20,+21,+22(7)/58,idemx2(3). Accordingly, the single-nucleotide polymorphism array profile, obtained with identical results from both a preoperative fine-needle biopsy sample and a sample from the excised tumor, displayed retained heterozygosity and relative gain of the same chromosomes, and complete loss of heterozygosity for the remaining autosomes. No imbalances due to structural rearrangements were detected by single-nucleotide polymorphism array analysis.
Single-nucleotide polymorphism array analysis on DNA extracted from a core-needle biopsy in case 7 displayed what was interpreted as a hyperdiploid karyotype with four copies of and retained heterozygosity for chromosomes 5 and 22, while the remaining autosomes and the X chromosome were present in two copies and showed complete loss of heterozygosity.
In summary, all four cases presented hyperhaploid clones and/or duplicates of such clones. Imbalances largely involved losses and gains of whole chromosomes. Structural rearrangements were confined to a hemizygous deletion in 13q in case 4 and marker chromosome formation in a subclone of case 2. Both parental copies of chromosomes 5 and 22 were present in all four cases. Relative gain and retained heterozygosity were also detected for chromosomes 18, 20, and 21 in cases 1, 4, and 6. All other autosomes displayed loss of heterozygosity as a result of chromosome loss with or without subsequent duplication of the remaining homolog. The cytogenetic and single-nucleotide polymorphism array data are summarized in Table 1.
Whole-Exome Sequencing
Achieving a mean coverage of × 50 in both blood and tumor samples with 90% of target bases having at least × 10 coverage and including only non-synonymous exonic mutations, a total of 49 somatic single-nucleotide variants, and 8 insertion/deletions were detected, none of which was recurrent (Supplementary Table 1). The number of mutations ranged from 5 to 34, median 6, per case. Only one gene was involved more than once: NF1 with two different mutations in cases 4 and 5, with variant allele frequencies of 50% and 22%, respectively.
The resequencing using a TruSeq Custom Amplicon panel panel achieved a coverage exceeding × 750 and confirmed the presence of the two NF1 mutations found at whole-exome sequencing in cases 4 and 5, with variant allele frequencies of 31% and 26%, respectively (Supplementary Table 1).
mRNA Sequencing
mRNA sequencing was performed on cases 2–6, no fusion transcripts were detected with either Fusioncatcher or Chimerascan (data not shown).
To extract genes that distinguish the expression signature of inflammatory leiomyosarcoma (n=5), different types of soft-tissue tumors (n=37) from which mRNA-sequencing data were available were used for comparison (11 sclerosing epithelioid fibrosarcomas, 8 ossifying fibromyxoid tumors, 6 myxoid liposarcomas, 3 myxoinflammatory fibroblastic sarcomas, 2 myoepitheliomas, and 1 case each of acral fibromyxoma, calcifying aponeurotic fibroma, dermatofibrosarcoma protuberans, low-grade fibromyxoid sarcoma, leiomyosarcoma, perineurioma, and solitary fibrous tumor). At unsupervised principal component analysis, the data were variance filtered until the 42 samples formed clusters, which corresponded to the different tumor types. This setting corresponded to a variance ratio F=0.3 and 1437 genes. A one-tailed test was performed to extract the 100 most upregulated genes in inflammatory leiomyosarcoma vs controls, listed in Supplementary Table 2. Of these, 27 genes are known to be involved in myogenesis or muscle function (marked in red in Supplementary Table 2). The expression levels of the latter genes in inflammatory leiomyosarcoma and leiomyosarcoma, as assessed by Affymetrix gene expression profiling, were extracted from previously published microarray data on 3 cases of inflammatory leiomyosarcoma and 15 cases of leiomyosarcoma,2 and shown in Table 2.
