
Abstract
Although almost any non-Hodgkin lymphoma can involve the spleen or an extranodal site as part of more widely disseminated disease, there is a group of small B-cell lymphomas that specifically arise in these locations. These are important to recognise as some appear to have a behaviour and prognosis that is distinct from their nodal counterparts. In addition, there are entities that are specific to extranodal locations (such as extranodal marginal zone lymphoma) and to the red or white pulp of the spleen. In this review, the characteristics of these entities will be presented as well as clues to help distinguish lymphoma from reactive infiltrates in extranodal sites and measure to distinguish between small B-cell lymphomas encountered in the spleen and at extranodal locations.
Similar content being viewed by others

Main
Extranodal lymphomas account for 24–48% of all non-Hodgkin lymphomas.1, 2, 3 Involvement of extranodal sites and the spleen by lymphoma is not an uncommon occurrence. A proportion of these cases will be secondary involvement by disseminated nodal lymphoma and all nodal small B-cell lymphomas have the potential to involve extranodal sites or the spleen.
In addition to secondary involvement of extranodal sites by small B-cell lymphoma such sites may be the primary site for development of the lymphoma. Some primary extranodal small B-cell lymphomas may be distinct to extranodal/splenic sites while others represent extranodal or splenic origin of their more common nodal counterparts. This review will concentrate on these groups of lymphomas, presenting the typical features of the lymphomas specifically associated with extranodal/splenic origin and highlighting similarities and differences of the extranodal/splenic lymphomas in those that have nodal counterparts. For the most part, the GI tract will be used as the paradigm for extranodal lymphomas as this is the location most frequently studied.
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT) lymphoma
The majority of small B-cell lymphomas arising in the gastrointestinal tract are extranodal marginal zone lymphoma of MALT (MALT lymphoma). MALT lymphoma is most frequently encountered in the gastrointestinal tract with the stomach the site being most often involved. Other common sites are the salivary glands, skin, orbit/ocular adnexae, lung and thyroid gland although MALT lymphomas have been described at almost all extranodal sites whether or not they have a true mucosa.4 The area in which MALT lymphoma is exceptionally rare is in the terminal ileum, which has the highest concentration of native MALT. It appears that acquired lymphoid tissue at extranodal sites has a morphology and organisation that mimics native MALT but is unstable and liable to the development of lymphoma. The first step in the pathway to development of MALT lymphoma is therefore acquisition of organised lymphoid tissue. In some organs, the stimulus for the acquisition of MALT is known such as Helicobacter pylori in the stomach,5 Hashimoto’s thyroiditis in the thyroid6 and Sjögren’s disease in the salivary glands.7 In other cases, the underlying stimulus is suspected for at least a proportion of the cases (Borrelia burgdorferi in the skin,8 Chlamydia psittaci in the conjunctive9) but for the remainder the underlying association remains enigmatic.
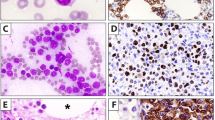
MALT lymphomas wherever they are encountered share the same organisation, morphology and immunophenotype. The neoplastic cells arise in the marginal zone and grow around and between the lymphoid follicles of the acquired lymphoid tissue (Figure 1). The cytology of MALT lymphoma can be variable. Typically the cells have irregular nuclei scanty cytoplasm resembling the centrocyte of the germinal centre (centrocyte-like cells) but the neoplastic cells may have round nuclei resembling small lymphocytes, have a monocytoid appearance or show plasmacytoid or plasmacytic features (Figure 1). Dutcher bodies may be seen. In most cases, many cytological variants are present in proximity to each other. Scattered activated large cells are ubiquitous. The lymphoma cells infiltrate any associated epithelial structures in a neoplastic mimic of the lymphoepithelium seen above Peyer’s patches in native MALT. This infiltration of the glandular epithelium to form lymphoepithelial lesions is associated with destruction of the epithelial cells by direct effect of the infiltrating neoplastic cells (Figure 1). In the stomach, neuroendocrine cells appear more resistant and scattered residual neuroendocrine cells can be seen within the neoplastic infiltrate in some cases.

Mucosa-associated lymphoid tissue (MALT) lymphoma. (a) Low power showing extensive infiltration of lung parenchyma with extension into bronchial epithelium; H&E. (b) Perifollicular infiltrate of cells with centrocyte-like morphology. A residual reactive germinal centre is present in the lower right of the image; H&E. (c) Characteristic morphology with moderate cytoplasm and slightly irregular nuclei. The cells are seen to infiltrate bronchiolar epithelium; H&E. (d) A lymphoepithelial lesion in stomach in gastric MALT lymphoma; H&E. (e) Extension of CD20-positive cells from the base of the mucosa around glands and into the superficial lamina propria.
