
Abstract
Scavenger receptor B-I (SR-BI) is a multirecognition receptor that regulates cholesterol trafficking and cardiovascular inflammation. Although it is expressed by neutrophils (PMNs) and lung-resident cells, no role for SR-BI has been defined in pulmonary immunity. Herein, we report that, compared with SR-BI+/+ counterparts, SR-BI−/− mice suffer markedly increased mortality during bacterial pneumonia associated with higher bacterial burden in the lung and blood, deficient induction of the stress glucocorticoid corticosterone, higher serum cytokines, and increased organ injury. SR-BI−/− mice had significantly increased PMN recruitment and cytokine production in the infected airspace. This was associated with defective hematopoietic cell-dependent clearance of lipopolysaccharide from the airspace and increased cytokine production by SR-BI−/− macrophages. Corticosterone replacement normalized alveolar neutrophilia but not alveolar cytokines, bacterial burden, or mortality, suggesting that adrenal insufficiency derepresses PMN trafficking to the SR-BI−/− airway in a cytokine-independent manner. Despite enhanced alveolar neutrophilia, SR-BI−/− mice displayed impaired phagocytic killing. Bone marrow chimeras revealed this defect to be independent of the dyslipidemia and adrenal insufficiency of SR-BI−/− mice. During infection, SR-BI−/− PMNs displayed deficient oxidant production and CD11b externalization, and increased surface L-selectin, suggesting defective activation. Taken together, SR-BI coordinates several steps in the integrated neutrophilic host defense response to pneumonia.
Similar content being viewed by others


INTRODUCTION
Community-acquired pneumonia is a serious condition that results when bacteria invade the distal airspaces and induce an innate immune response.1 Despite medical advances, pneumonia remains the most common cause of severe sepsis and the leading cause of death by infection.2 Although the lung is often considered in isolation during pneumonia, the pulmonary innate immune response arises from integrated communications between lung-resident cells and other organs including the bone marrow and adrenal glands. Whereas adrenal function correlates with pneumonia severity,3 its functional contribution to pulmonary host defense is undefined. Improved understanding of the molecular regulation that coordinates the integrated host response during pneumonia is needed in order to advance insight into why some patients develop bacteremia and septic shock while others do not.
Scavenger receptors (SRs) are a widely expressed family of pattern recognition receptors, categorized into eight classes (A–H) by domain architecture. SRs bind a very broad array of ligands, both endogenous (for example, oxidized lipids and amyloid) and exogenous (for example, pathogens and particulates), and have previously been implicated in several diseases, including atherosclerosis and Alzheimer’s disease.4 Although roles for class A SRs, including MARCO and SR-A, have been identified in the lung’s response to pathogens and particulates, little is known about the roles of other SR classes in pulmonary biology.5, 6, 7
SR class B type I (SR-BI) has been mostly studied in vascular biology as it mediates selective uptake of cholesterol ester from high-density lipoprotein (HDL) by hepatocytes and endothelium, regulating serum HDL and atherogenesis.8 However, HDL exerts wide-ranging actions, including suppression of leukocyte function,9 and SR-BI is expressed by several tissues that collaborate in host defense. Indeed, suggesting the potential for a role in the integrated response of tissues to environmental stress, SR-BI expression by several cell types has recently been shown to regulate the lipopolysaccharide (LPS) response (macrophages),10 clearance of plasma LPS (hepatocytes),11 chemotaxis (neutrophils (PMNs)),9 and stress glucocorticoid production (adrenal glands).12 The significance of these findings to humans is suggested by recent reports that SR-BI polymorphisms are associated with adrenal insufficiency, serum HDL, and atherosclerosis,13, 14 and serum HDL correlates with pneumonia severity in patients.15, 16
In addition to expression on alveolar macrophages, SR-BI is also expressed by alveolar epithelial (AE) cells, where it mediates vitamin E uptake.17 However, no further role for SR-BI has been defined in the lung. Herein, we report that SR-BI is critical to survival during bacterial pneumonia. SR-BI−/− mice recruit increased PMNs to the infected lung, associated with defective clearance of LPS from the airspace and increased alveolar cytokines. Alveolar neutrophilia is also promoted by adrenal insufficiency, as correction of glucocorticoid deficiency in infected SR-BI−/− mice normalizes airspace PMNs. However, despite increased airspace PMNs, SR-BI−/− mice suffer marked bacteremia, cytokine storm, and organ injury that derives, in part, from a dramatic defect in phagocytic killing. Taken together, we report an essential new role for SR-BI in the integrated physiologic response to pneumonia and in phagocyte antimicrobial function.
RESULTS
SR-BI deficiency compromises host defense during bacterial pneumonia
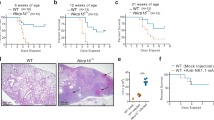
SR-BI has largely been studied in vascular biology because of its regulation of plasma HDL levels and endothelial responsiveness to HDL.18 Given that HDL suppresses leukocyte pro-inflammatory functions9 and SR-BI is expressed in the lung, we hypothesized that SR-BI-null mice would have an altered host defense response to bacterial pneumonia. To address this, SR-BI-sufficient (SR-BI+/+) and -deficient (SR-BI−/−) mice were dosed intratracheally (i.t.) with the clinically relevant Gram-negative bacterium Klebsiella pneumoniae, and mortality was monitored for 10 days. Remarkably, at an inoculum that was approximately an LD50 in SR-BI+/+ mice, all SR-BI−/− mice were dead by 48 h post inoculation (p.i.; Figure 1a).

