
Abstract
C1q nephropathy is an uncommon glomerular disease characterized by dominant or codominant mesangial staining for C1q in the absence of systemic lupus erythematosus. There are no series in the literature addressing the significance of C1q deposition in the renal allograft. We retrospectively analyzed 24 patients, most of whom were white (83%) and male (63%), with a mean age at transplant of 31 years. None of the patients were diagnosed with C1q nephropathy in the native kidney or had any features of systemic lupus erythematosus. The mean time from transplant to detection of mesangial C1q deposits was 37 months (>12 months in 71% of cases). Half of the patients had a preceding infection. The indication for biopsy was surveillance (63%) or graft dysfunction (37%). At biopsy, 52% had proteinuria (>1g/day in only 17%). The mean creatinine was 1.8 mg per 100 ml. Only 9% developed hematuria and none had hypoalbuminemia. The glomerular pattern on light microscopy was mesangial hypercellularity (46%), focal segmental glomerulosclerosis (21%), or no lesions (33%). All cases showed intense (≥2+) dominant (67%) or codominant (33%) mesangial staining for C1q on immunofluorescence. Mesangial electron-dense deposits were seen in 82% of cases. On follow-up (mean 1 year) of the 10 patients without rejection, most had stable creatinine with no or stable proteinuria, and none lost their graft. We conclude that C1q-dominant mesangial deposition in the renal allograft is a morphological pattern with no apparent clinical significance in the majority of patients. It is usually detected after the first year. The rate of preceding infection and the prevalence of proteinuria seem to be similar to the renal transplant recipients in general. Most cases show mesangial hypercellularity or no glomerular changes on light microscopy.
Similar content being viewed by others
Main
In 1982, Jones and Magil1 reported five patients with proteinuria and nonsystemic mesangiopathic glomerulonephritis, one of whom had C1q codominant mesangial deposits. In 1985, Jennette and Hipp2 introduced the term ‘C1q nephropathy’ and defined it as dominant or codominant mesangial staining for C1q in patients lacking clinical or serological evidence of systemic lupus erythematosus. In their report, which included 15 patients who presented in their second or third decade of life with nephrotic-range proteinuria, 100% of patients had proteinuria (range 1–24 g/day), 40% hematuria, 0% hypocomplementemia, and 0% positive antinuclear antibodies. The light microscopic findings varied from normal-appearing glomeruli to mesangial hypercellularity to endocapillary hypercellularity. On immunofluorescence (IF), all cases showed intense segmental or global mesangial staining for C1q, which was accompanied by equal or weaker staining for C3 and immunoglobulins. On electron microscopy, all cases showed mesangial electron-dense deposits, 20% revealed glomerular capillary wall deposits, and none revealed endothelial tubuloreticular inclusions. None of the nine patients treated with steroids had a resolution of proteinuria. The authors proposed that C1q nephropathy is a distinct clinicopathological entity that is associated with steroid-resistant nephrotic syndrome.