The presence of lymphoid follicles is a constant feature. In many cases these have been overrun by the neoplastic infiltrate and their presence can only be inferred by the demonstration of residual follicular dendritic cell meshworks. In some cases, the neoplastic cells are seen to specifically colonise residual lymphoid follicle centres. In these cases, the intrafollicular component may retain the morphology of the surrounding infiltrate or show plasmacytoid/cytic differentiation, but a proportion of the cells become enlarged and more activated in appearance. The latter cases may morphologically mimic follicular lymphoma.10 Studies that have examined gastric resection specimens in patients with MALT lymphoma have highlighted the presence of a small neoplastic infiltrate derived from the same neoplastic clone as the main lymphoma around most of the lymphoid follicles in the gastric mucosa distant from the main lesion.11, 12
The distinction between acquired lymphoid tissue with the characteristics of MALT and early MALT lymphoma can be problematic. In the stomach, Zukerberg et al13 found that the presence of prominent lymphoepithelial lesions, Dutcher bodies and moderate cytological atypia were present in neoplastic infiltrates but not reactive counterparts. Although infiltration or splitting of the muscularis mucosae by lymphoid nodules is not a helpful sign, the presence of small B cells infiltrating into the upper parts of the mucosa away from lymphoid follicles is a suspicious finding (Figure 1). In the salivary gland, the earliest finding for the diagnosis of MALT lymphoma is the widening of a zone of cells with monocytoid/marginal appearance between the outer margin of the follicle mantle and epithelial structures.7
Molecular studies should be used with caution. Monoclonal population have been found in cases of uncomplicated H. pylori-associated gastritis,14 although the frequency of detection of such clonal populations increases with an increasing certainty for a diagnosis of lymphoma.15
Immunophenotypically, the cells of MALT lymphoma show a rather non-descript B-cell phenotype. The cells express pan-B-cell markers (CD19, CD20, CD79a and PAX5) and BCL2. A proportion of cases are positive for CD43. They do not express CD10, BCL6, CD23 or cyclin D1. Although normally CD5 is negative, a proportion of CD5+ cases have been described. These appear to be encountered more in the head and neck and ocular regions. They appear to be more likely to present with a leukaemic component and bone marrow involvement and may be associated with more aggressive disease including early recurrence and more frequent dissemination.16, 17, 18, 19, 20, 21
The genetics of MALT lymphoma has been extensively investigated. In contrast to some other small B-cell lymphomas, there is no single genetic abnormality associated with the MALT lymphomas. However, many of the translocations identified (MALT1, BCL10 and FOXP1) have a common effect with dysregulation of the NF-kappa-B pathway. Certain translocations are associated with particular sites of origin. The t(11;18)(q21;q21), which juxtaposes API2 and MALT1 and results in the production of a functional chimeric protein is the most common translocation detected in the stomach, intestine and lung but is rare in other sites. Other translocations, including t(14;18)(q32;q21) (IGH@/MALT1), t(3;14)(p14.1q32) (FOXP1/IGH@) and t(1;14)(p22;q32) (BCL10/IGH@) are encountered with greater frequency at other sites.22, 23, 24, 25, 26
It is now well accepted that many cases of gastric MALT lymphoma, at least in the early stages, respond to H. pylori eradication therapy.27, 28 Antibiotic therapy has also proved effective in a subset of infection-associated MALT lymphomas at other sites. Conjunctival/orbital MALT lymphoma has been associated with Chlamydia psittaci9 and some cutaneous MALT lymphomas with Borrelia burgdorferi.8 In both cases, appropriated antibiotic therapy can result in tumour regression.
In the stomach, several factors have been shown to be associated with a lack of response to H. pylori eradication therapy alone. These include deep penetration of the gastric wall, a t(11;18)(q21;q21) strong nuclear staining for BCL10 and lack of underlying H. pylori infection.29, 30, 31, 32 Depth of invasion of the wall and the presence of the translocation may not be independent predictors of response as lymphomas with deep mural extension at presentation frequently are positive for the translocation.33
Following eradication therapy, examination of subsequent biopsies for regression of the tumour is essential. Regression of the lymphoma may be rapid (4 months following therapy) but in a proportion of cases may be delayed and in some may occur years after eradication without further intervention so an indication of response in reports of follow-up biopsies is required. Initial studies used a scoring system designed around the confidence of the presence of diagnostic lymphoma (Wotherspoon score).27 This system, although effective for presentation biopsies, has limitations in the post-eradication setting. The GELA group subsequently designed a system (Table 1), which combines assessment of lymphoid infiltrate, lamina propria fibrosis and the presence of lymphoepithelial lesions to divide cases into categories labelled complete response (absence of lymphoid infiltrate), probable minimal residual disease, responding disease and no change (no evidence of regressive changes).34 This system has been shown to have high inter-observer concordance. Correlation between the GELA category, other predictors of response (depth of wall invasion, genetics) and the endoscopic appearance will give an indication of the likelihood of response and predict when use of more conventional anti-lymphoma therapy should be adopted. Other studies have suggested the integration of the Wotherspoon score and assessment of stromal changes may be equally informative compared with the GELA system.35 The point at which intervention with conventional therapy is instituted remains controversial. No fixed time for further treatment has been established but lack of regressive changes after 18–24 months, particularly if associated with features that would indicate a low likelihood of response to Helicobacter eradication strategies alone, may indicate further intervention is necessary.
When anti-Helicobacter therapy is adjudged to have failed numerous conventional therapies have been applied with good and almost equal efficacy with both radiotherapy and chemotherapy having curative potential.36 MALT lymphomas are highly sensitive to radiation, which may be curative in localised stage IE and II1E disease. Different types of chemotherapy with or without immunotherapy have been suggested but efficacy and comparison data are limited and no standard chemotherapy has been defined so far.
The role of molecular studies for the detection of clonality has limited value in either the diagnostic or follow-up setting and is not recommended for routine use. At diagnosis, clonal populations have been detected to a variable degree using PCR in cases with lymphoid infiltrates that fail to fulfil the diagnostic criteria for lymphoma and in up to 3% of cases of uncomplicated H. pylori-associated gastritis.14 In the post-eradication follow-up setting, it has been demonstrated that many of the residual basal lymphoid aggregates (probable minimal residual disease in GELA system) harbour small residual clonal lymphoma cell populations.37 In addition, clonal populations reappear and disappear in follow-up biopsies over time and without therapeutic intervention38 indicating that the detection of a clonal population in isolation does not predict for relapsed disease, which should only be considered if the morphological features are those of overt MALT lymphoma (Wotherspoon score 4–5).