Scavenger receptor B-I −/− (SR-BI−/−) mice have a failure of host defense after pulmonary infection with K. pneumoniae. (a) SR-BI+/+ and SR-BI−/− mice (n=10 per group) were inoculated intratracheally (i.t.) with 200 colony forming units (CFUs) of K. pneumoniae, and survival was monitored. P<0.0001 by log-rank test. (b–h) SR-BI+/+ and SR-BI−/− mice were inoculated i.t. with 2,000 CFUs of K. pneumoniae, and necropsied 24 h post inoculation. Bronchoalveolar lavage fluid (BALF) total protein was quantified using Bradford assay (b), lung inflammation was scored (c), and bacterial CFUs in the lung (d), blood (e), and liver (f) were quantified. Serum levels of TNFα (tumor-necrosis factor-α), G-CSF (granulocyte-colony–stimulating factor), and CXCL2 (g), and lactate dehydrogenase (LDH) (h) were also quantified. Data shown in b, d–h are representative of two independent experiments involving N=5–8 per genotype. Panel c derives from one experiment of N=8 per genotype. *P<0.05; **P<0.01; ***P<0.001. CXCL2, chemokine (C-X-C motif) ligand 2.
Pneumonia can cause mortality through lung injury and consequent respiratory failure. In support of exacerbated acute lung injury, bronchoalveolar lavage fluid (BALF) protein, a surrogate measure of microvascular integrity in the lung,19 was higher in infected SR-BI−/− mice than in controls (Figure 1b). Histopathologic evaluation also revealed more severe neutrophilic inflammation in the lungs of infected SR-BI−/− mice than in the SR-BI+/+ counterparts (Figure 1c and Supplementary Figure 1 online). Bacteremia, arising from extrapulmonary dissemination of pathogens, is common in pneumonia and can lead to septic shock through dysregulated induction of the systemic innate immune response.2, 20 Compared with controls, infected SR-BI−/− mice had approximately threefold higher bacteria in lung tissue 24 h p.i. (Figure 1d), indicating defective pathogen clearance in the lung. More remarkably, at 24 h p.i., SR-BI−/− mice had an ∼2 log increase in bacteria in the blood (Figure 1e) and a nearly 3-log increase in bacteria in the liver (Figure 1f) compared with controls, suggesting more profound failure of host defense in the extrapulmonary compartment. Consistent with a robust sepsis syndrome, SR-BI−/− mice had much higher serum cytokines (tumor-necrosis factor-α (TNFα), granulocyte-colony–stimulating factor (G-CSF), and chemokine (C-X-C motif) ligand 2 (CXCL2) than controls (Figure 1g), as well as higher serum lactate dehydrogenase (Figure 1h), a marker of cell death denoting organ injury.
SR-BI deficiency increases innate immune responses in the infected airspace
We next sought to determine whether the innate immune response was defective in the SR-BI-null airway. Alveolar cytokines recruit PMNs into the airspace where they are pivotal bactericidal cells in the host response to infection.2 SR-BI−/− mice had increased PMNs in the airspace 24 h p.i. when compared with SR-BI+/+ controls (Figure 2a). BALF of infected SR-BI−/− mice also had higher levels of TNFα, a cytokine of established importance to PMN recruitment,21 and CXCL5/LIX, an AE-derived chemokine reported to have a key role in attraction of PMNs to the infected airspace (Figure 2b).22 By contrast, several other cyto-/chemokines (G-CSF, CXCL1, CXCL2, and interleukin (IL)-6) were induced to equivalent levels in SR-BI+/+ and SR-BI−/− BALF, suggesting that SR-BI has selective effects on innate immune responses in the infected airspace. BALF and serum IL-17 were also equivalent between SR-BI+/+ and SR-BI−/− mice 24 h post infection (data not shown).

The pulmonary response to K. pneumoniae infection is enhanced in Scavenger receptor B-I −/− (SR-BI−/−) mice. SR-BI+/+ and SR-BI−/− mice were inoculated intratracheally (i.t.) with 2,000 colony forming units (CFUs) of K. pneumoniae, and necropsied 24 h post inoculation. (a) Bronchoalveolar lavage fluid (BALF) macrophages (Mϕ) and neutrophils (PMN) were counted. (b) BALF levels of CXCL1, tumor-necrosis factor-α (TNFα), G-CSF (granulocyte-colony–stimulating factor), CXCL2, and interleukin (IL)-6 were quantified using Bioplex assay and CXCL5 using ELISA. Data shown are representative of two independent experiments involving N=5–8 mice per genotype. *P<0.05. CXCL2, chemokine (C-X-C motif) ligand 2.