Three subsequent reports from the United States by Iskandar et al,3 Markowitz et al,4 and Wong et al,5 which comprised a total of 43 patients, mostly children and young adults, found that the majority of patients with C1q nephropathy present with nephrotic syndrome and show either focal segmental glomerulosclerosis or minimal change disease pattern on light microscopy, although mesangial hypercellularity was common. In these reports, steroid responsiveness varied from unsatisfactory3 to intermediate.4, 5
More recent studies reported more heterogenous clinical presentations, histological findings, and outcomes of C1q nephropathy. Vizjak et al6 reported 72 cases of C1q nephropathy from Slovenia, representing the largest cohort described to date. In their study, light microscopy revealed no lesions (38%), focal segmental glomerulosclerosis (15%), proliferative glomerulonephritis (28%), or various other lesions (19%). Patients with no lesions had normal urine examination (7%), asymptomatic proteinuria or hematuria (22%), or nephrotic syndrome (63%). In contrast, all patients with focal segmental glomerulosclerosis presented with nephrotic syndrome. Of those with proliferative glomerulonephritis, 75% had chronic kidney disease and the remaining had asymptomatic urine abnormalities. Complete remission was achieved in three-fourth of the patients with minimal change disease, in only one-third of those with focal segmental glomerulosclerosis, and in just over half of those with proliferative glomerulonephritis. In another large study of 61 patients from Japan, the light microscopy pattern varied from no lesions (75%) to mesangial proliferative glomerulonephritis (12%), to focal segmental glomerulosclerosis (13%).7 Clinically, 36 patients had asymptomatic proteinuria (≤50 mg per 100 ml in most cases) and/or hematuria detected incidentally at urine screening tests, whereas the remaining 25 patients presented with nephrotic syndrome.7
There has been only a single case report of de novo ‘C1q nephropathy’ in the renal transplant.8 The patient presented with renal allograft dysfunction without proteinuria. Light microscopy showed acute cellular rejection; the glomeruli showed mesangial expansion and borderline mesangial hypercellularity. On IF, there was 2+ mesangial staining for C1q. Mesangial electron-dense deposits were detected ultrastructurally. Repeat biopsy 2 months later showed BK polyoma interstitial nephritis and persistent ‘C1q nephropathy’.8 Here, we report 24 patients whose renal allograft biopsies showed intense C1q-dominant or codominant mesangial deposits, fulfilling the diagnostic criteria for C1q nephropathy. Our aim was to report the presenting features, pathological findings, and significance of this enigmatic morphological pattern in the renal transplant.
Materials and methods
Reports of all renal allograft biopsies received and processed at the Renal Biopsy Laboratory at Mayo Clinic, Rochester, Minnesota, from January 1998 to October 2009 (N=17 747), were reviewed retrospectively for finding intense (≥2+) C1q-dominant or codominant mesangial deposits on IF. Biopsies from systemic lupus erythematosus patients and patients whose native disease was membranoproliferative glomerulonephritis were excluded. Of the 24 biopsies found, 22 were from patients who were followed up at the Mayo Clinic. In these patients, the posttransplant course was monitored by periodic determination of clinical and laboratory parameters, as well as renal histology, through both protocol and clinical allograft biopsies. Protocol (surveillance) biopsies were generally performed at implantation, 4 months, 1, 2, and 5 years after transplantation. The remaining two biopsies were from patients followed up at outside institutions.
For light microscopy, all cases were stained with hematoxylin and eosin, periodic acid-Schiff, Masson's trichrome, and Jones methenamine silver. For IF, 3-μm cryostat sections were stained using the Dako Autostainer with polyclonal FITC-conjugated antibodies to IgG, IgM, IgA, C3, C1q, kappa, lambda, fibrinogen, and albumin (Dako, Carpinteria, CA, USA). IF staining for C4d was performed in 20 biopsies (83%). Electron microscopy was performed in 22 biopsies (90%): on glutaraldehyde-fixed tissue in 15 and on deparaffinized tissue in 7 biopsies. No glomeruli were available for electron microscopy in the remaining two biopsies.
Clinical data, including demographic information, native kidney disease, time of transplant, clinical and laboratory findings, treatment, and follow-up, were obtained from the patients’ electronic medical records and the referral forms submitted at the time of biopsy. The following definitions were used: proteinuria, 24 h urine protein >150 mg/day or a spot urine protein to creatinine ratio >150 mg per 1 g of creatinine;9 hematuria, >5 red blood cells per high-power field on microscopic examination of the urinary sediment; and hypoalbuminemia, serum albumin <3.5 g per 100 ml.
Statistical analysis was performed using SPSS for Windows v. 15 (SPSS, Chicago, IL, USA) and StatXact v. 8 (Cytel, Cambridge, MA, USA). Continuous variables are reported as the mean±s.d. (or s.e.m. for Kaplan–Meier survival estimates). Analysis was carried out using exact nonparametric methods given the sample size. Tests performed included Wilcoxon–Mann–Whitney, Fisher–Freeman–Halton, Kruskal—Wallis, and Jonckheere–Terpstra, as appropriate for variable type (continuous, nominal, and ordinal). Renal survival was calculated by the method of Kaplan and Meier and using the Cox proportional hazards model. Statistical significance was assumed at P<0.05. The study was approved by the institutional review board of Mayo Clinic, Rochester.