Immunoproliferative small intestinal disease (IPSID) is a specific subtype of MALT lymphoma with a characteristic geographic distribution being found mainly in the Middle East, countries bordering the Mediterranean Sea and the Cape region of South Africa. It is characterised by florid plasma cell differentiation with production of IgA but lack of light chains. For many years, it has been appreciated that cases of IPSID in early stages respond to broad spectrum antibiotics39 and recently an association with Campylobacter jejuni has been demonstrated.40
Primary follicular lymphoma of the GI tract
Follicular lymphoma is most commonly associated with nodal disease with frequent presentation in stages III–IV, often with bone marrow involvement. The recent WHO classification of haemopoietic and lymphoid tumours has recognised the possibility of follicular lymphoma presenting with a primary extranodal location and has recognised primary intestinal lymphoma as a distinctive entity. Intestinal follicular lymphomas present either with polyps or as a diffuse obstructing lesion.
In a recent review of 249 cases (244 from the literature and 5 new cases) of GI follicular lymphoma,41 it was noted that the demographics were similar to nodal follicular lymphoma (M:F 1:1; median age 56 years). The majority occurred in the duodenum (usually as polyps rather than obstructing lesions) or small intestine and in contrast to nodal follicular lymphoma, the majority of GI follicular lymphomas present in clinical stages I/II (93.2%). Although earlier studies suggested a high proportion were unifocal in the GIT, the introduction of wireless capsule endoscopy and double balloon endoscopy have suggested that up to 73% may be multifocal.
The morphology of GI follicular lymphoma is identical to the nodal counterpart with a mixture of centroblasts and centrocytes within the neoplastic follicles but the majority of cases are grade 1–2 with only 4.3% being grade 3 (compared with approximately 20% for nodal follicular lymphoma).41, 42 Morphologically the infiltrate consists of either a single abnormal follicle or small groups of neoplastic follicles, which may be limited to the mucosa or extend more deeply through the wall (Figure 2). The immunophenotype of GI follicular lymphoma is identical to the nodal counterpart with expression of pan-B-cell markers, CD10 and BCL6 (Figure 2). The majority, 90%, are positive for BCL2 protein (Figure 2). They are negative for CD5 and cyclin D1. Many cases express IgA. Expression of α4β7 integrin, which mediates migration to intestinal mucosa by binding to Mad-CAM-1 on mucosal vascular endothelium, has been demonstrated43, 44 which may explain the tendency for these tumours to remain localised to the GIT. Genetic studies have shown the presence of the typical t(14;18)(q32;q21) BCL2/IGH@ in a similar proportion of cases to nodal lymphoma.

Follicular lymphoma. There are discrete follicles in the lamina propria. The follicle centres contain a predominant centrocyte population with expression of CD10 and bcl-2 protein. A population of neoplastic cells around the follicle are highlighted by staining for CD10. (a–c) H&E. (d) CD10. (e) bcl-2.
The prognosis is generally good45, 46 and, in common with the nodal counterpart, a decision to treat is usually determined by the extent of the disease and patient symptomatology.
Mantle cell lymphoma
Primary mantle cell lymphoma is encountered almost as frequently as MALT lymphoma in the intestine. It is rare in the stomach and can be encountered infrequently at other extranodal sites including endocrine glands and the ocular adnexa. In the intestine, mantle cell lymphoma frequently presents with multiple polyps that may be present throughout the intestine and in the stomach (multiple lymphomatous polyposis) but this presentation is not unique to mantle cell lymphoma. Primary extranodal mantle cell lymphoma shows essentially identical features to its nodal counterpart and has similar demographics.
The cell morphology is usually that of typical mantle cell lymphoma with irregular/clefted nuclei and scanty cytoplasm (Figure 3). The nuclei have absent or indistinct nucleoli. Immunophenotypically, the cells express pan-B-cell markers, CD5, CD43 and cyclin D1 (Figure 3). Exceptions to this typical phenotype include a subset of cases that lack CD5 and or CD43 expression and even rare cyclin D1-negative cases. At the genetic level the extranodal lymphomas show the same t(11;14)(q13;q32) as the nodal counterpart.

Mantle cell lymphoma in colonic biopsy showing infiltration of the lamina propria by a sheet of small cells with irregular nuclei with expression of cyclin D1. (a) Low power showing nodular infiltrate in lamina propria; H&E. (b) Intermediate power showing edge of a nodule with mild interstitial infiltration around glands but no lymphoepithelial lesion; H&E. (c) High power showing the characteristic cellular morphology with scanty cytoplasm, irregular/indented nuclear contours and absent nucleoli; H&E. (d) Nuclear staining for cyclin D1 is present in the lymphoma cells with epithelial and endothelial cell staining acting as internal control.
Differential diagnosis of extranodal small B-cell lymphomas
The morphological features of small B-cell lymphomas at extranodal sites can overlap and the distinction between specific entities may be difficult. MALT lymphomas showing prominent follicular colonisation may mimic follicular lymphoma, whereas mantle cell lymphoma with a nodular growth may be confused with grade 1 follicular lymphoma.