SR-BI−/− mice have increased and sustained airway immune responses to inhaled LPS
Despite the higher alveolar neutrophilia in SR-BI−/− lungs at 24 h p.i., bacteria were also increased in SR-BI−/− lungs at this time point (Figure 1), thus confounding conclusions as to cause vs. effect, and also obscuring the degree to which BALF cytokines derive from airway-resident vs. recruited immune cells. In order to examine the pulmonary innate immune response with more precise kinetics, we conducted a 30-min exposure of SR-BI+/+ and SR-BI−/− mice to aerosolized LPS, the major immunostimulatory glycolipid of the outer cell wall of Gram-negative bacteria. In response to inhaled LPS, SR-BI−/− mice had enhanced airspace recruitment of leukocytes (Figure 3a, Supplementary Figure 2) and PMNs specifically (Figure 3b). Increased alveolar neutrophilia was seen by 8 h post exposure and was sustained out to 48 h. Similar to the case with K. pneumoniae, SR-BI−/− mice had elevations in BALF TNFα and CXCL5, but not other cyto-/chemokines including IL-17 2 h after LPS exposure, when compared with controls (Figure 3c, data not shown). This time point largely reflects cytokine production by resident alveolar cells as it precedes substantial recruitment of PMNs into the airspace (Figure 3b). At 8 h after LPS, TNFα remained relatively elevated in SR-BI−/− BALF, but G-CSF and IL-6 were elevated compared with controls (Figure 3d). Sustained alveolar cytokines and neutrophilia in SR-BI−/− mice suggested to us the possibility that SR-BI might mediate clearance of LPS from the airspace.

The pulmonary response to inhaled lipopolysaccharide (LPS) is increased in Scavenger receptor B-I −/− (SR-BI−/−) mice. SR-BI+/+ and SR-BI−/− mice were challenged with aerosolized LPS. Bronchoalveolar lavage fluid (BALF) total leukocytes (WBCs) (a) and neutrophils (PMN) (b) were counted at 2, 8, 24, and 48 h post exposure. (c–d) BALF CXCL1, tumor-necrosis factor-α (TNFα), G-CSF (granulocyte-colony–stimulating factor), CXCL2, and interleukin (IL)-6 proteins were quantified using Bioplex assay and CXCL5 using ELISA at 2 h (c) and 8 h (d), respectively, post exposure. Data shown are representative of two independent experiments involving N=5–8 mice per genotype per time point. *P<0.05. CXCL2, chemokine (C-X-C motif) ligand 2.
Hematopoietic cell SR-BI mediates LPS clearance and regulates toll-like receptor (TLR) responses in the airway
SR-BI is reported to mediate cellular uptake of LPS and to have a role in clearance of LPS from plasma;11, 23, 24 thus, SR-BI−/− mice have sustained induction of serum cytokines during endotoxemia due to delayed LPS clearance by the liver.11 No reports, to our knowledge, have addressed how/whether LPS is cleared by the lung. In order to address this, BALF LPS was quantified with the limulus amebocyte lysate assay at various time points following LPS inhalation. Compared with controls, SR-BI−/− mice had equivalent BALF LPS 2 h following exposure, but significantly higher BALF LPS 8 and 24 h after exposure (Figure 4a). In control experiments, an LPS standard was spiked ex vivo into cell-free BALF collected from naive SR-BI+/+ and SR-BI−/− mice. Equal levels of LPS were detected in the two BALF types, verifying that SR-BI+/+ and SR-BI−/− BALFs have equivalent LPS neutralization capacity (data not shown).

Decreased clearance of, and enhanced responsiveness to, lipopolysaccharide (LPS) in the Scavenger receptor B-I −/− (SR-BI−/−) airway. (a) SR-BI+/+ and SR-BI−/− mice (N=5–8 per group) were challenged with aerosolized LPS (300 μg ml−1, 30 min) and LPS clearance from the airspace at various time points post exposure was measured with the LAL assay on the bronchoalveolar lavage fluid (BALF). (b) SR-BI bone marrow chimeras, generated by bone marrow reconstitution of lethally irradiated SR-BI+/+ (WT) recipients with bone marrow from WT or SR-BI−/− mice were exposed to aerosolized LPS. Mice (N=7 per genotype) were killed 24 h post exposure for LAL measurement of BALF LPS. (c) Peritoneal exudate macrophages from SR-BI+/+ and SR-BI−/− mice were incubated with biotin-labeled LPS and both surface-bound and total (internalized plus surface-bound) LPS were quantified using flow cytometry. Histograms are shown at left and pooled and quantified the mean fluorescence intensity (MFI) data at right. N=6 per genotype. (d) SR-BI+/+ and SR-BI−/− peritoneal exudate macrophages were incubated with media, Pam3CSK4 (100 ng ml−1), Poly I:C (10 μg ml−1), LPS (10 ng ml−1), or CpG oligodeoxynucleotide (10 μg ml−1) for 24 h, and TNFα (tumor-necrosis factor-α) was measured in cell supernatants using ELISA. (e) TLR4 cell-surface display on SR-BI+/+ and SR-BI−/− peritoneal-elicited macrophages was quantified using flow cytometry. Histograms are shown at left (shaded trace is unstained cells) and pooled and quantified mean fluorescence intensity (MFI) data at right. N=3 mice per genotype run in technical duplicate. **P<0.01. (f) SR-BI bone marrow chimeras as in b (N=7 per genotype) were exposed to aerosolized LPS and BALF macrophages (Mϕ) and neutrophils (PMN) were counted 24 h post exposure. Data shown are representative of two independent experiments. *P<0.05; **P<0.01.