Results
Pathological Findings
Intense C1q-dominant or codominant mesangial deposits were detected in 0.4% of allograft biopsies in which full IF staining was performed (24 of 5955 biopsies). The renal biopsy findings are detailed in Table 1. Sampling for light microscopy included a mean of 16 glomeruli (range 6–39 glomeruli) and a mean of 9% of glomeruli were globally sclerotic. The most common glomerular pattern of injury on light microscopy, seen in 11 cases (46%), was mesangial hypercellularity, which varied from mild and focal (in the majority of cases) to moderate and diffuse (Figure 1a). Five cases (21%) showed a pattern of focal segmental glomerulosclerosis, not otherwise specified, with (three cases) or without (two cases) mesangial hypercellularity (Figure 1b). In only one of these five cases, the original disease was focal segmental glomerulosclerosis. In the remaining eight cases (33%), glomeruli were normocellular with no lesions of focal segmental glomerulosclerosis (designated as no lesions). None of the cases showed endocapillary hypercellularity, collapsing features, crescents, or necrosis.

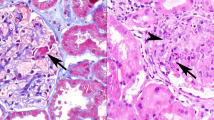
(a) There is moderate global mesangial hypercellularity. Glomerular capillary lumina are patent and the glomerular basement membranes appear normal in thickness (hematoxylin and eosin, × 400). (b) This glomerulus shows a lesion of focal segmental glomerulosclerosis, with an increase in matrix material, inframembranous hyaline and lipid insudation, and adhesion to Bowman's capsule. The nonsclerotic portion of the tuft exhibits mild segmental mesangial hypercellularity (trichrome stain, × 200). (c) There is intense granular global mesangial staining for C1q (immunofluorescence micrograph, × 400). (d) Electron micrograph shows abundant mesangial electron-dense deposits present primarily subjacent to the reflection of the glomerular basement membrane over the mesangium (arrows), accompanied by mild mesangial hypercellularity. No electron-dense deposits are identified involving the glomerular basement membranes, which appear mildly thickened. There is segmental podocyte foot process effacement ( × 3400).
In addition to the C1q deposition, three biopsies showed borderline acute cellular rejection, one showed acute cellular- and antibody-mediated rejection, two showed transplant glomerulopathy (one with a pattern of focal segmental glomerulosclerosis and one with mesangial hypercellularity pattern), one showed acute tubular necrosis, and one showed acute tacrolimus toxicity. None of the patients had histological evidence of BK polyoma nephropathy (including one patient with BK polyoma viremia) or cytomegalovirus nephritis. Most patients had no or only mild tubular atrophy and interstitial fibrosis (Table 1). Similarly, the degree of arteriosclerosis and arteriolar hyalinosis was absent or only mild in the majority of patients.
The number of nonsclerotic glomeruli sampled for IF ranged from 1 to 16 (mean 6 samples). IF revealed dominant (16 cases) or codominant (8 cases) intense (≥2+) staining for C1q in all cases, on a scale of 0–3+ (Table 2; Figure 1c). The pattern of staining was granular to semilinear mesangial (segmental or global). The mean C1q staining intensity was 2.4+. Positivity for C3 was seen in two-thirds of cases with a mean positivity of 0.8+. In only two cases, C3 intensity was similar to C1q (3+). IgG was positive in 83% of cases with a mean intensity of 1.4+, including four cases with a staining intensity similar to C1q. IgM was positive in 83% of cases with a mean intensity of 1.3+, including two cases with staining intensity similar to C1q. IgA positivity was infrequent, detected in 29% of cases (mean intensity 1.1+), and was weaker than C1q in all positive cases. The vast majority of cases showed weak staining for both κ-light chain (mean intensity 1.2+) and λ-light chain (mean intensity 1.3+). IF staining for C4d, performed in 20 cases, was positive in the mesangium in all patients and in peritubular capillaries in three patients (including two with crossmatch-positive transplants).