Lymphoepithelial lesions are no longer considered pathognomonic of MALT lymphoma. Typical lymphoepithelial lesions can be seen in cases of primary thyroid follicular lymphoma47 and mimics of lymphoepithelial lesions can be seen with mantle cell lymphoma in the colon and in pulmonary infiltrates associated with rheumatoid lung.
Immunophenotypic studies are key to the distinction between the various small B-cell lymphomas and in most cases will enable the correct diagnosis to be made. There are, however, some pitfalls and it has become evident that in some cases neoplastic cells may up or downregulate antigens dependent on the cellular compartment that they occupy. An example of this is the widely recognised finding of downregulation of CD10 on the surface of neoplastic cells of follicular lymphoma when they are found in the interfollicular compartment.
Distinguishing follicular lymphoma from MALT lymphoma may be difficult in some cases, particularly if the biopsy material is suboptimal. In these cases, a useful indicator is the degree of extrafollicular infiltration with MALT lymphomas usually showing a significant degree of perifollicular infiltration.
Lymphomatous intestinal infiltration presenting with polyps and termed multiple lymphatous polyposis was first described in 1961,48 this condition was initially considered almost synonymous with mantle cell lymphoma. However, it is now evident that follicular hyperplasia, follicular lymphoma and MALT lymphoma may have indistinguishable endoscopic appearance with multiple small polyp/mucosal nodules over long segments of intestine.49, 50
Splenic lymphoma pathology
The key to assessment of splenic histology is good fixation. Ideally the spleen should be received in the laboratory fresh. After appropriate weighing and measuring, the spleen should be sliced and the appearance of the cut surface described. Most of the tissue will be surplus to diagnostic requirements and so it is best to concentrate on areas that will be selected for histological assessment and ensure that this tissue is ideally fixed. Selection of block-sized segments and placing them in adequate fixative will then ensure perfect histological preservation. Material can be prepared for flow cytometry, molecular genetics and cytogenetics as required. It is also helpful to sample hilar lymph nodes as these are frequently involved by the lymphomas that involve the white pulp and most histopathologists are more comfortable with the appearance of lymphoma in lymph nodes than in the spleen.
Splenic marginal zone lymphoma
There is very close relationship between splenic lymphoma with villous lymphocytes and the histological diagnosis of splenic marginal zone lymphoma (SMZL). However, not all cases of SMZL are associated with circulating lymphocytes that have classical villous morphology and some cases with circulating villous lymphocytes may have splenic lymphomas of other subtypes including mantle cell lymphoma.51
Classically SMZL presents with predominantly white pulp disease. The white pulp nodules may contain central residual reactive follicle centres but in many areas these are regressed or absent (Figure 4). In the typical cases, there is an outer zone of cells with a marginal zone appearance where there is moderately abundant cytoplasm and round nuclei (Figure 4). In this zone there are frequently scattered large transformed cells. Plasma cell differentiation may be seen. There is often a zone of smaller cells with scanty cytoplasm and nuclei with more condensed chromatin. This inner zone abuts the follicle centre if it is present and there is no residual mantle zone seen. The combination of these two zones resembles the normal mantle and marginal zone appearance of the normal splenic white pulp but in SMZL both zones are composed of neoplastic cells. Epithelioid histiocytes may be present.52 Although the classical biphasic pattern is the most common pattern encountered in SMZL monophasic patterns have been described with the uniform cell population being either of marginal (outer zone) type or small cell (inner zone) type.53 The red pulp is infiltrated by a similar population of cells, which are present diffusely with invasion of the sinusoids and as small nodules, which may be composed of either small and/or marginal zone type cells.

Splenic marginal zone lymphoma. (a) Predominant white pulp infiltration with interstitial red pulp disease and small red pulp nodules. The white pulp contains a regressed germinal centre; H&E. (b) Central germinal centres may reactive; H&E. (c) The infiltrate around the residual follicles has an inner zone of small cells and an outer zone of cells with more abundant cytoplasm; H&E. (d) Bone marrow infiltration shows an interstitial population of small cells with an intrasinusoidal component; CD20.
In many cases, the hilar lymph nodes may show nodular infiltration by a population that includes both cell types. Often the zoned pattern is absent, although zonation may be seen in some cases. In other cases, the nodal infiltrate may be nodular but monophasic with either small or marginal zone type cells. The sinuses are usually patent.
In the bone marrow, there is usually a nodular and interstitial infiltrate of lymphoid cells. The nodules may mimic the splenic white pulp and in some cases central reactive germinal centres may be evident. The interstitial infiltrate frequently includes an intrasinusoidal component (Figure 4).
Immunophenotypically, the cells express pan-B-cell markers, are bcl-2 positive and usually express for IgM and IgD. The cells usually lack CD5 and CD43 and are negative for cyclin D1. Staining for CD10 and bcl-6 highlights residual follicle centres but the lesional cells are negative. Proliferation markers show a targetoid pattern with a central area with high proliferation in residual follicle centres where present, low proliferation in the inner small cell zone and a higher proliferation in the outer marginal zone. Staining for tartrate-resistant acid phosphatase (TRAP) and DBA44 (CD76) is usually, but not always, negative. The cells are negative for annexin A1. Although usually negative, a proportion (20–25%) of cases may express CD5. In these cases, there may be co-expression of CD23 and/or CD43. In a proportion of CD5 positive cases, there is discordance between the detection of CD5 on cells in the peripheral blood by flow cytometry and in the splenic tissue by immunohistochemistry. This may be explained by differences in the sensitivity of the two techniques but may also be the consequence of local factors such as the presence of cytokines (eg, IL4) or may be associated with chemokines and altered BCR signalling. Although CD5+ cases may be associated with higher lymphocytosis and diffuse marrow involvement, there is no influence on prognosis.54, 55, 56 No unique genetic abnormality has so far been associated with SMZL although deletion of 7q31–32 is frequent being found in up to 40% of cases.57
Differential diagnosis and pitfalls in the diagnosis of SMZL
Major pitfalls in the diagnosis of SMZL include mistaking reactive proliferations with prominent marginal zone expansions for a SMZL and not recognising that other small B-cell lymphomas can mimic SMZL when they involve the splenic marginal zone. Demonstration of clonality and correlation with the clinical setting is important in the distinction between lymphoma and florid benign marginal zone expansion.