Speculating that SR-BI expressed by airway-resident macrophages (that is, alveolar macrophages) might mediate LPS clearance from the airspace, we transferred SR-BI+/+ or SR-BI−/− bone marrow to lethally irradiated SR-BI+/+ recipients in order to generate mice chimeric for SR-BI expression in hematopoietic cells. Mice with isolated hematopoietic cell SR-BI deficiency (that is, receiving SR-BI−/− marrow) had higher BALF LPS 24 h post LPS inhalation than their SR-BI-sufficient counterparts (Figure 4b), confirming that SR-BI expression by bone marrow-derived cells mediates LPS clearance from the airway. In order to address this further, peritoneal-elicited macrophages from SR-BI+/+ and SR-BI−/− mice were incubated with biotin-labeled LPS to assess intracellular uptake of LPS. Consistent with a role for macrophage SR-BI in LPS clearance from the airway, SR-BI−/− macrophages displayed a significant reduction in both surface-bound LPS and in the sum of surface-bound plus internalized LPS (Figure 4c).
LPS is internalized by leukocytes after activation of TLR4 on the cell surface;24 however, whether mass internalization of LPS serves as an activation or as a detoxification pathway remains unclear. Despite their reduced uptake of LPS, SR-BI−/− macrophages were hyper-responsive to LPS, producing elevated levels of TNFα (Figure 4d). Interestingly, SR-BI−/− macrophages also produced elevated TNFα after ligation of TLR2 (Pam3CSK4)—like TLR4, a plasmalemmal TLR—whereas ligation of the endosomal receptors TLR3 (polyI:C) and TLR9 (CpG DNA) induced equivalent TNFα in SR-BI+/+ and SR-BI−/− macrophages (Figure 4d). Consistent with their heightened responsiveness to LPS, SR-BI−/− macrophages displayed increased cell-surface TLR4 (Figure 4e).
Taken together, these findings suggest an important role for hematopoietic cell, and potentially macrophage, SR-BI in clearance of LPS from the airway and in suppression of the innate cytokines that drive alveolar neutrophilia. Indeed, suggesting the importance of hematopoietic cell SR-BI to regulation of alveolar neutrophilia in vivo, chimeric mice with isolated deficiency of SR-BI in hematopoietic cells had higher airspace neutrophils 24 h post-LPS inhalation than SR-BI-sufficient counterparts (Figure 4f).
SR-BI deficiency unveils a role for adrenal function in pulmonary innate immunity
Having found that airway-infected SR-BI−/− mice appear to die from failure of host defense, we next sought an explanation. SR-BI−/− mice reportedly display stress-induced adrenal insufficiency because of absence of HDL-mediated, SR-BI-dependent delivery of cholesterol, the precursor for steroid hormones, to the adrenals.12 Although systemic glucocorticoids are well known to regulate airway and systemic inflammation in vivo,3 the role of native adrenal function in pulmonary host defense remains undefined.
We next addressed the possibility that adrenal insufficiency might contribute to the pneumonia phenotype of the SR-BI-null mouse. Although SR-BI+/+ and SR-BI−/− mice had comparable serum corticosterone in the naive state, 24 h after K. pneumoniae, SR-BI+/+ mice had a significant increase in corticosterone that was absent in SR-BI−/− mice (Figure 5a), indicating adrenal insufficiency in the latter strain. We established conditions for i.p. corticosterone supplementation, commenced before induction of pneumonia, that achieved equivalent serum levels in infected SR-BI+/+ and SR-BI−/− mice (Figure 5b). Corticosterone replacement induced a modest rightward shift in the survival curves of both strains during bacterial pneumonia but did not rescue the increased mortality of SR-BI−/− mice (Figure 5c) nor the increased bacterial burden in the airspace or blood of SR-BI−/− mice (Figure 5d and e). Corticosterone replacement did not normalize the augmented BALF levels of TNFα or CXCL5 of SR-BI−/− mice (Figure 5f and g), suggesting, consistent with our ex vivo macrophage LPS exposure studies, that increased induction of cytokines in the SR-BI−/− airway does not simply arise because of disinhibition from glucocorticoids. Interestingly, despite the failure of corticosterone supplementation to normalize cytokines in the infected SR-BI−/− airway, it abolished the relative alveolar neutrophilia of SR-BI−/− mice (Figure 5h). Taken together, these studies suggest that, whereas adrenal insufficiency does not underlie the increased mortality or pathogen dissemination of SR-BI−/− mice during pneumonia, it does have an impact on the innate immune response of the SR-BI−/− airway by disinhibiting alveolar neutrophilia through a cytokine-independent mechanism.