Discussion
The current study provides further evidence for inflammatory leiomyosarcoma being a distinct nosologic entity, with specific morphologic and genetic features. Including the four new cases, 10 out of 11 inflammatory leiomyosarcomas display near-haploidization, with or without subsequent polyploidization. More specifically, they display loss of heterozygosity for most chromosomes but always show retained heterozygosity for chromosomes 5 and 22, and often for chromosomes 18, 20, and 21. As far as we are aware, haploidization is exceedingly rare in other soft-tissue tumors, and none of those few other cases displays the same pattern of retained heterozygosity for chromosomes 5 and 22. Thus, the genetic profile of inflammatory leiomyosarcoma shows very high specificity and sensitivity, well at par with the strong correlation that exists between most gene fusions and distinct subtypes of soft-tissue sarcoma.12 Haploidization with or without polyploidization has, however, also been reported in acute lymphoblastic leukemia, chondrosarcoma, and rare cases of other tumors but with different patterns of retained disomies.5
In the present study, we attempted to reveal other somatic events that may contribute to tumor development. mRNA sequencing did not disclose any potential fusion transcripts, and whole-exome sequencing on five cases of inflammatory leiomyosarcoma identified no consistently mutated gene. Only a single gene, NF1, was involved in more than one case; two different mutations were found in cases 4 and 5. Both NF1 mutations were frameshift deletions, thus resulting in a truncated protein product. NF1 is located on chromosome 17, which displays loss of heterozygosity in all cases investigated so far; however, no intra-chromosomal deletion targeting the NF1 locus was detected in any of the seven single-nucleotide polymorphism array profiles studied. Still, analysis of expression levels from previously published microarray data (performed on 3 inflammatory leiomyosarcoma and 15 leiomyosarcoma2) and from the mRNA-sequencing data presented here shows that NF1 is downregulated about twofold both in inflammatory leiomyosarcoma vs leiomyosarcoma and in inflammatory leiomyosarcoma vs control tumors (Supplementary Figure 1); however, it should be emphasized that NF1 was expressed at low levels in most tumors. Thus, although we cannot exclude mutations occurring outside the coding part of the genome, it seems as if the haploidization as such is the main tumorigenic event in inflammatory leiomyosarcoma. As for most numerical neoplasia-associated chromosome aberrations, it is unclear how the ploidy shift affects neoplastic transformation; possibly, the distorted gene expression profile achieved by specific combinations of lost and retained chromosomes is sufficient. In line with this, differential expression for genes on retained chromosomes vs genes on monoallelic chromosomes has been reported also in acute lymphoblastic leukemia.13, 14
Not only the consequences but also the causes of haploidization are unclear. Because the single-nucleotide polymorphism array data did not indicate subclonality for any of the numerical changes, the near-haploid state is most likely reached through massive loss of chromosomes in a single mitotic catastrophe, in line with findings in acute lymphoblastic leukemia.13 Subsequent polyploidization then occurs by whole-genome doubling. Although NF1 is recurrently mutated also in near-haploid acute lymphoblastic leukemia13 as well as in adrenocortical carcinoma and rhabdomyosarcoma, all of which may display both haploidization and whole-genome doubling,7, 13, 15 we do not believe that distorted NF1 function is the cause of the haploidization. First, the prototypical NF1-associated malignancies—malignant peripheral nerve sheath tumor and optical glioma—only rarely display haploidization. Second, the allelic frequencies of NF1 mutations in the two cases of inflammatory leiomyosarcoma should have been higher than 22–26% and 31–50%, respectively, detected at deep sequencing if they preceded haploidization. We cannot exclude, however, that reduced NF1 activity facilitates whole-genome doubling; both cases with NF1 mutation showed whole-genome doubling but, on the other hand, so did five out of six other inflammatory leiomyosarcomas analyzed by G-banding, including three without NF1 mutation.
The nonrandom retention of both parental copies of certain chromosomes, notably chromosomes 5 and 22, implies that these chromosomes harbor key regulatory genes essential for cell survival. However, we have no explanation to why these particular chromosomes are retained biparentally and, to our knowledge, none of them has any imprinted region. Loss or retention of imprinted loci might, on the other hand, account for some of the monosomies, but support for this hypothesis is lacking. Indeed, when assessing the expression levels at mRNA sequencing of two imprinted genes—the maternally expressed H19 and the paternally expressed IGF2—on chromosome 11, which consistently shows complete loss of heterozygosity in inflammatory leiomyosarcoma, cases 2 and 3 had high expression of H19 but low expression of IGF2, whereas the opposite expression pattern was seen in cases 4–6 (Supplementary Figure 2). Thus, at least for chromosome 11, no consistent pattern regarding retained maternal or paternal chromosomes could be discerned.
