
Abstract
Smad nuclear interacting protein 1 (SNIP1) has been implicated in the pathogenesis of inflammatory bowel disease (IBD). However, the mechanisms involved are still largely unknown. Our results demonstrated that SNIP1 was markedly decreased in intestinal epithelial cells (IEC) from IBD patients compared with healthy controls. Impaired expression of SNIP1 caused a significant decrease of transepithelial electrical resistance but an increase of fluorescein isothiocyanate-dextran flux in Caco-2 monolayers, whereas overexpression of SNIP1 reversed such effects. Overexpression of SNIP1 also inhibited the activity of NF-κB p65 and proinflammatory cytokine production (e.g., TNF-α, IL-1β, and IL-8) by IEC. Importantly, supplementation of exogenous SNIP1 significantly ameliorated intestinal mucosal inflammation in experimental colitis, characterized by less-severe intestinal epithelial barrier damage and decreased proinflammatory cytokine production. Our data thus demonstrated a novel mechanism whereby SNIP1 regulates intestinal inflammation through modulating intestinal epithelial barrier function. Targeting SNIP1 may provide a therapeutic approach for the treatment of IBD.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), are chronic inflammatory conditions that are classically characterized by periods of remission and relapse.1, 2 Although the etiology and pathogenesis of IBD remain elusive, emerging evidences have shown that IBD are resulted from the combination of genetic polymorphisms, environmental factors, and dysregulated immune responses to intestinal commensal microbiota, characterized by an infiltration of varieties of activated immune cells, increased expression of proinflammatory cytokines, and a disruption in the intestinal mucosa barrier.3, 4
Among multiple mechanisms, abnormal immune responses to gut microbiota are crucial to the pathogenesis of IBD. Intestinal homeostasis in healthy individuals is ensured by a complex system called “intestinal barrier”, a dynamic structure that separates intestinal contents from the host tissues, regulates nutrient absorption, and allows interactions with the resident mucosal immune system.5, 6, 7 A monolayer of IEC not only forms a physical barrier separating host from luminal environment, but also interacts with other immune cells, such as T cells and dendritic cells.8, 9, 10 An impaired epithelial barrier provides the opportunity for gut microbiota invasion and causes intestinal inflammation. However, it is still not completely clear how intestinal epithelial barrier is regulated.
Smad nuclear interacting protein 1 (SNIP1), primarily described as a mediator of TGF-β-induced gene expression,11 has been demonstrated as an anti-inflammatory molecule, which binds to the C/H1 domain of the transcriptional co-activators p300 and CREB-binding protein (CBP), a binding site for various transcriptional molecules, including nuclear factor-kappa B (NF-κB).12 It has been shown that SNIP1 protects pressure overload-induced cardiac hypertrophy, fibrosis, and contractile dysfunction through inhibition of NF-κB signaling.13 In our recent study, we have demonstrated that SNIP1 is significantly downregulated in the peripheral blood mononuclear cells and lamina propria mononuclear cells of patients with active IBD, and inhibits Th17 cell differentiation and TNF-α production.14 However, whether and how SNIP1 regulates intestinal mucosal homeostasis and the pathogenesis of IBD are still largely unknown.
In this report, we found that SNIP1 was markedly decreased in IEC of IBD patients and murine colitis model. Stimulation with TNF-α markedly decreased SNIP1 expression in both human primary IEC and colonic epithelial cell lines by inhibiting vitamin D receptor (VDR) expression. SNIP1 promoted the intestinal epithelial barrier function both in vivo and in vitro, and protected the intestines from inflammation in an experimental colitis model in mice.
Results
SNIP1 is significantly decreased in the IEC from IBD patients and DSS-induced murine colitis
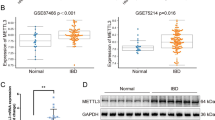
Our recent data have demonstrated that SNIP1 expression is markedly decreased in inflamed intestinal mucosa of IBD patients compared with that in healthy controls.14 However, SNIP1 expression in IEC from IBD patients and its role in the pathogenesis of IBD remain unclear. As shown in Figure 1a, immunohistochemical staining revealed that IEC was a major source of SNIP1 in addition to lamina propria mononuclear cells. The frequencies of SNIP1+ IEC were significantly decreased in inflamed mucosa of active IBD patients compared with healthy controls. To further examine SNIP1 expression, IEC were isolated from freshly isolated colonic tissues of IBD patients and healthy individuals underwent colonoscopy or colectomy. quantitative reverse transcription PCR (qRT-PCR) analysis revealed that SNIP1 was markedly decreased in the IEC of inflamed mucosal tissues from both active CD and UC patients compared with healthy controls (Figure 1b). No statistical difference was observed between active CD and UC groups. To exclude the interference of leukocyte-expressing SNIP1 in the experimental results, IEC were sorted using anti-CD45 flow cytometric antibody (Supplementary Figure S1a online), and SNIP1 mRNA expression in the purified IEC was measured by qRT-PCR. Similar results were also observed (Supplementary Figure S1b). In addition, SNIP1 protein expression was also significantly decreased in IEC of active IBD patients as evidenced by western blot (Figure 1c and d).

SNIP1 is decreased in IEC of IBD patients and DSS-induced colitis in mice. (a) Representative sections were prepared from colonic mucosa of a healthy control (HC) (left panels), and inflamed intestinal mucosa of an active CD (A-CD) patient (middle panels), and an active UC (A-UC) patient (right panels), and stained for SNIP1 by immunohistochemistry (× 200 magnification). (b) IEC were isolated from colonic biopsies of HC (n=29), inflamed mucosa of A-CD patients (n=32), A-UC patients (n=28), CD patients with remission (R-CD, n=27), and UC patients with remission (R-UC, n=26). Expression of SNIP1 was analyzed by qRT-PCR. ***P<0.001. (c,d) The protein levels of SNIP1 in IEC from healthy controls and active CD and UC patients were assessed by western blot. Data are representative of three independent experiments.