The number of glomeruli studied ultrastructurally ranged from 1 to 7 (mean 3.5). Mesangial electron-dense deposits were detected in 82% of patients, predominantly subjacent to reflection of the glomerular basement membrane over the mesangium (Figure 1d). The deposits were segmental in 55% of cases and global in 45% of cases. The deposits were more global in the focal segmental glomerulosclerosis and mesangial hypercellularity groups compared with the no lesions group (P=0.014). The texture of deposits was granular without substructure. None of the cases showed subendothelial deposits, subepithelial deposits, or endothelial tubuloreticular inclusions. The percentage of podocyte foot process effacement ranged from none in 6 cases, to mild (<25%) in 12 cases, to moderate (25–50%) in 1 case, to marked (>50%) in 3 cases. The three cases with marked foot process effacement (70–80%) included two cases with recurrent focal segmental glomerulosclerosis and one case with concurrent transplant glomerulopathy.
Before the biopsy that showed intense C1q-dominant or codominant mesangial deposits, 14 patients had had one or more transplant biopsies of the same allograft. On light microscopy, 13 showed no lesions and 1 showed mesangial hypercellularity. Glomerular staining for C1q was negative in 12 patients and trace to 1+ in 2 patients.
Following the biopsy that showed intense C1q-dominant or codominant mesangial deposits, seven patients underwent one to five biopsies of the same allograft. C1q-dominant mesangial deposits persisted in five patients in whom the latest biopsy was performed 0.5, 5, 12, 35, and 36 months after the first biopsy and disappeared in the remaining two in whom the latest biopsy was carried out 6 and 14 months after the first biopsy. The glomerular pattern seen on light microscopy on subsequent biopsies was similar to that seen on the first biopsy.
Clinical Features
The cohort of 24 patients with intense C1q-dominant or codominant mesangial deposits in the renal allograft included 15 males and 9 females (Table 3). In total, 20 patients (83%) were Caucasian, 2 Asian, 1 Hispanic, and 1 American Indian. The mean age at transplantation was 31 years (range 5–72 years; including 6 children (≤15 years) and 18 adults). The native kidney disease was polycystic kidney disease (29% of patients), biopsy-proven focal segmental glomerulosclerosis (17%), hypertensive arteriosclerosis (8%) (one biopsy-proven), biopsy-proven IgA nephropathy/Henoch–Schönlein Purpura (8%), biopsy-proven diabetic nephropathy (4%), biopsy-proven pauci-immune necrotizing and crescentic glomerulonephritis (4%), biopsy-proven anti-GBM disease (4%), solitary kidney (4%), renal hypoplasia (4%), primary hyperoxaluria (4%), reflux nephropathy (4%), biopsy-proven chronic cyclosporine toxicity following liver transplant (4%), and unknown disease (4%). IF and electron microscopy were performed in all 11 patients in whom the native kidney had been biopsied. None of the patients had a diagnosis of C1q nephropathy, lupus nephritis, or membranoproliferative glomerulonephritis in the native kidney. The kidney source was a living-related donor in 38% of patients, a living-unrelated donor in 38% of patients, and a deceased donor in 25% of patients. Three patients were presensitized, including two with positive flow cytometry crossmatch transplant and one with an ABO-incompatible transplant. Of the four patients with end-stage kidney disease secondary to focal segmental glomerulosclerosis, two had recurrent disease, manifesting clinically as heavy proteinuria, and histologically as diffuse podocyte foot process effacement with (one patient) or without (one patient) focal segmental glomerulosclerosis lesions.
The intense C1q-dominant or codominant mesangial deposits were detected in the first allograft in 19 patients. The remaining five patients had one previous transplant. Three of these five patients lost the first transplant owing to ‘chronic allograft nephropathy’, although there are no available data regarding the renal histology. The fourth patient lost his first transplant because of rejection, with no C1q deposits detected on multiple biopsies. In the fifth patient, whose original disease was focal segmental glomerulosclerosis, the first allograft was lost because of recurrent focal segmental glomerulosclerosis. This patient had had an allograft biopsy of his first allograft (performed 15 years before his second transplant) that showed focal segmental glomerulosclerosis and intense C1q deposition.