Many types of small B-cell lymphomas including those that usually present in lymph nodes or other extranodal sites can be encountered with primary/dominant presentation in the spleen. Although usually easily distinguished because they often show similar morphological and immunophenotypic characteristics to their more typical nodal counterparts, some cases may mimic SMZL. Often examination of the hilar nodes is very useful as they may show the classic features of one other types of small B-cell lymphoma.
Although the biphasic pattern of infiltration is characteristic of SMZL it is not restricted to this entity with follicular lymphoma, mantle cell lymphoma and chronic lymphocytic leukaemia/small lymphocytic lymphoma all occasionally showing an inner small cell zone surrounded by an outer paler zone.58 In the bone marrow, intrasinusoidal infiltration is not restricted to SMZL and may be seen in many other types of small B-cell lymphoma, particularly those with splenic involvement.59
Mantle cell lymphoma presenting with massive splenomegaly and a leukaemic phase shows typical morphology but may be associated with a more indolent course with initial response to splenectomy alone and delayed requirement for chemotherapy.60 A variant of mantle cell lymphoma where the cells are larger and the nuclei are round with small central eosinophilic nucleoli has been described which does not correspond to either of the conventionally recognised blastoid or pleomorphic variants. This nucleolated variant may be confused with B-prolymphocytic leukaemia but the cells stain for cyclin D1 and show the characteristic t(11;14) translocation.61 In most cases, foci with more typical mantle cell morphology are found often at the centre of some of the white pulp nodules.
Hairy Cell Leukaemia
In the spleen, there is usually heavy infiltration of the red pulp with filling of the cords. Blood lakes consisting of areas of haemorrhage surrounded by hairy cells are typically present. The white pulp is usually atrophic. The cells appear spaced apart due to the presence of abundant clear cytoplasm. The cell borders in tissue sections are well defined. The nuclei are round or bean shaped.
The bone marrow shows a patchy interstitial infiltrate of similar cells with variable density. There may be some preservation of fat and haemopoietic marrow in early stages. The areas infiltrated by hairy cells are associated with increased reticulin deposition, which results in dry aspirate taps in many cases. The peripheral blood contains a variable number of cells with irregular hairy cytoplasmic projections. There is invariably monocytopenia. Lymph node involvement is rare and may be subtle or more florid. In the more subtle cases, there is a focal/patchy infiltrate of typical cells with pale/clear cytoplasm in the interfollicular area.
Immunophenotypically, the hairy cells express pan-B-cell antigens and are positive for DBA44 (CD76), CD25 and with antibodies to TRAP. The cells express CD11c and CD123 and in frozen tissue or by flow cytometry can be shown to express CD103. More recently, staining with annexin A1 has been shown to be highly specific for hairy cell leukaemia (HCL).62 The cells are positive for cyclin D1 in a significant proportion of cases but these are usually CD5 negative. A smaller proportion expresses CD10.63, 64 Genetic studies have identified BRAF V600E mutation to be present in all cases of HCL studies whereas this is rare in other splenic B-cell lymphomas.65, 66
Splenic B-cell lymphoma/leukaemia, unclassifiable
The current WHO classification of lymphoid tumours recognises that there are small B-cell lymphomas that infiltrate the splenic red pulp but do not fall into any of the standard lymphoma types. Various terms have been applied to these and the two lesions that have been most investigated are splenic diffuse red pulp small B-cell lymphoma (SDRPSBL) (formally referred to as SMZL, diffuse variant) and HCL-variant (HCL-v). These are included in the classification as provisional entities until more data have been accumulated. The relationship between these two entities remains uncertain and controversial and the relationship to other small B-cell lymphomas remains to be delineated.67 The putative cell of origin is unknown. Lymphomas that fail to fulfil the features of either of these two provisional entities are best left as ‘unclassifiable’.
Splenic diffuse red pulp small B-cell lymphoma
Clinically patients present with splenomegaly that may be massive. Lymphocytosis is usually low and there is often thrombocytopenia but rarely anaemia.
Characteristically SDRPSBL shows diffuse infiltration of the splenic red pulp with involvement of the cords and the sinusoids (Figure 5). The white pulp is atrophic. The cells are small and tend to show some plasmacytoid-type morphology with clumped chromatin. There may be blood lakes.68, 69 In the bone marrow, there is usually striking intrasinusoidal infiltration usually with an interstitial component (Figure 5). The peripheral blood shows small cells that have scanty cytoplasm and nuclei with clumped chromatin. There may be villous type surface projections.

Splenic red pulp lymphomas. (a) Splenic diffuse red pulp small B-cell lymphoma (SDRPSBL) showing diffuse infiltration of red pulp with no residual white pulp; H&E. (b) SDRPSBL: the cells have plasmacytoid features and infiltrate the cords and sinusoids; H&E. (c) SDRPSBL in the bone marrow showing interstitial and intrasinusoidal pattern of infiltration; CD20. (d) Hairy cell leukaemia-variant (HCL-v) in the spleen showing infiltration of red pulp by intermediate sized cells; H&E. (e) The cells have prominent central nucleoli; H&E. (f) HCL-v in the bone marrow showing pronounced intrasinusoidal pattern of infiltration; CD20.