As dextran sulfate sodium (DSS)-induced colitis in mice is a well-established experimental model of acute intestinal mucosal inflammation, we sought to compare SNIP1 expression in colonic epithelial cells from DSS-induced colitis and control mice. We observed that SNIP1 mRNA expression was also markedly decreased in the purified colonic epithelial cells from DSS-induced colitis mice compared with that from control mice (Supplementary Figure S1c,d). Taken together, these results indicate that SNIP1 is downregulated in the IEC from both active IBD patients and DSS-induced colitis mice.
TNF-α downregulates SNIP1 expression in IEC
We next investigated the mechanisms whereby SNIP1 expression was downregulated in the IEC from inflamed intestinal mucosa of IBD patients. As accumulating evidences have shown that elevated proinflammatory cytokines (e.g., TNF-α, IFN-γ, IL-1β, IL-6, and IL-17A) are common features of intestinal mucosal inflammation and play a crucial role in the pathogenesis of IBD,14, 15 we detected the expression of TNF-α, IL-1β, IL-6, IFN-γ, and IL-17A mRNA in inflamed mucosa of active IBD patients. Linear cor-relation analyses revealed that IEC-derived SNIP1 mRNA expression was inversely correlated with expression of TNF-α, IL-1β, and IL-6 mRNA, respectively, in the inflamed mucosa from active CD (Supplementary Figure S2a-e) and UC patients (Supplementary Figure S2f-j). In light of these findings, we asked whether SNIP1 downregulation might be associated with the increased levels of proinflammatory cytokines as mentioned above. Human primary IECs were isolated from fresh colonic mucosal biopsies of healthy donors and then stimulated in vitro with TNF-α, IFN-γ, IL-1β, IL-6, and IL-17A, respectively, for 6 h. As shown in Figure 2a, TNF-α significantly, whereas IL-1β and IL-6 modestly inhibited expression of SNIP1 mRNA. As human primary IEC are short-lived after isolation ex vivo, Caco-2 and HCT-116 cells were utilized and exhibited relatively higher levels of SNIP1 expression than other colonic epithelial cell lines tested (i.e., SW480, LoVo, HT-29) (Figure 2b and c). These cells were then treated in vitro with TNF-α, IL-1β, IL-6, IFN-γ, and IL-17A, respectively. Inclusion of TNF-α markedly decreased SNIP1 mRNA expression in both Caco-2 cells (Figure 2d) and HCT-116 cells (Figure 2e). We next determined the kinetics of TNF-α-induced SNIP1 inhibition. Decreased SNIP1 mRNA expression was observed by 12 h after TNF-α treatment, and reached the trough levels by 24 h, indicating that TNF-α inhibits SNIP1 mRNA expression in both a time- (Figure 2f and g; Supplementary Figure S3a, b) and a dose-dependent manner (Figure 2h and i; Supplementary Figure S3c, d).

TNF-α inhibits SNIP1 expression in IEC. (a) Primary IEC (1 × 106) obtained from healthy donors (n=10) were stimulated with various cytokines (20 ng ml−1) as indicated for 6 h. Expression of SNIP1 mRNA was determined by qRT-PCR. *P<0.05, **P<0.01 versus IEC cultured in medium alone. (b) The mRNA levels of SNIP1 in different intestinal epithelial cell lines were measured by qRT-PCR. *P<0.05, **P<0.01 compared with SW480 cells. (c) The protein levels of SNIP1 in different intestinal epithelial cell lines were analyzed by western blot. (d,e) Expression of SNIP1 mRNA in Caco-2 cells (d) and HCT-116 cells (e) treated with different cytokines (20 ng ml−1) as indicated for 48 h was analyzed by qRT-PCR. *P<0.05, **P<0.01 versus cells cultured in medium alone. (f,g) Caco-2 (f) and HCT-116 cells (g) were treated with TNF-α (20 ng ml−1) at different time points as indicated, and expression of SNIP1 mRNA was analyzed by qRT-PCR. *P<0.05, **P<0.01, versus untreated cells. (h,i) Caco-2 (h) and HCT-116 cells (i) were stimulated with different doses of TNF-α as indicated for 48 h, and expression of SNIP1 mRNA was analyzed by qRT-PCR. *P<0.05, **P<0.01 versus untreated cells. Gene expression was normalized to GAPDH in each sample. Data are representative of three independent experiments.
VDR directly induces SNIP1 expression in human colonocytes
To investigate the mechanisms by which TNF-α regulates SNIP1 expression in IEC, we used the MEME-chromatin immunoprecipitation (ChIP) data sets for the motif scanning from large nucleotide and JASPAR database searching for transcription factor binding sites of SNIP1 motif. We found that VDR, which has been reported to be associated with the pathogenesis of IBD and host factors influencing the gut microbiota,16, 17 acts as one of the candidate transcription factors, which might be able to bind to SNIP1 promoter. We transduced VDR-expressing lentivirus (LV-VDR) into Caco-2 cells, and VDR expression was verified in Figure 3a. As expected, SNIP1 mRNA expression was significantly increased in Caco-2 cells after VDR overexpression (Figure 3b). To determine whether VDR directly binds the promoter of SNIP1, we performed ChIP assay using anti-VDR antibody and qRT-PCR using primers specific for the SNIP1 promoter area. Consistently, we found that VDR directly bound to the SNIP1 promoter (Figure 3c). To further confirm this point, the putative VDR-binding site in the promoter region of SNIP1 gene and its site-specific mutant were cloned into a reporter vector upstream of the luciferase gene (Supplementary Figure S4), respectively, and then co-transduced with LV-VDR into Caco-2 cells. As shown in Figure 3d, SNIP1 promoter luciferase activity analysis demonstrated that VDR directly activated SNIP1 expression.