The mean time from transplant to the detection of intense C1q-dominant or codominant mesangial deposits was 37 months (range 0.1–241 months). One patient with end-stage renal disease due to hypertension was found to have intense C1q codominant mesangial deposits in a protocol biopsy performed three days posttransplant. Both the recipient and the donor (who was his cousin) did not have proteinuria, and the donor had a normal serum creatinine. Of the remaining 23 patients, the intense C1q-dominant or codominant mesangial deposits were detected 4–12 months posttransplant in 6 patients and ≥12 months posttransplant in 17 patients.
The maintenance immunosuppression regimen consisted of tacrolimus, mycophenolate mofetil, and prednisone (63% of patients); tacrolimus and mycophenolate mofetil (13%); tacrolimus, micophenolic acid, and prednisone (8%); cyclosporine, mycophenolate mofetil, and prednisone (4%); cyclosporine and prednisone (4%), tacrolimus, sirolimus, and prednisone (4%); and prednisone and azathioprine (4%). A total of 12 patients (50%) had preceding infection: bacterial urinary tract infection in 4, pneumonia in 1, viral gastroenteritis in 1, cytomegalovirus viremia in 1, BK polyoma viremia in 1, hepatitis C viremia in 1, Epstein Barr viremia in 1, varicella zoster in 1, and onychomycosis in 1. The mean time from infection to biopsy was 1.5 months (range 0–6 months). None of the patients had clinical features of systemic lupus erythematosus. Antinuclear antibody and anti-double-stranded antibody titers were negative in all 11 patients tested, and none of the 9 patients tested had low serum complement (C)3 or C4.
The indication for biopsy was surveillance (63% of patients), worsening creatinine (21%), proteinuria and worsening creatinine (8%), and proteinuria (8%). At biopsy, 52% of patients had proteinuria. The proteinuria was >1 g/day in four patients (17%). Two patients had nephrotic-range proteinuria; 1 of whom with a 24-h urine protein of 4.2 g had recurrent focal segmental glomerulosclerosis and the other one with a 24-h urine protein of 4 g had concurrent transplant glomerulopathy. None of the patients had full nephrotic syndrome. The mean 24-h urine protein for the entire cohort was 572 mg, which was higher in patients with the focal segmental glomerulosclerosis pattern (1900 mg) compared with those with no lesions (367 mg) or with pure mesangial hypercellularity (242 mg) (P=0.015). Seven patients (29%) had worsening serum creatinine from baseline value (>20% increase in creatinine from baseline value). On biopsy, six of these seven patients had concurrent significant pathological findings that might explain the worsening renal function, including transplant glomerulopathy (two patients), borderline acute cellular rejection (one patient), type IB acute cellular rejection and acute antibody-mediated rejection (one patient), acute tubular necrosis (one patient), and acute tacrolimus toxicity (one patient). The mean serum creatinine at biopsy was 1.8 mg per 100 ml (range, 0.9–4.0). Only 9% of patients developed hematuria and none had hypoalbuminemia. The mean serum albumin was 4.2 g per 100 ml (range, 3.5–5.1).
Clinical follow-up data was available in 18 of 24 patients (75%), with a mean follow-up period of 19 months (range 1–60 months). Four patients (22%) were treated with pulse steroids for borderline or acute cellular rejection diagnosed concurrently with the C1q mesangial deposits (two patients) or 2–13 months after the detection of C1q mesangial deposits (two patients). One patient (6%) who had recurrent focal segmental glomerulosclerosis was treated with plasmapheresis and an angiotensin-converting enzyme inhibitor. One patient who had evidence of tacrolimus toxicity was managed by lowering the dose of the medication. One patient who had subnephrotic proteinuria (2.5 g/day) was treated with an angiotensin-converting enzyme inhibitor. The remaining 11 patients (61%) were not treated.