Including the 4 new cases presented here, there are now 10 reported inflammatory leiomyosarcomas with confirmed near-haploidization (refs 2, 3, 4 and present study), the clinical features of which seem to be distinct from those in other forms of leiomyosarcoma. First, there is a skewed sex distribution, with only one woman among the inflammatory leiomyosarcoma cases;4 this feature was not observed in the first report of inflammatory leiomyosarcoma.1 Second, inflammatory leiomyosarcoma patients are younger, median age 41 years (range, 18–64 years), than conventional leiomyosarcoma patients. Third, the outcome seems better for inflammatory leiomyosarcoma patients; of eight informative patients, seven did not show any type of relapse after a median follow-up of 26 months (range, 3–120 months), whereas one developed lung metastases after 22 months, but then remained in second remission until the last follow-up at 41 months (refs 2, 3, 4 and present study). The excellent outcome is in line with the fact that all tumors for which such data were reported were classified as low-grade. Whether the favorable outcome is due to the genetic features of inflammatory leiomyosarcoma, or whether other factors, such as the strong intrinsic inflammatory component, might contribute, is not known. A strong inflammatory component has been shown to be associated with good prognosis in, eg, osteosarcoma16 but in other malignancies, such as thyroid cancer,17 it predicts poor outcome while in yet others, such as breast cancer, the prognostic impact of inflammatory cells is context-dependent.18
Inflammatory leiomyosarcoma originally derived its name from the morphological resemblance of the tumor cells to smooth muscle cells and the positive staining for α-smooth muscle actin and desmin in the majority of cases.1 This conclusion was questioned by Chang et al.,4 who in their study of three patients found poor expression of both α-smooth muscle actin and desmin. They thus speculated whether inflammatory leiomyosarcoma should belong to the group of myofibroblastic tumors.4 It could be pointed out, however, that only one of their three cases had cytogenetic data supporting near-haploidy. Myofibroblasts are operationally defined as fibroblasts that have developed stress fibers and achieved contractile properties.19 Apart from the expression of α-smooth muscle actin—encoded by the ACTA2 gene—which is shared with smooth muscle cells, few characteristic, let alone specific, immunomarkers have been identified.19 When looking at the expression levels for genes implicated in myofibroblasts,19, 20 there was no strong support for tumor cells in inflammatory leiomyosarcoma being myofibroblasts. The only highly expressed gene that has been suggested as myofibroblast-specific20 was THY1, a.k.a. CD90. However, this gene was abundantly expressed also in the single case of conventional leiomyosarcoma, as well as in 17 of the 36 other tumors included (Supplementary Figure 3). Furthermore, according to the Human Protein Atlas, THY1 mRNA is well expressed also in smooth muscle tissue (http://www.proteinatlas.org/ENSG00000154096-THY1/tissue). Interestingly, THY1 has also been implicated as a cancer stem cell marker, and it was recently shown that co-expression of THY1 and Integrin α7 (ITGA7), a transmembrane protein functioning as a receptor for laminin, is important for stemness and epithelial–mesenchymal transition in esophageal squamous cell carcinomas.21 However, ITGA7 is also strongly expressed in different types of muscle tissue, its importance in myogenesis is emphasized by inactivating mutations resulting in congenital myopathies.22, 23 ITGA7 showed relatively high expression also in inflammatory leiomyosarcoma (Supplementary Figure 3).
Our gene expression data are in excellent agreement with a myogenic differentiation. First, all tumors seen by us showed high expression of both ACTA2 and DES, in line with the consistent expression of their corresponding proteins α-smooth muscle actin and desmin at immunohistochemistry analysis2 (present study; Supplementary Figure 3). Second, among the 100 genes showing the strongest differential expression in inflammatory leiomyosarcoma compared to control tumors, 27 are known to be involved in muscle development or function (Table 2; Supplementary Table 2). Interestingly, though, several of these differentially expressed genes, notably MYF5, MYF6, MYOD1, MYOG, and PAX7, are known to be crucial for the differentiation of satellite cells, ie, adult muscle stem cells, into skeletal muscle cells,24 and mRNA expression in normal tissues is restricted to skeletal muscle (http://www.proteinatlas.org). This raises the interesting possibilities that inflammatory leiomyosarcoma cells either represent primitive myogenic cells or that haploidization of a myogenic precursor cell allows for concomitant expression of genes associated with both smooth muscle and striated muscle development.
In summary, we believe that it is now established beyond doubt that inflammatory leiomyosarcoma is a distinct tumor entity with a highly characteristic genetic profile and a favorable clinical outcome. Furthermore, our protein and gene expression data provide support for keeping inflammatory leiomyosarcoma among myogenic neoplasms.
References
Merchant W, Calonje E, Fletcher CDM . Inflammatory leiomyosarcoma: a morphological subgroup within the heterogeneous family of so-called inflammatory malignant fibrous histiocytoma. Histopathology 1995;27:525–532.
Nord KH, Paulsson K, Veerla S et al. Retained heterodisomy is associated with high gene expression in hyperhaploid inflammatory leiomyosarcoma. Neoplasia 2012;14:807–812.
Dal Cin P, Sciot R, Fletcher CDM et al. Inflammatory leiomyosarcoma may be characterized by specific near-haploid chromosome changes. J Pathol 1998;185:112–115.
Chang A, Schuetze SM, Conrad EU 3rd et al. So-called "inflammatory leiomyosarcoma'': a series of 3 cases providing additional insights into a rare entity. Int J Surg Pathol 2005;13:185–195.