VDR directly promotes SNIP1 expression in IEC. (a–d) Caco-2 cells were transfected with LV-VDR and LV-NC, respectively, and cultured in medium alone for 48 h. Expression of VDR (a) and SNIP1 (b) mRNA was assessed by qRT-PCR. (c) ChIP analysis of VDR occupancy on the SNIP1 promoter in these cells was performed by qRT-PCR. (d) Luciferase activity of SNIP1 gene promoter was detected in each group. (e–g) Caco-2 cells were transduced with LV-VDR and LV-NC, respectively, for 48 h, and then stimulated with TNF-α (20 ng ml−1) for 48 h. (e) The mRNA levels of VDR were detected by qRT-PCR. ChIP analysis (f) and luciferase assay (g) were performed to confirm the effect of VDR on SNIP1 gene promoter. **P<0.01, ***P<0.001. Data are expressed as mean±s.e.m. Data are representative of three independent experiments.
As previous studies have shown that TNF-α inhibits VDR expression in IEC,18 we then investigated whether TNF-α could downregulate SNIP1 expression by inhibiting VDR expression. Caco-2 cells were transduced with LV-VDR and control lentivirus (LV-NC), respectively, followed by stimulation with TNF-α in vitro. VDR expression was verified in Figure 3e. As expected, TNF-α significantly inhibited SNIP1 expression, whereas LV-VDR abrogated such an effect (Figure 3f), which was further confirmed by SNIP1 luciferase activity (Figure 3g). Collectively, these findings suggest that TNF-α inhibits SNIP1 expression in inflamed intestinal mucosa of IBD by downregulation of VDR expression.
Knockdown of SNIP1 induces intestinal epithelial barrier dysfunction through inhibiting TJ protein expression
It has been shown that compromised intestinal mucosal barrier function causes the invasion of luminal pathogens into the lamina propria, which further triggers the dysregulation of immune responses, leading to chronic colonic inflammation.19 Moreover, aberrant gene expression is also associated with the pathogenesis of IBD through affecting intestinal epithelial tight junction (TJ) permeability.20, 21 We asked whether downregulated SNIP1 expression might be involved in the epithelial barrier dysfunction in the pathogenesis of IBD. To this end, Caco-2 monolayers were transduced with SNIP1 siRNA lentivirus (LV-SNIP1 siRNA), and the changes of barrier permeability were detected by transepithelial electrical resistance (TEER) and FITC-dextran flux assays in Transwell polyester membrane filters. As illustrated in Figure 4a, treatment with LV-SNIP1 siRNA significantly decreased TEER compared to monolayers transduced with control lentivirus (LV-NC siRNA) or cultured in medium alone. Consistently, FITC-dextran flux was found to be significantly increased in monolayers after SNIP1 knockdown (Figure 4b). TJ proteins (e.g., ZO-1, occluding, and claudins) have been shown to have a crucial role in maintaining epithelial barrier function. Figure 4c shows that expression of ZO-1, occludin and claudin-1 was markedly decreased after knockdown of SNIP1. We then investigated whether overexpression of SNIP1 could enhance the barrier function of Caco-2 monolayers. We transduced SNIP1-expressing lentivirus (LV-SNIP1) or control vectors into Caco-2 monolayers and found that the value of TEER was markedly increased in SNIP1-overexpressed monolayers (Figure 4d). Consistently, a decrease of FITC-dextran flux was observed in monolayers after LV-SNIP1 transduction compared with controls (Figure 4e). Moreover, western blot analysis further revealed that overexpression of SNIP1 significantly increased the protein levels of ZO-1, occluding, and claudin-1 (Figure 4f). Taken together, these data suggest that SNIP1 maintains intestinal epithelial barrier function possibly through upregulating TJ proteins expression.

SNIP1 promotes epithelial barrier function and tight junction protein expression in vitro. (a,b) Caco-2 monolayers were established using LV-SNIP1 siRNA- or LV-NC siRNA-transduced cells, and epithelial barrier function was determined by TEER (a) and FITC-dextran flux assay (b). **P<0.01 compared with untransduced monolayers. (c) Expression of SNIP1, ZO-1, Claudin-1, and Occludin was determined by western blot. (d,e) Caco-2 monolayers were established using LV-SNIP1- or LV-NC-transduced cells, and epithelial barrier function was determined by TEER (d) and FITC-dextran flux assay (e). *P<0.05 compared with untransduced monolayers. (f) Expression of SNIP1, ZO-1, Claudin-1, and Occludin was determined by western blot. Data are representative of three independent experiments.