Scavenger receptor B-I −/− (SR-BI−/−) mice reveal a role for adrenal function in regulation of alveolar neutrophilia during pneumonia. (a) Serum corticosterone was measured in SR-BI+/+ and SR-BI−/− mice at baseline and 24 h post infection with K. pneumoniae (2,000 colony forming units (CFUs)). N=5–8 per genotype. (b) SR-BI+/+ and SR-BI−/− mice were dosed i.p. with vehicle (dimethyl sulfoxide/corn oil) or corticosterone (10 mg kg−1) 1 h before intratracheally (i.t.) K. pneumoniae (2,000 CFUs). Serum corticosterone levels were measured 24 h post inoculation. (c) SR-BI+/+ and SR-BI−/− mice (N=10 per group) were dosed daily with i.p. vehicle or corticosterone (10 mg kg−1) starting 1 h before i.t. K. pneumoniae (200 CFUs) and survival was monitored. (d, e) Bacterial CFUs in lung homogenates (d) and blood (e) were quantified 24 h post i.t. K. pneumoniae in mice pretreated i.p. with vehicle or corticosterone. (f–h) Bronchoalveolar lavage fluid (BALF) TNFα (tumor-necrosis factor-α) (f), CXCL5 (g), and PMNs (h) were quantified 24 h post i.t. K. pneumoniae in mice pretreated with i.p. vehicle or corticosterone. Data shown are representative of one experiment. *P<0.05; **P<0.01.
SR-BI−/− mice have impaired clearance of bacteremia
Given the marked increase in bacterial burden in the blood of SR-BI−/− mice, we next pursued the possibility that defective clearance of pathogens in the bloodstream, once seeded from the lung, might underlie the severe failure of host defense in this strain. In order to address this directly, we bypassed the lung, and injected K. pneumoniae intravenously. Bacterial burden in both the blood and the liver was significantly higher in SR-BI−/− mice than in SR-BI+/+ controls 4 h after intravenous inoculation (Figure 6a and b), confirming defective pathogen clearance in the blood in vivo. PMNs have a key role in bacterial killing in the bloodstream.25 Excluding relative neutropenia as a cause for defective clearance of bacteremia in SR-BI−/− mice, circulating PMNs (as well as lymphocytes, monocytes, and eosinophils) were equivalent in number between naive SR-BI+/+ and SR-BI−/− mice (Figure 6c). Moreover, the two strains mounted a similar increase in circulating PMNs 24 h following i.t. K. pneumoniae (Figure 6d), suggesting an intact bone marrow response to lung infection. The normal circulating PMN numbers and augmented alveolar PMN numbers in SR-BI−/− mice, taken together with our finding of defective bacterial clearance in these two compartments, suggested that PMN bacterial killing function might be defective in SR-BI−/− mice.

Scavenger receptor B-I −/− (SR-BI−/−) mice have decreased clearance of bacteremia. SR-BI+/+ and SR-BI−/− mice (N=5–8 per group) were inoculated intravenously with 70,000 colony forming units (CFUs) of K. pneumoniae. Bacterial CFUs in blood (a) and liver homogenates (b) were quantified 4 h post inoculation. (c, d) Peripheral blood leukocyte and differential counts in SR-BI+/+ and SR-BI−/− mice (N=5–7 per group) at baseline (c) or 24 h post intratracheally (i.t.) K. pneumoniae (2,000 CFUs; d). Data shown are representative of one experiment. **P<0.01; ***P<0.001.
SR-BI deficiency impairs phagocytic killing
SR-BI is expressed by human PMNs and mediates the suppressive effect of HDL on CD11b display and chemotaxis by these cells.9 In order to confirm SR-BI expression by murine PMNs, bone marrow PMNs were purified by Percoll gradient and further fluorescence-activated cell sorting (FACS)-sorted for Ly6G+CD11b+F4/80neg7AADneg, yielding a population that was >95% PMNs, with no detectable contamination by (F4/80+) macrophages (Figure 7a). Purified WT PMNs did express SR-BI message (Ct value 25.8), whereas no expression was detected in SR-BI−/− PMNs (Figure 7b). However, PMN expression of SR-BI was considerably lower than that in either macrophages or dendritic cells derived from murine bone marrow (Figure 7c).

Scavenger receptor B-I −/− (SR-BI−/−) neutrophils display decreased phagocytosis and intracellular killing of bacteria. (a) Bone marrow neutrophils (PMNs) were isolated by Percoll gradient from SR-BI+/+ and SR-BI−/− mice and further purified by fluorescence-activated cell sorting (FACS) sorting for Ly6G+ CD11b+7AAD− F4/80−. Representative plots indicating FACS scheme and post-sort purity are shown. (b, c) SR-BI expression relative to GAPDH (real-time PCR) in purified bone marrow PMNs from SR-BI+/+ and SR-BI−/− mice (b), and in SR-BI+/+ bone marrow PMNs relative to bone marrow-derived macrophages (Mϕ) and bone marrow-derived dendritic cells (DCs) (c). (d) Phagocytosis of E. coli bioparticles by SR-BI+/+ and SR-BI−/− peritoneal-elicited Gr-1+ PMNs was assessed (N=4–6 mice per condition). (e, f) SR-BI+/+ and SR-BI−/− mice were injected i.p. with thioglycollate, followed 4 h (e) or 96 h (f) later by i.p. K. pneumoniae (1 × 108 colony forming units (CFUs)). Peritoneal cells were lavaged (HBSS with 100 μg ml−1 gentamicin) 30 min later, washed, and then incubated (37 °C) for varying durations, followed by lysis for intracellular CFU quantification. N=5–8 per genotype per time point. (g) Phagocytic intracellular killing was tested as in e, except that bone marrow chimeric mice (SR-BI+/+ recipients reconstituted with SR-BI+/+ or SR-BI−/− bone marrow) were used (N=2–4 per genotype per time point). Data shown are representative of one experiment. *P<0.05.