Immunophenotypically, the cells express pan-B-cell markers, DBA44 (CD76) and they may be positive for CD11c. Staining for CD5, CD25, CD123, CD10 and CD23 is usually negative. Staining for annexin A1 has not been described.
Hairy cell leukaemia-variant
As with SDRPSBL patients with HCL-v most frequently present with massive splenomegaly but in these cases lymphocytosis is often pronounced. About 50% of patients show thrombocytopenia with only about a quarter presenting with anaemia.70
HCL-v has a similar pattern of infiltration of the red pulp as SDRPSBCL. There may be blood lakes and the white pulp is atrophic. The cells are slightly larger than those of SDRPSBCL and the nuclei contain small eosinophilic nucleoli70, 71 (Figure 5). The plasmacytoid features seen in DSRPSBCL are missing. In the bone marrow, the infiltrate is also strikingly intrasinusoidal and may be entirely so (Figure 5). In the peripheral blood, the cells have been described as having morphology that is intermediate between HCL and prolymphocytic leukaemia as the cells show irregular cytoplasmic projections and the nuclei have distinct nucleoli. In contrast to HCL, there is no monocytopenia associated with HCL-v.
Immunophenotypically, the cells are positive for pan B-cell markers, usually positive for CD11c and DBA44 (CD76) and CD103 can be detected by flow cytometry or in frozen sections. Staining for CD5, CD23, CD25 and CD123 is usually negative. Annexin A1 is not present nor are BRAF V600E mutations, consistent with the current belief that these cases are not variants of HCL.
Conclusion
Extranodal and splenic small B-cell lymphomas share many features with their nodal counterparts but there are differences both within the spectrum of morphological appearances and in clinical behaviours. Some lymphomas are characteristically found at extranodal sites or in the spleen and these have distinct pathogenetic, morphological and clinical features. The distinction between the various small B-cell lymphomas can be achieved by careful morphological study with appropriate immunohistochemical stains. In the spleen, there is considerable morphological overlap in lymphomas that involve the white pulp preferentially and a marginal zone pattern is not restricted to splenic marginal zone B-cell lymphoma. Small B-cell lymphomas that preferentially infiltrate the red pulp form a distinct group of lymphomas that as yet remain poorly understood and are the subject of on-going study.
References
Zucca E, Roggero E, Bertoni F et al Primary extranodal non-Hodgkin’s lymphomas. Part 1: gastrointestinal, cutaneous and genitourinary lymphomas. Ann Oncol 1997;8:727–737.
Zucca E, Roggero E, Bertoni F et al Primary extranodal non-Hodgkin’s lymphomas. Part 2: head and neck, central nervous system and other less common sites. Ann Oncol 1999;10:1023–1033.
Krol AD, le Cessie S, Snijder S et al Primary extranodal non-Hodgkin’s lymphoma (NHL): the impact of alternative definitions tested in the Comprehensive Cancer Centre West population-based NHL registry. Ann Oncol 2003;14:131–139.
Thieblemont C, Coiffier B . MALT lymphomas: sites of presentations, clinical features and staging procedures In: Zuuca E, Bertoni F, (eds) MALT Lymphoma. Kluwier Academic/Plenum Publishers: New York, 2004, pp 60–80.
Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR et al Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991;338:1175–1176.
Hyjek E, Isaacson PG . Primary B cell lymphoma of the thyroid and its relationship to Hashimoto’s thyroiditis. Hum Pathol 1988;19:1315–1326.
Hyjek E, Smith WJ, Isaacson PG . Primary B-cell lymphoma of salivary glands and its relationship to myoepithelial sialadenitis. Hum Pathol 1988;19:766–776.
Cerroni L, Zöchling N, Pütz B et al Infection by Borrelia burgdorferi and cutaneous B-cell lymphoma. J Cutan Pathol 1997;24:457–461.
Chanudet E, Zhou Y, Bacon CM et al Chlamydia psittaci is variably associated with ocular adnexal MALT lymphoma in different geographical regions. J Pathol 2006;209:344–351.
Isaacson PG, Wotherspoon AC, Diss T et al Follicular colonization in B-cell lymphoma of mucosa-associated lymphoid tissue. Am J Surg Pathol 1991;15:819–828.
Wotherspoon AC, Doglioni C, Isaacson PG . Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992;20:29–34.
Du MQ, Diss TC, Dogan A et al Clone-specific PCR reveals wide dissemination of gastric MALT lymphoma to the gastric mucosa. J Pathol 2000;192:488–493.
Zukerberg LR, Ferry JA, Southern JF et al Lymphoid infiltrates of the stomach. Evaluation of histologic criteria for the diagnosis of low-grade gastric lymphoma on endoscopic biopsy specimens. Am J Surg Pathol 1990;14:1087–1099.
de Mascarel A, Dubus P, Belleannée G et al Low prevalence of monoclonal B cells in Helicobacter pylori gastritis patients with duodenal ulcer. Hum Pathol 1998;29:784–790.
Hummel M, Oeschger S, Barth TF et al Wotherspoon criteria combined with B cell clonality analysis by advanced polymerase chain reaction technology discriminates covert gastric marginal zone lymphoma from chronic gastritis. Gut 2006;55:782–787.