SNIP1 inhibits proinflammatory signaling in colonic epithelial cells through suppressing NF-κB activation
It has been shown that increased expression of NF-κB p65 and its target gene (e.g., TNF-α, IL-1β, and IL-8) plays critical roles in the pathogenesis of IBD,22, 23 and that SNIP1 is able to block the activity of NF-κB via inhibiting the effect of translational factors co-activator CBP/p300 on p65 and directly binding to NF-κB p65 submit,12 we then investigated whether SNIP1 could regulate the activity of NF-κB p65 and expression of proinflammatory cytokines (e.g., TNF-α, IL-1β, and IL-8) in IEC. To this end, HCT-116 cells were transduced with LV-SNIP1 as indicated, and NF-κB p65 activation was determined by western blot. Overexpression of SNIP1 significantly inhibited the activity of NF-κB p65 compared with cells cultured in medium alone (Figure 5a). We also measured the levels of proinflammatory cytokines in these cells. As shown in Figure 5b, expression of TNF-α, IL-1β, and IL-8 mRNA was markedly decreased after LV-SNIP1 transduction. Similar data were also obtained using Caco-2 cells, in which NF-κB p65 activity and mRNA levels of proinflammatory cytokine (e.g., TNF-α, IL-1β, and IL-8) were significantly decreased in LV-SNIP1-transduced cells compared with LV-NC (Figure 5c and d). To further determine the role of SNIP1 in regulation of activation of NF-κB and proinflammatory cytokine production, we then investigated whether knockdown of SNIP1 could promote activation of NF-κB and proinflammatory cytokine production. HCT-116 cells were transduced with LV-SNIP1 siRNA and LV-NC siRNA, respectively. Figure 5e illustrates that silencing of SNIP1 expression promoted the activity of NF-κB p65 compared with controls. The expression of TNF-α, IL-1β, and IL-8 at both mRNA and protein levels was increased in cells transduced with LV-SNIP1 siRNA (Figure 5f; Supplementary Figure S5a–c). Similar data were also obtained in Caco-2 cells after transduction with LV-SNIP1 siRNA compared with cells transduced with LV-NC siRNA (Figure 5g and h; Supplementary Figure S5d-f). Collectively, these data indicate that SNIP1 inhibits expression of proinflammatory cytokines and NF-κB activation in IEC.

SNIP1 inhibits expression of proinflammatory cytokines through downregulating NF-κB p65 activation. (a–d) HCT-116 and Caco-2 cells were transduced with LV-SNIP1 and LV-NC, respectively, for 48 h. The phosphorylation of NF-κB p65 was detected by western blot as indicated (a,c). Expression of TNF-α, IL-1β, and IL-8 mRNA was analyzed by qRT-PCR (b,d). *P<0.05 compared with cells cultured in medium alone. (e–h) HCT-116 and Caco-2 cells were transduced with LV-SNIP1 siRNA and LV-NC siRNA, respectively, for 48 h. Phosphorylation of NF-κB p65 was detected by western blot (e,g). Expression of TNF-α, IL-1β, and IL-8 mRNA was analyzed by qRT-PCR (f,h). *P<0.01 compared with cells transduced with LV-NC. Data are pooled from three independent experiments.
SNIP1 suppresses intestinal inflammation in DSS-induced colitis in mice
To further determine the role of SNIP1 in the pathogenesis of IBD, we asked whether supplementation of SNIP1 could inhibit intestinal mucosal inflammation in vivo. We established acute colitis in C57BL/6 mice by treating them with 2% DSS in drinking water for 7 days, followed by 3 days of regular water. The in vivo-jetPEI-mediated SNIP1-DNA was injected intraperitoneally into these colitis mice starting on day 4–9 after DSS exposure as indicated in Figure 6. The mice without receiving DSS treatment were also administrated intraperitoneally with SNIP1-DNA or control DNA as controls. The clinical signs of colitis, including body weight, stool consistency and rectal bleeding, were monitored daily.24 On day 10, all mice were killed and primary IEC were isolated for further analysis. SNIP1-DNA treatment significantly promoted the expression of SNIP1 in primary IEC from DSS-treated mice compared with controls (Figure 6a). Interestingly, SNIP1-DNA treatment suppressed DSS-induced weight loss and colon shortening, resulting in lowering the disease activity index (Figure 6b–d). Histological analysis further revealed that SNIP1-DNA administration led to a significant protection of the colon crypt structures and mild histological damage in the colon compared with controls (Figure 6e and f).

Administration of SNIP1-DNA ameliorates DSS-induced colitis in mice. Two groups of DSS-exposed mice (n=15) were administered intraperitoneally with SNIP1-DNA or control DNA (4 mg kg−1 of body weight) daily starting on day 4–9 (arrow indicates) after DSS induction. Two groups of mice without receiving DSS treatment (n=15) were also administered with SNIP1-DNA or control DNA (4 mg kg−1 of body weight) on the same schedule as negative controls. (a) Effects of SNIP1-DNA or control DNA on the expression of SNIP1 in IEC isolated from different experimental groups. The protein levels of SNIP1 were measured by western blot. (b) The changes of body weight were expressed as the percentage of initial weight at the start of the experiments. *P<0.05 versus the DSS-exposed counterparts receiving control DNA at the same time points. (c) The changes of DAI in each group. *P<0.05 versus mice receiving control DNA in DSS-treated group. (d) Representative colons of mice after the DSS induction are shown. (e) Colonic sections from different experimental groups were performed with H&E staining (original magnification: × 200), and quantitation of histological scores (f) was calculated and presented in bar chart. *P<0.05. Data are representative of three independent experiments.