Pursuing the effect of SR-BI deficiency upon PMN host defense functions, we found that SR-BI−/− PMNs were defective at phagocytosis of fluor-labeled bacterial bioparticles ex vivo (Figure 7d). In order to better model phagocytic killing as it occurs at inflammatory foci in vivo,19 K. pneumoniae was next injected i.p. into SR-BI+/+ and SR-BI−/− mice 4 h after thioglycollate elicitation of PMNs to the peritoneum; peritoneal cells (∼80% PMNs for both genotypes) were then harvested 30 min post-peritoneal infection and assayed for intracellular bacterial killing during a time course ex vivo. This model allows for precisely timed examination of the initial PMN-bacterial encounter as it occurs in vivo. As shown in Figure 7e, SR-BI−/− phagocytes had significantly lower intracellular K. pneumoniae colony forming units (CFUs) than SR-BI+/+ counterparts at time 0 (that is, immediately upon harvest from the inoculated peritoneum), consistent with reduced bacterial internalization. More strikingly, whereas SR-BI+/+ phagocytes, as expected, displayed a time-dependent intracellular killing curve (that is, reduction in intracellular CFUs), SR-BI−/− phagocytes displayed a time-dependent increase in intracellular CFUs (that is, effectively, an intracellular bacterial growth curve), indicating a marked defect in intracellular killing of bacteria.
In order to more specifically address killing function of SR-BI−/− macrophages, we repeated the killing assay, but this time injected K. pneumoniae i.p. 96 h post thioglycollate, near the peak of peritoneal macrophage accumulation. As shown in Figure 7f, SR-BI−/− macrophages had reduced CFUs at time 0, consistent with defective phagocytosis. However, SR-BI+/+ and SR-BI−/− macrophages reduced intracellular CFUs roughly equivalently, suggesting that SR-BI−/− macrophages have intact killing function. The inflammatory milieu of the peritoneum 4 and 96 h post thioglycollate is very different, and direct comparisons between killing assays at the two time points must thus be performed with caution. Nonetheless, taken together, these findings may suggest that SR-BI−/− PMNs account for the marked phagocytic killing deficit seen in the 4 h post thioglycollate assay.
SR-BI−/− mice have elevated plasma levels of enlarged HDL particles because of defective clearance of HDL by the liver.8 Both HDL and glucocorticoids can suppress PMN functions.9, 26 Thus, we next sought to determine whether or not the killing impairment observed in phagocytes of SR-BI−/− mice is secondary to the SR-BI−/− plasma microenvironment. The HDL and adrenal phenotypes of the SR-BI−/− mice both derive from SR-BI deficiency in radioresistant cells (hepatocytes and adrenocortical cells, respectively). Thus, as expected, lethally irradiated SR-BI+/+ recipient mice transplanted with either SR-BI+/+ or SR-BI−/− bone marrow had equivalent serum cholesterol and equivalent plasma corticosterone after induction of peritonitis (Supplementary Figure 3). When phagocytic killing function was compared between these two chimeras 4 h post thioglycollate, phagocytes from mice with isolated hematopoietic cell SR-BI deficiency again displayed a profound defect in intracellular killing (Figure 7g), suggesting that PMN dysfunction in the SR-BI-null mouse is not secondary to HDL excess (that is, acting through a receptor other than SR-BI). Taking a genetic approach to further address whether excess HDL drives the host defense phenotype of SR-BI−/− mice during pneumonia, we infected HDL-deficient apoA-I−/− mice with i.t. K. pneumoniae, aiming to see whether they would display a phenotype opposite to that of SR-BI−/− mice. Rather, apoA-I+/+ and apoA-I−/− mice had comparable survival and alveolar neutrophilia 24 h after infection, whereas, similar to SR-BI−/− mice, apoA-I−/− mice had a significant increase over WT counterparts in systemic bacterial burden, as assessed by splenic CFUs (Supplementary Figure 4). Taken together, these results indicate that HDL may have an impact on subphenotypes of host defense during bacterial pneumonia, but that the profound sepsis and PMN-killing deficit observed in SR-BI−/− mice does not arise from HDL excess.
SR-BI deficiency decreases neutrophil antimicrobial function
PMNs at the site of infection internalize pathogens and then kill them by oxidant-dependent and -independent mechanisms. Bacteria are internalized into the PMN phagosome that then matures, as NADPH oxidase–dependent reactive oxygen species are released into it. In order to address the effect of SR-BI deficiency upon PMN bacterial–killing mechanisms, intracellular reactive oxygen species were measured with flow cytometry in both alveolar and peripheral blood PMNs 24 h after i.t. K. pneumoniae, a time point at which infected SR-BI−/− mice had much higher bacterial stimulus in both locations (Figure 1). Despite the higher bacterial burden, SR-BI−/− PMNs in both locations had significantly lower intracellular reactive oxygen species signal than SR-BI+/+ controls (Figure 8a and b). Moreover, consistent with lower activation/maturation status of SR-BI−/− PMNs during infection, circulating SR-BI−/− PMNs had significantly higher surface display of CD62L/L-selectin (Figure 8c), consistent with lesser L-selectin shedding, and lower surface display of the activation marker CD11b (Figure 8d).