Ferry JA, Yang WI, Zukerberg LR et al CD5+ extranodal marginal zone B-cell (MALT) lymphoma. A low grade neoplasm with a propensity for bone marrow involvement and relapse. Am J Clin Pathol 1996;105:31–37.
Ueda G, Oka K, Matsumoto T et al Primary hepatic marginal zone B-cell lymphoma with mantle cell lymphoma phenotype. Virchows Arch 1996;428:311–314.
Ballesteros E, Osborne BM, Matsushima AY . CD5+ low-grade marginal zone B-cell lymphomas with localized presentation. Am J Surg Pathol 1998;22:201–207.
Wenzel C, Dieckmann K, Fiebiger W et al CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001;42:823–829.
Heuring AH, Franke FE, Hütz WW . Conjunctival CD5+ MALT lymphoma. Heuring Br J Ophthalmol 2001;85:498–499.
Tasaki K, Shichishima A, Furuta M et al CD5-positive mucosa-associated lymphoid tissue (MALT) lymphoma of ocular adnexal origin: usefulness of fluorescence in situ hybridization for distinction between mantle cell lymphoma and MALT lymphoma. Pathol Int 2007;57:101–107.
Ott G, Katzenberger T, Greiner A et al The t(11;18)(q21;q21) chromosome translocation is a frequent and specific aberration in low-grade but not high-grade malignant non-Hodgkin’s lymphomas of the mucosa-associated lymphoid tissue (MALT-) type. Cancer Res 1997;57:3944–3948.
Dierlamm J, Baens M, Wlodarska I et al The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood 1999;93:3601–3609.
Murga Penas EM, Hinz K, Röser K et al Translocations t(11;18)(q21;q21) and t(14;18)(q32;q21) are the main chromosomal abnormalities involving MLT/MALT1 in MALT lymphomas. Leukemia 2003;17:2225–2229.
Streubel B, Simonitsch-Klupp I, Müllauer L et al Variable frequencies of MALT lymphoma-associated genetic aberrations in MALT lymphomas of different sites. Leukemia 2004;18:1722–1726.
Streubel B, Vinatzer U, Lamprecht A et al T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia 2005;19:652–658.
Wotherspoon AC, Doglioni C, Diss TC et al Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993;342:575–577.
Neubauer A, Thiede C, Morgner A et al Cure of Helicobacter pylori infection and duration of remission of low-grade gastric mucosa-associated lymphoid tissue lymphoma. J Natl Cancer Inst 1997;89:1350–1355.
Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A et al Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001;357:39–40.
Ye H, Gong L, Liu H . Strong BCL10 nuclear expression identifies gastric MALT lymphomas that do not respond to H pylori eradication. Gut 2006;55:137–138.
Ruskoné-Fourmestraux A, Lavergne A, Aegerter PH et al Predictive factors for regression of gastric MALT lymphoma after anti-Helicobacter pylori treatment. Gut 2001;48:297–303.
Nakamura S, Matsumoto T, Suekane H et al Predictive value of endoscopic ultrasonography for regression of gastric low grade and high grade MALT lymphomas after eradication of Helicobacter pylori. Gut 2001;48:454–460.
Liu H, Ruskone-Fourmestraux A, De Jong D et al \t\911;18) is a marker for stage gastric MALT lymphomas that will not respond to H. pylori eradication. Gastroenterology 2002;122:1286–1294.
Copie-Bergman C, Gaulard P, Lavergne-Slove A et al Proposal for a new histological grading system for post-treatment evaluation of gastric MALT lymphoma. Gut 2003;52:1656.
ShiozawaE Norose T, Kaneko K et al Clinicopathological comparison of the World Health Organisation/Wotherspoon score to the Groupe d’Etude des Lymphomes de l’Adult grade for the post-treatment evaluation of gastric mucosa-associated lymphoid tissue lymphoma. J Gastroenterol Hepatol 2009;24:307–315.
Ruskone-Fourmestraux A, Fischbach W, Aleman BM et al EGILS consensus report. Gastric extranodal marginal zone B-cell lymphoma of MALT. Gut 2011;60:747–758.
Alpen B, Thiede C, Wündisch T et al Molecular diagnostics in low-grade gastric marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue type after Helicobacter pylori eradication therapy. Clin Lymphoma 2001;2:103–108.
Bertoni F, Conconi A, Capella C et al Molecular follow-up in gastric mucosa-associated lymphoid tissue lymphomas: early analysis of the LY03 cooperative trial. Blood 2002;99:2541–2544.
Ben-Ayed F, Halphen M, Najjar T et al Treatment of alpha chain disease. Results of a prospective study in 21 Tunisian patients by the Tunisian-French intestinal Lymphoma Study Group. Cancer 1989;63:1251–1256.
Lecuit M, Abachin E, Martin A et al Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med 2004;350:239–248.
Yamamoto S, Nakase H, Yamashita K et al Gastrointestinal follicular lymphoma: review of the literature. J Gastroenterol 2010;45:370–388.
Schmatz AI, Streubel B, Kretschmer-Chott E et al Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol 2011;29:1445–1451.
Drillenburg P, van der Voort R, Koopman G et al Preferential expression of the mucosal homing receptor integrin alpha 4 beta 7 in gastrointestinal non-Hodgkin’s lymphomas. Am J Pathol 1997;150:919–927.
Bende RJ, Smit LA, Bossenbroek JG et al Primary follicular lymphoma of the small intestine: alpha4beta7 expression and immunoglobulin configuration suggest an origin from local antigen-experienced B cells. Am J Pathol 2003;162:105–113.