After confirming that SNIP1 regulated IEC barrier function in vitro (Figure 4), we then investigated how SNIP1 affects intestinal mucosal barrier dysfunction in DSS-colitic mice in vivo. As shown in Figure 7a and b, TEER was found to be increased in the distal and proximal colon of SNIP1-DNA-treated mice compared with that from control DNA-treated counterparts under exposure to DSS. Consistently, DSS administration seriously increased the permeability of gut mucosa in control DNA-treated mice, whereas SNIP1-DNA treatment significantly suppressed intestinal permeability in DSS-induced colitis mice (Figure 7c). Importantly, expression of ZO-1, claudin-1, and occludin was observed to be significantly decreased after DSS exposure, which was markedly reversed by SNIP1-DNA treatment (Figure 7d–g). As previous work has demonstrated that IEC-derived antimicrobial peptides (S100A8 and Muc2) play an important protective role in maintaining intestinal epithelial barrier in DSS-induced colitis,25, 26 we observed that DSS-exposed mice displayed reduced expression of S100A8 and Muc2, whereas the administration of SNIP1-DNA significantly restored their expression in IEC (Supplementary Figure S6a,b). We also analyzed the expression of TNF-α, IL-1β, IL-6, IL-17A, and MCP-1 in colonic tissues, and found that SNIP1-DNA treatment significantly downregulated proinflammatory cytokine expression at both mRNA and protein levels (Supplementary Figure S6c–l). These data thus demonstrate that SNIP1 inhibits intestinal inflammation through regulating intestinal barrier function and intestinal homeostasis.

SNIP1 promotes intestinal epithelial barrier function in vivo. (a,b) Colonic tissues were obtained from mice as described in Figure 6, and then cut into two distal (a) and proximal (b) segments. The changes of TEER in each segment were detected using the Ussing chamber. (c) In vivo permeability before and after DSS treatment was detected by gavage with FITC-dextran. (d,e) Inflamed colonic tissues were collected, and expression of ZO-1, occluding, and claudin-1 at both mRNA and protein levels was measured by qRT-PCR (d–f) and western blot (g), respectively. *P<0.05, **P<0.01 versus control mice. #P<0.05 versus mice receiving control DNA in DSS-treated group. Data are representative of three independent experiments.
Discussion
SNIP1 has been found to express in a variety of cells and tissues and regulate different biological progress.27, 28 Our previous studies demonstrated that SNIP1 is significantly downregulated in peripheral blood mononuclear cells and lamina propria mononuclear cells of patients with active IBD, and suppresses Th17 differentiation and TNF-α secretion.14 In the current study, we demonstrated that expression of SNIP1 was significantly lower in intestinal epithelia from inflamed mucosa of both active CD and UC patients compared with that from healthy controls. Downregulation of SNIP1 impaired IEC barrier function. Furthermore, treatment with SNIP1 promoted IEC barrier integrity and protected the intestines from inflammation, suggesting that SNIP1 promotes intestinal homeostasis and that decreased expression of SNIP1 in IBD patients contributes to the disease progression.
Proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6, and IL-17A) have been reported to be involved in the development of a variety of autoimmune diseases, including IBD.29, 30 TNF-α, a cytokine critical to the pathogenesis of IBD, is found to suppress aberrant gene expression through targeting TNFR in IEC.18, 22 Recent studies have demonstrated that TNF-α-induced downregulation of TJ proteins contributes to the disruption of intestinal epithelial barrier function in IBD.31, 32 In addition, a TNF-α-induced decrease of DMT1 expression was also found to be associated with anemia in active IBD patients, and anti-TNF-α therapy successfully reversed such an effect.33 In the current study, our data revealed that IEC-derived SNIP1 inversely correlated with the expression of TNF-α in active IBD biopsies. Treatment with TNF-α could inhibit the expression of SNIP1 in colonic epithelial cell lines. TNF-α has been shown to inhibit IEC expression of VDR,18 which is associated with IBD and host factors influencing the gut microbiota.16, 17 Interestingly, we identified that VDR directly binds the promoter of SNIP1 and promotes SNIP1 expression in IEC. Ectopic expression of VDR abrogated TNF-α inhibition of SNIP1 expression. Our data thereby indicate that TNF-α inhibits IEC expression of SNIP1 by downregulating VDR expression.
IEC are able to express TJ proteins, including ZO-1, occludin, and claudins, which form a natural intestinal barrier that prevents microbiota antigens and toxins penetrating into the lamina propria.34, 35 Disrupting the apical junction complex between IEC induces intestinal barrier dysfunction. It has been shown that decreased expression of TJ proteins contributes to the pathogenesis of experimental colitis.20, 36 In this study, we found that knockdown of SNIP1 in Caco-2 monolayer resulted in increased intestinal permeability and decreased expression of ZO-1, occludin and claudin-1 protein, whereas overexpression of SNIP1 had an opposite effect. NF-κB activation is involved in TNF-α-induced decrease of TJ proteins expression in IEC.31, 37 It has been shown that SNIP1 inhibits NF-κB signaling by competing for its binding to the C/H1 domain of CBP/p300 transcriptional co-activators.12 We observed that knockdown of SNIP1 in IEC enhanced the activity of NF-κB, while overexpression of SNIP1 in IEC had an opposite effect. Thus, SNIP1 regulates IEC TJ proteins expression and barrier function possibly through downregulation of NF-κB activation.