Infected Scavenger receptor B-I −/− (SR-BI−/−) mice display deficient neutrophil antimicrobial functions in vivo. SR-BI+/+ and SR-BI−/− mice (N=5–8 per group) were inoculated intratracheally (i.t.) with 2,000 colony forming units (CFUs) of K. pneumoniae and necropsied 24 h post inoculation. Intracellular reactive oxygen species (iROS) generation was quantified in Ly6G+ bronchoalveolar lavage fluid (BALF) neutrophils (PMNs) (a) and blood PMNs (b) by flow cytometry (N=5 per genotype). (c) Left panel displays representative flow cytometry histograms of CD62L on blood PMNs of K. pneumoniae-infected SR-BI+/+ and SR-BI−/− mice. Shaded trace is isotype control, black line is SR-BI+/+, and red line is SR-BI−/−. The mean fluorescence intensity (MFI; right panel) of CD62L on Ly6G+ blood PMNs. (d) Left panel displays representative flow cytometry histograms of CD11b on blood PMNs of K. pneumoniae-infected SR-BI+/+ and SR-BI−/− mice. Shaded trace is isotype control, black line is SR-BI +/+, and red line is SR-BI −/−. MFI (right panel) of CD11b on Ly6G+ blood PMNs. Data shown are representative of one experiment. *P<0.05.
DISCUSSION
Pneumonia is a serious condition that arises when bacteria invade the lower airway and induce an innate immune response.2, 20 As the host antimicrobial response is not restricted to cells of the airspace in isolation, and pneumonia often kills through complications of the systemic host response (that is, severe sepsis), advances in pneumonia management will require an improved understanding of the integrated physiology of host defense.
In this report, we show that SR-BI, an SR heretofore largely studied in vascular biology in its role as a receptor for HDL, is pivotal to host survival during bacterial pneumonia and has an important role in integrating the responses of several cell types both within and outside the lung. We propose that this arises from expression of SR-BI by several distinct, but interacting, cell types in the host, having an impact on both cell-intrinsic functions (for example, macrophage TLR responses, adrenocortical stress hormone synthesis, and PMN bacterial killing) and intercellular communications across organs that govern the host response (for example, corticosteroid regulation of PMN trafficking to the lung). Whereas some of these roles of SR-BI are HDL-dependent (adrenal stress hormone synthesis12), we show that others (that is, phagocytic killing) are independent, in part, of the plasma HDL excess phenotype of SR-BI deficiency. In the end, several of the consequences of SR-BI deletion conspire to amplify alveolar neutrophilia, including enhanced alveolar cytokine production, reduced alveolar LPS clearance, and adrenal insufficiency. Despite this, impaired phagocytic killing enhances bacterial overgrowth in the lung (and bloodstream), providing yet further stimulus for alveolar neutrophilia.
Adrenal insufficiency worsens outcomes in human sepsis.3 Supplemental glucocorticoids have been tested in the therapy of a wide array of diseases, including acute lung injury and pneumonia.27 In our study, corticosterone supplementation normalized the relative alveolar neutrophilia of infected SR-BI−/− mice without correcting the elevated BALF levels of TNFα or CXCL5. This suggests that the endogenous stress corticosterone “burst” that occurs in WT mice (and is absent in SR-BI−/− mice) shortly after infection represses alveolar accumulation of PMNs in a manner not simply because of attenuation of alveolar cytokines. We speculate that this may result from a direct repressive effect on PMN migration, as glucocorticoids inhibit PMN chemotaxis.26 Derepressed PMN migration may explain why the relative increase in alveolar neutrophilia in SR-BI−/− mice was out of proportion to the modest and selective increase in alveolar cytokines. Given that the BALF cytokine increases were modest, transient (CXCL5 was normalized by 8 h post LPS (Figure 3d)), and dissociable from the airspace neutrophilia, we posit that derepression of PMN migration by glucocorticoid deficiency may be the major driver of alveolar neutrophilia in the SR-BI−/− mouse.
Although SRs recognize partially overlapping sets of ligands (for example, SR-A, CD36, and SR-BI all recognize oxidized LDL), critical differences among SRs have been identified, indicating that they are not functionally redundant. To date, SRs studied in lung biology have largely been of class A. SR-A−/− mice display enhanced pulmonary clearance of both Cryptococcus and Pneumocystis,5, 6 whereas MARCO−/− mice have impaired clearance of Streptococcus pneumoniae.7 Unlike our findings for SR-BI−/− mice, mice deficient in LOX1, a class E SR, display reduced lung neutrophilia after LPS exposure.28 SR-BI has been little studied in the context of infection beyond its well-described role in cellular internalization of hepatitis C virus.29 One report identified SR-BI as a receptor for Mycobacterium tuberculosis but found no impact of SR-BI deletion on M. tuberculosis pneumonia in vivo,30 whereas two other reports have identified a role for SR-BI in Plasmodium infection of the liver.31
Parallel to the diverse roles of different SRs in pulmonary immunity, important differences among SRs have been identified in relation to TLR signaling at the cellular level. Similar to our finding for SR-BI−/− macrophages, SR-A−/− macrophages produce increased TNFα in response to LPS.6 On the other hand, CD36 promotes cellular responses to both LPS and TLR2 ligands.32, 33 It will be of interest in future studies to discern the mechanism of enhanced TLR signaling in SR-BI knockout cells. Possible mechanisms include the following: (i) deletion of a cellular ligand disposal (that is, uptake) pathway that normally tempers TLR responses by competing for (that is, clearing) extracellular ligand; (ii) absence of tonic suppressive effects of HDL on the cell; (iii) alterations in plasma membrane lipids; and/or (iv) other metabolic alterations in the macrophage. Whereas the increased cell-surface TLR4 on SR-BI−/− macrophages may conceivably contribute to their heightened LPS response, caution is warranted in gauging the physiologic significance of this finding.