Shia J, Teruya-Feldstein J, Pan D et al Primary follicular lymphoma of the gastrointestinal tract: a clinical and pathologic study of 26 cases. Am J Surg Pathol 2002;26:216–224.
Damaj G, Verkarre V, Delmer A et al Primary follicular lymphoma of the gastrointestinal tract: a study of 25 cases and a literature review. Ann Oncol 2003;14:623–629.
Bacon CM, Diss TC, Ye H et al Follicular lymphoma of the thyroid gland. Am J Surg Pathol 2009;33:22–34.
Cornes JS . Multiple lymphomatous polyposis of the gastrointestinal tract. Cancer 1961;14:249–257.
Ruskoné-Fourmestraux A, Delmer A, Lavergne A et al Multiple lymphomatous polyposis of the gastrointestinal tract: prospective clinicopathologic study of 31 cases. Groupe D’étude des Lymphomes Digestifs. Gastroenterology 1997;112:7–16.
Kodama T, Ohshima K, Nomura K et al Lymphomatous polyposis of the gastrointestinal tract, including mantle cell lymphoma, follicular lymphoma and mucosa-associated lymphoid tissue lymphoma. Histopathology 2005;47:467–478.
Isaacson PG, Matutes E, Burke M et al The histopathology of splenic lymphoma with villous lymphocytes. Blood 1994;84:3828–3834.
Schmid C, Kirkham N, Diss T et al Splenic marginal zone cell lymphoma. Am J Surg Pathol 1992;16:455–466.
Papadaki T, Stamatopoulos K, Belessi C et al Splenic marginal-zone lymphoma: one or more entities? A histologic, immunohistochemical, and molecular study of 42 cases. Am J Surg Pathol 2007;31:438–446.
Kojima M, Sato E, Oshimi K et al Characteristics of CD5-positive splenic marginal zone lymphoma with leukemic manifestation; clinical, flow cytometry, and histopathological findings of 11 cases. J Clin Exp Hematop 2010;50:107–112.
Baseggio L, Traverse-Glehen A, Petinataud F et al CD5 expression identifies a subset of splenic marginal zone lymphomas with higher lymphocytosis: a clinico-pathological, cytogenetic and molecular study of 24 cases. Haematologica 2010;95:604–612.
Giannouli S, Paterakis G, Ziakas PD et al Splenic marginal zone lymphomas with peripheral CD5 expression. Haematologica 2004;89:113–114.
Watkins AJ, Huang Y, Ye H et al Splenic marginal zone lymphoma: characterization of 7q deletion and its value in diagnosis. J Pathol 2010;220:461–474.
Piris MA, Mollejo M, Campo E et al A marginal zone pattern may be found in different varieties of non-Hodgkin’s lymphoma: the morphology and immunohistology of splenic involvement by B-cell lymphomas simulating splenic marginal zone lymphoma. Histopathology 1998;33:230–239.
Pich A, Fraire F, Fornari A et al Intrasinusoidal bone marrow infiltration and splenic marginal zone lymphoma: a quantitative study. Eur J Haematol 2006;76:392–398.
Ruchlemer R, Wotherspoon AC, Thompson JN et al Splenectomy in mantle cell lymphoma with leukaemia: a comparison with chronic lymphocytic leukaemia. Br J Haematol 2002;118:952–958.
Ruchlemer R, Parry-Jones N, Brito-Babapulle V et al Prolymphocytic leukaemia with t(11;14) revisited: a splenomegalic form of mantle cell lymphoma evolving with leukaemia. Br J Haematol 2004;125:330–336.
Falini B, Tiacci E, Liso A et al Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet 2004;363:1869–1870.
Chen YH, Tallman MS, Goolsby C et al Immunophenotypic variations in hairy cell leukemia. Am J Clin Pathol 2006;125:251–259.
Jasionowski TM, Hartung L, Greenwood JH et al Analysis of CD10+ hairy cell leukemia. Am J Clin Pathol 2003;120:228–235.
Tiacci E, Trifonov V, Schiavoni G et al BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–2315.
Blombery P, Wong SQ, Hewitt CA et al Detection of BRAF mutations in patients with hairy cell leukemia and related lymphoproliferative disorders. Haematologica 2012;97:780–783.
Traverse-Glehen A, Baseggio L, Callet-Bauchu E et al Hairy cell leukaemia-variant and splenic red pulp lymphoma: a single entity? Br J Haematol 2010;150:113–116.
Mollejo M, Algara P, Mateo MS et al Splenic small B-cell lymphoma with predominant red pulp involvement: a diffuse variant of splenic marginal zone lymphoma? Histopathology 2002;40:22–30.
Kanellis G, Mollejo M, Montes-Moreno S et al Splenic diffuse red pulp small B-cell lymphoma: revision of a series of cases reveals characteristic clinico-pathological features. Haematologica 2010;95:1122–1129.
Matutes E, Wotherspoon A, Catovsky D . The variant form of hairy-cell leukaemia. Best Pract Res Clin Haematol 2003;16:41–56.
Jones G, Parry-Jones N, Wilkins B et al Revised guidelines for the diagnosis and management of hairy cell leukaemia and hairy cell leukaemia variant. Br J Haematol 2012;156:186–195.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
About this article
Cite this article
Wotherspoon, A. Extranodal and splenic small B-cell lymphoma. Mod Pathol 26 (Suppl 1), S29–S41 (2013). https://doi.org/10.1038/modpathol.2012.183
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.183