To date, the role of NF-κB in colitis remains controversial. Recent study has reported that IEC-specific inhibition of NF-κB through conditional deletion of NEMO causes a spontaneous development of severe colitis in mice, characterized by increased rate of IEC apoptosis and a decreased production of antimicrobial peptides,38 indicating an anti-inflammatory overall function of NF-κB in colonic epithelial cells under physiological conditions. However, during the development of IBD and DSS-induced colitis, NF-κB activation turns out to be involved in some proinflammatory epithelial signaling cascades, resulting in a compromised integrity of the epithelial barrier and an enhanced mucosal immune response.23, 39 Our data showed that SNIP1 could inhibit IEC proinflammatory cytokine production through downregulation of NF-κB activation, in which knockdown of SNIP1 promoted, whereas ectopic expression of SNIP1 inhibited expression of TNF-α, IL-1β, and IL-8 in HCT-116 and Caco-2 cell lines in vitro. These data thus indicate that SNIP1 could potentially regulate the pathogenesis of IBD through promoting IEC barrier function and inhibiting proinflammatory cytokine production. Indeed, in DSS-induced colitis model, treatment with SNIP1-DNA using the in vivo-jetPEI reagent demonstrated to inhibit colitis development, characterized by less-severe colitis with improved colonic mucosal inflammation, less disruption of intestinal barrier function, and decreased production of proinflammatory cytokines in inflamed mucosa. Moreover, IEC-derived antimicrobial peptides (e.g., S100A8, Muc2), critical for maintaining intestinal epithelial integrity during colitis,25, 26 were also markedly increased in SNIP1-DNA-treated mice. Collectively, these findings indicate that SNIP1 plays a critical role in maintaining the integrity of the intestinal epithelial barrier and reducing the inflammation of the intestinal mucosa.
In summary, our study demonstrated a critical role of SNIP1 in regulating mucosal homeostasis during the development of IBD. As administration of SNIP1-DNA using the in vivo-jetPEI reagent has proven to prevent an experimental colitis in mice, it may provide a novel therapeutic approach for the treatment of IBD.
Methods
Patients and samples
Colonoscopic and ileoscopic biopsies were collected from inflamed and unaffected sites of the colons from 77 CD patients (42 patients with active CD, 35 in remission), 76 UC patients (39 patients with active UC, 37 in remission), and 41 healthy controls. All patients enrolled in this study were from the Department of Gastroenterology, the Shanghai Tenth People’s Hospital (Shanghai, China). The baseline characteristics are described in Supplementary table 1. The diagnosis of CD or UC was based on clinical, radiological, and histological findings and endoscopic examination. The severity of diseases was assessed according to international standard criteria such as Mayo scores for UC patients and CD activity index for the diagnosis of CD patients. The study was approved by the Institutional Review Board for Clinical Research of the Shanghai Tenth People’s Hospital of Tongji University. Written informed consent was also obtained from all subjects before the study protocol.
Mice and DSS-induced colitis
Female C57BL/6 mice were purchased from Shanghai SLAC Laboratory Animal (Shanghai, China). All mice were maintained at Animal Facility of the Shanghai Tenth Hospital, Tongji University under specific pathogen-free conditions. All animal experiments in this study were approved by the Animal Care and Use Committee at Tongji University. Colitis was induced as reported previously.40 In brief, the experiments enrolled 4 groups of 8 to 10-week-old female C57BL/6 mice (15 mice per group), and two of which were treated with 2% DSS (molecular mass, 36000–50000, MP Biomedicals, LLC; Solon, OH, USA) in drinking water for 7 days, followed by drinking water for additional 3 days. A group of DSS-exposed mice were injected intraperitoneally with in vivo-jetPEI (Polyplus; Illkirch, France)-mediated SNIP1-DNA (4 mg kg−1 of body weight) starting on day 4–9 according to the manufacturer’s instructions.41 The control group was administered with control vector DNA (4 mg kg−1 of body weight). Two groups of non-DSS-exposed mice received drinking water for 10 days, and were administered intraperitoneally with SNIP1-DNA or control DNA as negative controls. The severity of colitis was scored daily by recording standard parameters including body weight, diarrhea, and bloody stools. Colonic tissues were removed, fixed in 10% formalin overnight, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) for histological analysis. The histological scores were evaluated as ranking grade 0–4 as follows: 0, normal intestinal mucosa; (1), loss of one-third of the crypts; (2), loss of two-thirds of the crypts; (3), complete loss of the crypts and the lamina propria is covered with a single layer of epithelium and mild inflammatory cell infiltration is present; and (4), erosions and marked inflammatory cell infiltration are present as previously described.24
Antibodies and reagents
Dulbecco’s modified Eagle’s minimum essential medium (DMEM), RPMI 1640 medium, penicillin, streptomycin, fetal bovine serum, and L-gentamycin were all purchased from HyClone (Logan, UT, USA). Recombinant TNF-α, IFN-γ, IL-1β, IL-6, and IL-17A were purchased from BioLegend (San Diego, CA, USA). All primary antibodies for western blot, immunohistochemical and immunofluorescence staining were purchased from Abcam (Cambridge, UK), and secondary antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody to β-actin was purchased from Abcam.
Isolation of IEC
IEC were isolated as described previously.20 In brief, the colon was removed from the killed mice, cut into 0.5-cm pieces, and placed in cold phosphate-buffered saline (PBS) to remove debris. Colonic tissues were obtained from IBD patients underwent endoscopic examination or colectomy. After incubating at 37 °C for 20 × 2 min in PBS with 2 mM DTT and 1 mM ethylenediaminetetraacetic acid under gently shaking to isolate primary IECs. To exclude the interference of mixed leukocytes, these cells were collected and then purified using flow cytometric sorting with anti-CD45 antibody.
Cell culture, treatment, and transduction
Human IEC lines (HCT-116, HT-29, SW480, Caco-2, and LoVo) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HT-29, SW480, and LoVo cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in 5% CO2. HCT-116 and Caco-2 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. HCT-116 and Caco-2 cells were stimulated with TNF-α (20 ng ml−1), IFN-γ (20 ng ml−1), IL-1β (20 ng ml−1), IL-6 (20 ng ml−1), and IL-17A (20 ng ml−1), respectively, or cultured in medium alone for 48 h, followed by isolation of total RNA for further analyses.