On follow-up, six patients (33%) had no proteinuria with stable creatinine (<20% increase in final creatinine from the value at biopsy), two (11%) had ≥50% decline in proteinuria with stable creatinine, six (33%) had <50% decline in proteinuria or worsening creatinine, and four (22%) resumed hemodialysis or were retransplanted. Of the latter four patients, three had transplant glomerulopathy (diagnosed concurrently with the mesangial C1q deposition in two patients and 15 months later in one patient) and one had severe tubular atrophy and interstitial fibrosis, concurrent with the mesangial C1q deposition. One patient died of pneumonia 7 months post-biopsy.
Patients with no lesions or with mesangial hypercellularity were more likely to have ≥50% decline in proteinuria with stable creatinine, compared with those with the focal segmental glomerulosclerosis pattern (P=0.04). By Kaplan–Meier survival estimates (end point=graft loss), predictors of graft loss were the presence of a secondary glomerular lesion (concurrent transplant glomerulopathy or tacrolimus toxicity), with progression to end-stage kidney disease at a mean of 18.25±11.16 vs 60.00±0.00 months, P=0.002, and deceased donor (vs living donor) kidney transplant, 17.81±6.68 months vs 53.00±8.08, P=0.011. The percentage of global glomerulosclerosis was a borderline predictor (HR 1.08; P=0.081), as was baseline serum creatinine posttransplant (HR 86.76; P=0.078). Serum creatinine at biopsy, proteinuria at biopsy, and histological pattern on light microscopy (no lesion (48.15±14.99 months) vs mesangial hypercellularity (41.40±5.90 months) vs focal segmental glomerulosclerosis (39.00±0.00 months); P=0.85) did not predict graft survival.
Of the 10 patients with intense C1q-dominant or codominant mesangial deposits, who did not have any other significant pathological finding in the first biopsy or thereafter (that is, no transplant glomerulopathy, acute tubular necrosis, acute rejection, or tacrolimus toxicity) and after a mean follow-up of 1 year, 5 continued to have no proteinuria including 2 with no lesions and 3 with mesangial hypercellularity. Two patients (both with focal segmental glomerulosclerosis) had improvement of proteinuria, from 4200 to 833 mg/day in 1 and from 760 to 190 mg/day in the other. The remaining three patients (one with no lesions, one with mesangial hypercellularity, and one with focal segmental glomerulosclerosis) had slight worsening of proteinuria, from 95 to 155 mg/day, 70 to 210 mg/day, and from 300 to 340 mg/day, respectively. Serum creatinine remained stable in 9 of these 10 patients and slightly increased (from 1.0 to 1.4 mg/dl) in the remaining patient. No specific treatment was given for the intense C1q-dominant or codominant mesangial deposits in nine patients, whereas the remaining one who had recurrent focal segmental glomerulosclerosis was treated with plasmapheresis and an angiotensin-converting enzyme inhibitor, which resulted in improvement of proteinuria.
Discussion
None of our 24 patients had a diagnosis of C1q nephropathy in the native kidney, and the disease was detected after the first year posttransplantation in most patients, suggesting that C1q deposition in the renal allograft is likely a de novo process in most cases. However, the native kidney was not examined histologically in 13 (53%) of our patients (mainly those with end-stage kidney disease secondary to polycystic kidney disease or congenital urological anomalies); therefore, in these patients, recurrent C1q deposits remain a possibility. The prevalence of C1q nephropathy among native kidney biopsies ranges from 0.2 to 1.9%.2, 4, 6, 10 The prevalence of intense C1q-dominant or codominant mesangial deposits among our renal allograft biopsies that underwent full IF analysis was 0.4%. This prevalence suggests that de novo ‘C1q nephropathy’ likely represents the third most common de novo morphological glomerular pattern after de novo focal segmental glomerulosclerosis (seen in 10–20% of allografts) and de novo membranous glomerulopathy (seen in 2–9% of allograft biopsies).11, 12 None of our patients were African-Americans, which is in contrast to the cohorts of patients with C1q nephropathy in the native kidney reported by Jennette and Hipp2 and Markowitz et al,4 which included mainly (68%) African-American patients.