Mandahl N, Johansson B, Mertens F et al. Disease-associated patterns of disomic chromosomes in hyperhaploid neoplasms. Genes Chromosomes Cancer 2012;51:536–544.
McGowan-Jordan J, Simons A, Schmid M . An International System for Human Cytogenetic Nomenclature. Karger: Basel, Switzerland, 2016.
Walther C, Mayrhofer M, Nilsson J et al. Genetic heterogeneity in rhabdomyosarcoma revealed by SNP array analysis. Genes Chromosomes Cancer 2016;55:3–15.
Walther C, Hofvander J, Nilsson J et al. Gene fusion detection in formalin-fixed paraffin-embedded benign fibrous histiocytomas using fluorescence in situ hybridization and RNA sequencing. Lab Invest 2015;95:1071–1076.
Iyer MK, Chinnaiyan AM, Maher CA . ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 2011;27:2903–2904.
Nicorici D, Satalan M, Edgren H et al. FusionCatcher—a tool for finding somatic fusion genes in paired-end MRNA sequencinguencing data. bioRxiv 2014.
Trapnell C, Williams BA, Pertea G et al. Transcript assembly and quantification by MRNA sequencing reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–515.
Mertens F, Antonescu CR, Mitelman F . Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016;55:291–310.
Safavi S, Olsson L, Biloglav A et al. Genetic and epigenetic characterization of hypodiploid acute lymphoblastic leukemia. Oncotarget 2015;6:42793–42802.
Holmfeldt L, Wei L, Diaz-Flores E et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013;45:242–252.
Zheng S, Cherniack AD, Dewal N et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016;29:723–736.
Buddingh EP, Kuijjer ML, Duim RA et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res 2011;17:2110–2119.
Cunha LL, Marcello MA, Ward LS . The role of the inflammatory microenvironment in thyroid carcinogenesis. Endocr Relat Cancer 2014;21:R85–R103.
Karn T, Pusztai L, Rody A et al. The influence of host factors on the prognosis of breast cancer: stroma and immune cell components as cancer biomarkers. Curr Cancer Drug Targets 2015;15:652–664.
Hinz B, Phan SH, Thannickal VJ et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 2012;180:1340–1355.
Hsia LT, Ashley N, Ouaret D et al. Myofibroblasts are distinguished from activated skin fibroblasts by the expression of AOC3 and other associated markers. Proc Natl Acad Sci USA 2016;113:E2162–E2171.
Ming XY, Fu L, Zhang LY et al. Integrin alpha7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat Commun 2016;7:13568.
Hayashi YK, Chou FL, Engvall E et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 1998;19:94–97.
Castiglioni A, Hettmer S, Lynes MD et al. Isolation of progenitors that exhibit myogenic/osteogenic bipotency in vitro by fluorescence-activated cell sorting from human fetal muscle. Stem Cell Reports 2014;2:92–106.
Sincennes MC, Brun CE, Rudnicki MA . Concise review: epigenetic regulation of myogenesis in health and disease. Stem Cells Transl Med 2016;5:282–290.
Acknowledgements
This study was supported by grants from the Swedish Cancer Society, The Swedish Childhood Cancer Foundation, and Region Skåne.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Arbajian, E., Köster, J., Vult von Steyern, F. et al. Inflammatory leiomyosarcoma is a distinct tumor characterized by near-haploidization, few somatic mutations, and a primitive myogenic gene expression signature. Mod Pathol 31, 93–100 (2018). https://doi.org/10.1038/modpathol.2017.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.113
This article is cited by
-
Pulmonary inflammatory leiomyosarcoma represents a potential diagnostic pitfall of DNA methylation-based classification of sarcomas: a case report
BMC Pulmonary Medicine (2023)
-
Myxoid pleomorphic liposarcoma is distinguished from other liposarcomas by widespread loss of heterozygosity and significantly worse overall survival: a genomic and clinicopathologic study
Modern Pathology (2022)
-
Toward a unifying entity that encompasses most, but perhaps not all, inflammatory leiomyosarcomas and histiocyte-rich rhabdomyoblastic tumors
Modern Pathology (2021)
-
“Inflammatory Leiomyosarcoma” and “Histiocyte-rich Rhabdomyoblastic Tumor”: a clinicopathological, immunohistochemical and genetic study of 13 cases, with a proposal for reclassification as “Inflammatory Rhabdomyoblastic Tumor”
Modern Pathology (2021)
-
Histiocyte-rich rhabdomyoblastic tumor: a report of two cases and a review of the differential diagnoses
Virchows Archiv (2021)

{kind=link}
{kind=link}
{kind=link}