The lentivirus vectors encoding SNIP1 (LV-SNIP1) or SNIP1 siRNA used to knockdown SNIP1 expression (LV-SNIP1 siRNA), and the empty vector as a control (LV-NC siRNA) were purchased from Genechem (Shanghai, China). Cells were transduced with LV-SNIP1, LV-SNIP1 siRNA, or LV-control (LV-NC) at a multiplicity of infection of 10:1 and cultured in medium for 48 h. These cells were used for RNA isolation and western blot.
Measurement of intestinal barrier function
To test the intestinal barrier function, Caco-2 cells (1 × 105) were seeded in the apical side of Transwell inserts (4.0 μm pore size; Corning, Tewksbury, MA, USA). After formatting a complete monolayer, the integrity of cell monolayer was detected by measuring TEER using an epithelial Millicell ERS-2 Voltohmmeter (Merck Millipore; Bedford, MA, USA) as described previously.42 To detect the permeability of cell monolayers, FITC-dextran (FD-40, 4 kDa, 1 mg/ml, Sigma-Aldrich; St. Louis, MO, USA) was added to the apical chamber and incubated for 2 h. The concentrations of FITC-dextran in the basal solutions were determined using Synergy H2 microplate reader (Biotek Instruments; Winooski, VT, USA) at an excitation wavelength of 490 nm and an emission wavelength of 520 nm. In DSS-induced colitis model, intestinal barrier function was measured by detecting colonic TEER in freshly obtained tissues using Ussing chamber as described previously.20 To test the permeability, SNIP1-DNA- or control DNA-treated mice were administrated orally with FITC-dextran (600 mg kg−1 of body weight). After 4 h, blood samples were collected though eye bleeding and then centrifuged at 3500 rpm to obtain the sera. FITC-dextran content in each serum sample was detected by Synergy H2 microplate reader.
RNA isolation and qRT-PCR
Total RNA was extracted from primary IEC, IEC lines, and colonic tissues using GenElute Single Cell RNA Purification Kit (Sigma-Aldrich) according to the manufacturer’s instructions. Total RNA was performed to generate complementary cDNA using a 5 × All-In-One RT MasterMix kit (Applied Biological Materials; Richmond, BC, Canada). The relative levels of gene transcription were detected by qRT-PCR using a SYBR Green PCR kit (TaKaRa; Dalian, China) according to the manufacturer’s protocol. GAPDH was used as a housekeeping gene, and each example was run in triplicate. The relative amount of transcript was analyzed by the 2−ΔΔCT method.
Immunohistochemical staining
Fresh colonic biopsies were fixed with 10% formalin overnight, processed and embedded in paraffin, and then cut into 5-μm sections. To localize SNIP1 expression, sections were stained with anti-SNIP1 mAb (Abcam) as primary antibody, followed by staining with fluorescein isothiocyanate-conjugated anti-IgG as a second antibody.
Statistical analysis
Data were expressed as mean±s.e.m., and analyzed using Prism 5.0 software (Graphpad Software; San Diego, CA, USA). Statistical comparisons were performed using an unpaired two-tailed Student’s t-test or one-way analysis of variance. Statistical significance was set at P<0.05. Linear cor-relation was performed to analyze the correlation of the mRNA levels of inflammatory cytokines (TNF-α, IL-β, IL-6, IL-17A, and IFN-γ) and SNIP1 mRNA expression in IEC from IBD patients.
References
Boyapati, R.K., Rossi, A.G., Satsangi, J. & Ho, G.T. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol. 9, 567–582 (2016).
He, C. et al. MicroRNA 301A promotes intestinal inflammation and colitis-associated cancer development by inhibiting BTG1. Gastroenterology 152, 1434–1448 (2017).
Wadwa, M. et al. IL-10 downregulates CXCR3 expression on Th1 cells and interferes with their migration to intestinal inflammatory sites. Mucosal Immunol. 9, 1263–1277 (2016).
Calderon-Gomez, E. et al. Commensal-specific CD4(+) cells from patients with crohn's disease have a T-helper 17 inflammatory profile. Gastroenterology 151, 489–500 (2016).
Nalle, S.C. & Turner, J.R. Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal Immunol. 8, 720–730 (2015).
Goto, Y. & Kiyono, H. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol. Rev. 245, 147–163 (2012).
Chellappa, K. et al. Opposing roles of nuclear receptor HNF4α isoforms in colitis and colitis-associated colon cancer. eLife 5, e10903 (2016).
Harrison, O.J. et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol. 8, 1226–1236 (2015).
Westendorf, A.M. et al. CD4+Foxp3+ regulatory T cell expansion induced by antigen-driven interaction with intestinal epithelial cells independent of local dendritic cells. Gut 58, 211–219 (2009).
Maggio-Price, L. et al. Lineage targeted MHC-II transgenic mice demonstrate the role of dendritic cells in bacterial-driven colitis. Inflamm. Bowel Dis. 19, 174–184 (2013).
Kim, R.H. et al. A novel smad nuclear interacting protein, SNIP1, suppresses p300-dependent TGF-beta signal transduction. Genes Dev. 14, 1605–1616 (2000).
Kim, R.H. et al. SNIP1 inhibits NF-kappa B signaling by competing for its binding to the C/H1 domain of CBP/p300 transcriptional co-activators. J. Biol. Chem. 276, 46297–46304 (2001).