In the initial reports of C1q nephropathy in the native kidney, most patients presented with nephrotic syndrome and exhibited focal segmental glomerulosclerosis or minimal change disease patterns on biopsy.2, 3, 4, 5 In more recent reports, patients presenting with asymptomatic proteinuria or hematuria outnumbered those presenting with nephrotic syndrome.6, 7, 10 In a recent study by Hisano et al, from Japan, 59% of C1q nephropathy cases were detected by urine screening tests, most commonly for school children.7 A third of all their patients had mild proteinuria (≤50 mg per 100 ml), but only one had no proteinuria. Most cases with C1q deposition in the renal allograft in our cohort were diagnosed as an incidental finding on surveillance biopsy. Although proteinuria was detected in over half of the patients, it was typically mild (<1 g/day) in most of cases. In a recent study from our center, 45% of transplant recipients in general were found to have proteinuria (>150 mg/day), including 7% with proteinuria >1.5 g/day.13 Therefore, C1q deposition may not be responsible for proteinuria in our patients. The milder clinical and pathological features associated with C1q deposition in the allograft compared with the native kidney are possibly due to the disease detection at early stages by protocol biopsies, the therapeutic effects of the maintenance immunosuppression regimens administered to prevent graft rejection, and/or the hemodynamic effects of calcineurin inhibitors. In contrast to recurrent IgA nephropathy,14 microscopic hematuria is a rare manifestation of C1q deposition, found in only 9% of our cases.
After a mean follow-up of 1 year and without any specific therapy, most of our 10 patients who had C1q deposition without any other significant pathology had stable creatinine with no or stable proteinuria, and none lost their graft. The impact of C1q deposition on long-term graft survival requires additional studies with a larger number of patients and a longer duration of follow-up. In the absence of proteinuria, no specific therapy is needed. Antiproteinuric measures are recommended in patients with proteinuria. This recommendation is based on recent data indicating that proteinuria after kidney transplantation, even at low levels (<500 mg/day), is associated with reduced graft survival, independent of glomerular pathology, graft function, and acute rejection.13, 15
In the recent study by Vizjak et al,6 of 72 cases of C1q nephropathy in the native kidney, 37.5% of patients had underlying infections, most commonly upper respiratory tract infection. Furthermore, the single previously reported patient with de novo ‘C1q nephropathy’ in the renal allograft had concurrent BK polyoma interstitial nephritis.8 In our study, half of patients had preceding infections, including bacterial (five patients), viral (7 patients) and fungal (1 patient) infections. This rate of infection does not seem to be higher than that reported in renal transplant recipients in general.16 The possibility that ‘C1q nephropathy’ in some patients represent an atypical form of infectious glomerulonephritis cannot be excluded, although the normal serum complement, the absence of endocapillary hypercellularity, the weaker staining for C3 than C1q, and the absence of subepithelial humps are all against this possibility.
In the study by Hisano et al,7 of 61 cases of C1q nephropathy in the native kidney, 8 patients underwent repeat biopsies. C1q deposits disappeared in three patients including two who had a minimal change disease pattern in the first biopsy and a focal segmental glomerulosclerosis pattern on the second biopsy and persisted in five patients. On repeat biopsies in seven patients of our cohort (performed 0.5–36 months after the first biopsy), C1q mesangial deposits disappeared in two and persisted in five patients, but the pattern of injury on light microscopy remained the same. Therefore, in the transplant setting, it is unlikely that the focal segmental glomerulosclerosis pattern represents a later stage of disease.
Electron-dense mesangial deposits were detected in 75–100% of C1q nephropathy cases in the native kidney4, 6, 17 and in 82% of our cases. The absence of electron-dense deposits in a minority of cases of C1q nephropathy is likely due to sampling bias. Similar to what Jennette and Hipp2 described in their original report of C1q nephropathy in the native kidney, many of our cases showed segmental, although intense (≥2+), staining for C1q on IF. Therefore, examining more than two full glomeruli ultrastructurally may be needed to detect the deposits.