Lu, Y.Y. et al. Smad nuclear interacting protein 1 acts as a protective regulator of pressure overload-induced pathological cardiac hypertrophy. J. Am. Heart Assoc. 5, e003943 (2016).
He, C. et al. miR-301a promotes intestinal mucosal inflammation through induction of IL-17A and TNF-α in IBD. Gut 65, 1938–1950 (2016).
Yu, L. et al. Infliximab preferentially induces clinical remission and mucosal healing in short course Crohn's disease with luminal lesions through balancing abnormal immune response in gut mucosa. Mediators Inflamm. 2015, 793764 (2015).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Wang, J. et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 48, 1396–1406 (2016).
Chen, Y. et al. MicroRNA-346 mediates tumor necrosis factor alpha-induced downregulation of gut epithelial vitamin D receptor in inflammatory bowel diseases. Inflamm. Bowel. Dis. 20, 1910–1918 (2014).
Michielan, A. & D'Inca, R. Intestinal permeability in inflammatory bowel disease: pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflamm. 2015, 628157 (2015).
Liu, W. et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Invest. 123, 3983–3996 (2013).
Ye, D., Guo, S., Al-Sadi, R. & Ma, T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 141, 1323–1333 (2011).
Wu, W. et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut 64, 1755–1764 (2015).
Law, I.K. et al. Neurotensin—regulated miR-133α is involved in proinflammatory signalling in human colonic epithelial cells and in experimental colitis. Gut 64, 1095–1104 (2015).
Wang, Z. et al. Syndecan-1 acts in synergy with tight junction through Stat3 signaling to maintain intestinal mucosal barrier and prevent bacterial translocation. Inflamm. Bowel Dis. 21, 1894–1907 (2015).
Zindl, C.L. et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl Acad. Sci. USA 110, 12768–12773 (2013).
Kissoon-Singh, V., Moreau, F., Trusevych, E. & Chadee, K. Entamoeba histolytica exacerbates epithelial tight junction permeability and proinflammatory responses in Muc2(-/-) mice. Am. J. Pathol. 182, 852–865 (2013).
Roche, K.C., Wiechens, N., Owen-Hughes, T. & Perkins, N.D. The FHA domain protein SNIP1 is a regulator of the cell cycle and cyclin D1 expression. Oncogene 23, 8185–8195 (2004).
Yu, B. et al. The FHA domain proteins DAWDLE in Arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc. Natl Acad. Sci. USA 105, 10073–10078 (2008).
Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14, 329–342 (2014).
Chen, M.L. & Sundrud, M.S. Cytokine networks and T-cell subsets in inflammatory bowel diseases. Inflamm. Bowel Dis. 22, 1157–1167 (2016).
Ma, T.Y. et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G367–G376 (2004).
Slebioda, T.J. & Kmiec, Z. Tumour necrosis factor superfamily members in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 2014, 325129 (2014).
Wu, W. et al. Divalent metal-ion transporter 1 is decreased in intestinal epithelial cells and contributes to the anemia in inflammatory bowel disease. Sci. Rep. 5, 16344 (2015).
Vivinus-Nebot, M. et al. Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut 63, 744–752 (2014).
Sun, M., He, C., Cong, Y. & Liu, Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. 8, 969–978 (2015).
Hu, G.Q. et al. AIM2 contributes to the maintenance of intestinal integrity via Akt and protects against Salmonella mucosal infection. Mucosal Immunol. 9, 1330–1339 (2016).
Chen, S.W. et al. Protective effect of hydrogen sulfide on TNF-α and IFN-γ-induced injury of intestinal epithelial barrier function in Caco-2 monolayers. Inflamm. Res. 64, 789–797 (2015).
Nenci, A. et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007).
Nata, T. et al. MicroRNA-146b improves intestinal injury in mouse colitis by activating nuclear factor-kappaB and improving epithelial barrier function. J. Gene Med. 15, 249–260 (2013).
Spalinger, M.R. et al. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 8, 918–929 (2015).
Wirtz, S., Billmeier, U., McHedlidze, T., Blumberg, R.S. & Neurath, M.F. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 141, 1875–1886 (2011).
Liu, H., Wang, P., Cao, M., Li, M. & Wang, F. Protective role of oligomycin against intestinal epithelial barrier dysfunction caused by IFN-gamma and TNF-α. Cell Physiol. Biochem. 29, 799–808 (2012).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81630017, 81470822, 91740117).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Author contributions
Y.S. and C.H. performed the experiments. C.M., T.Y., Y.C., and W.C. analyzed the data; Z.L. diagnosed the patients and provided clinical information and samples; Y.S. and Z.L. wrote the manuscript. Z.L. conceived the project, designed and supervised the experimental work and data analyses. All authors participated in revising the manuscript and agreed to the final version.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Shi, Y., He, C., Ma, C. et al. Smad nuclear interacting protein 1 (SNIP1) inhibits intestinal inflammation through regulation of epithelial barrier function. Mucosal Immunol 11, 835–845 (2018). https://doi.org/10.1038/mi.2017.95
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.95
This article is cited by
-
MKRN1 promotes colorectal cancer metastasis by activating the TGF-β signalling pathway through SNIP1 protein degradation
Journal of Experimental & Clinical Cancer Research (2023)
-
MAdCAM-1 mediates retinal neuron degeneration in experimental colitis through recruiting gut-homing CD4+ T cells
Mucosal Immunology (2021)
-
MicroRNA-219a-5p suppresses intestinal inflammation through inhibiting Th1/Th17-mediated immune responses in inflammatory bowel disease
Mucosal Immunology (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}