The pathogenesis of C1q nephropathy is still unknown. Because of the its resemblance to minimal change disease and focal segmental glomerulosclerosis in some patients, Markowitz et al4 speculated that C1q may bind to immunoglobulins and become entrapped nonspecifically in the paramesangial region as a result of increased mesangial trafficking in the setting of glomerular proteinuria. Others favor that C1q mesangial deposits reflects immune complex deposition.6 It is possible that C1q, the first component of the complement cascade and an extremely basic immune reactant, binds to a polyanionic antigen present within the immune complexes deposited from the circulation or formed in situ.2 The polyanionic antigen may be an intrinsic mesangial antigen or an extrinsic one (such as a viral nucleic acid or bacterial antigen). It is unlikely that de novo C1q deposition is mediated by an antibody-mediated reaction to donor antigens, as only three patients (13%) in our cohort were presensitized and only one (4%) had evidence of antibody-mediated rejection on light microscopy.
We conclude that C1q-dominant mesangial deposition in the renal allograft is a morphological pattern, with no apparent clinical significance in the majority of patients. Most cases show mesangial hypercellularity or no glomerular changes on light microscopy.
References
Jones E, Magil A . Nonsystemic mesangiopathic glomerulonephritis with ‘full house’ immunofluorescence. Pathological and clinical observation in five patients. Am J Clin Pathol 1982;78:29–34.
Jennette JC, Hipp CG . C1q nephropathy: a distinct pathologic entity usually causing nephrotic syndrome. Am J Kidney Dis 1985;6:103–110.
Iskandar SS, Browning MC, Lorentz WB . C1q nephropathy: a pediatric clinicopathologic study. Am J Kidney Dis 1991;18:459–465.
Markowitz GS, Schwimmer JA, Stokes MB, et al. C1q nephropathy: a variant of focal segmental glomerulosclerosis. Kidney Int 2003;64:1232–1240.
Wong CS, Fink CA, Baechle J, et al. C1q nephropathy and minimal change nephrotic syndrome. Pediatr Nephrol 2009;24:761–767.
Vizjak A, Ferluga D, Rozic M, et al. Pathology, clinical presentations, and outcomes of C1q nephropathy. J Am Soc Nephrol 2008;19:2237–2244.
Hisano S, Fukuma Y, Segawa Y, et al. Clinicopathologic correlation and outcome of C1q nephropathy. Clin J Am Soc Nephrol 2008;3:1637–1643.
Isaac J, Shihab FS . De novo C1q nephropathy in the renal allograft of a kidney pancreas transplant recipient: BK virus-induced nephropathy? Nephron 2002;92:431–436.
Guy M, Borzomato JK, Newall RG, et al. Protein and albumin-to-creatinine ratios in random urines accurately predict 24 h protein and albumin loss in patients with kidney disease. Ann Clin Biochem 2009;46:468–476.
Fukuma Y, Hisano S, Segawa Y, et al. Clinicopathologic correlation of C1q nephropathy in children. Am J Kidney Dis 2006;47:412–418.
Truong L, Gelfand J, D’Agati V, et al. De novo membranous glomerulonephropathy in renal allografts: a report of ten cases and review of the literature. Am J Kidney Dis 1989;14:131–144.
Antignac C, Hinglais N, Gubler MC, et al. De novo membranous glomerulonephritis in renal allografts in children. Clin Nephrol 1988;30:1–7.
Amer H, Fidler ME, Myslak M, et al. Proteinuria after kidney transplantation, relationship to allograft histology and survival. Am J Transplant 2007;7:2748–2756.
Ponticelli C, Traversi L, Feliciani A, et al. Kidney transplantation in patients with IgA mesangial glomerulonephritis. Kidney Int 2001;60:1948–1954.
Amer H, Cosio FG . Significance and management of proteinuria in kidney transplant recipients. J Am Soc Nephrol 2009;20:2490–2492.
Snyder JJ, Israni AK, Peng Y, et al. Rates of first infection following kidney transplant in the United States. Kidney Int 2009;75:317–326.
Kersnik Levart T, Kenda RB, Avgustin Cavić M, et al. C1Q nephropathy in children. Pediatr Nephrol 2005;20:1756–1761.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Said, S., Cornell, L., Valeri, A. et al. C1q deposition in the renal allograft: a report of 24 cases. Mod Pathol 23, 1080–1088 (2010). https://doi.org/10.1038/modpathol.2010.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.92