The AE has an important role in the innate immune response during bacterial pneumonia, producing the key cytokines and chemokines such as GM-CSF and CXCL5 that recruit PMNs to the airspace.20 AE cells are thought to be the solitary source of CXCL5 in the lung,22 producing this chemoattractant in a NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)-dependent manner in response to LPS or S. pneumoniae.34 Our finding that SR-BI−/− mice have elevated BALF CXCL5 implicates altered AE function. SR-BI is expressed by AE cells, wherein it is reported to mediate HDL-dependent uptake by AE cells of the antioxidant vitamin E.17 SR-BI deficiency in AEs may conceivably enhance their direct response to LPS, as observed in macrophages. Alternatively, higher CXCL5 induction could be an indirect consequence of higher airspace TNFα, which is reported to synergize with IL-17 to promote CXCL5 production by the AE.35 As NF-κB activation and pro-inflammatory cytokine induction are promoted by oxidative stress in AEs,36 increased CXCL5 induction could also result in part from vitamin E deficiency.37
A notable finding of our report is that SR-BI regulates phagocytic killing of bacteria. SR-BI−/− mice displayed defective bacterial clearance both in the lung and in the bloodstream. Whereas our peritoneal phagocytic killing assay has the advantage of allowing precisely timed examination of the initial PMN-pathogen encounter in the in vivo context, we cannot strictly exclude contributions from phagocytes other than PMNs to our results (for example, peritoneal macrophages). Nonetheless, we provide evidence of intact killing function in SR-BI−/− macrophages and show that the bacterial killing deficit is associated with defects in PMN generation of oxidants, as well as in additional signs of PMN activation (L-selectin shedding, CD11b display). As both SR-A and LIMP2 are reported to regulate phagolysosomal fusion,38, 39 we speculate that SR-BI may possibly regulate steps in post-internalization processing of microbes. PMN SR-BI was also recently reported to mediate vitamin E entry into PMNs,40 and vitamin E supplementation enhances bacterial killing by PMNs.41 Thus, it is possible that functional deficits in SR-BI−/− PMNs may derive, in part, from vitamin E deficiency. Whereas the changes in CD11b and CD62L may suggest a less mature/activated PMN phenotype in SR-BI−/− mice, we cannot exclude the possibility that this is secondary to accelerated release of immature PMNs from the SR-BI−/− bone marrow due to exacerbated sepsis.
We are unaware of any past reports that have defined mechanisms by which LPS is cleared from the airspace. Our finding that hematopoietic cell, and, likely, macrophage SR-BI mediates LPS clearance from the airspace is noteworthy, although future studies outside the scope of the present report will be required to define the physiological significance of this finding in different settings. Given that LPS was cleared by 48 h post exposure in the SR-BI−/− mouse, and BALF cytokines were only modestly and selectively increased in the SR-BI−/− airway, we speculate that defective LPS clearance was not the major driver of increased alveolar neutrophilia in our studies. However, LPS clearance could have a role not only in termination of pro-inflammatory signaling, but also in LPS tolerance in the lung and in regulation of disease phenotypes driven by chronic or recurrent pulmonary exposure to LPS, such as bronchiectasis and byssinosis.
As aging42 as well as several environmental exposures (cigarette smoke,43 ozone,42 and LPS24) downregulate SR-BI, it is possible that environmental modulation of SR-BI is a common immune-modifying event in the human lung. Conversely, there are several common systemic exposures/conditions reported to upregulate SR-BI, including statins,44 vitamin E,40 nifedipine,45 aspirin,46 and diabetes mellitus.47 Finally, of interest, a growing number of genetic variants of SR-BI in humans have been described that phenocopy several findings in SR-BI−/− mice.13, 14 Taken together, this suggests that genetic and environmental modulation of SR-BI may be a common modifier of immune responses in humans.
The recognition that SR-BI and HDL regulate pulmonary host defense now raises new and exciting questions, both fundamental and translational, about lung biology. Given that additional HDL cargo beyond vitamin E, such as microRNAs,48 are also delivered into cells via SR-BI, it will be of interest to examine the extent of additional circulating immunomodulatory molecules for which SR-BI may serve as a gateway to the lung. The composition of HDL is now known to be altered in several common disease states that have been associated with increased risk for lung disease, including obesity and diabetes mellitus.48 Future studies are warranted to define whether modifications of HDL are communicated to, and modify, the lung via SR-BI, and whether these interactions can be manipulated to therapeutic benefit.
METHODS
Reagents. Escherichia coli 0111:B4 LPS, penicillin, and streptomycin were from Sigma (St Louis, MO). K. pneumoniae 43816 (serotype 2), DMEM, and fetal bovine serum were from American Type Culture Collection (Rockville, MD). The Limulus amebocyte lysate assay was from Lonza (Basel, Switzerland).
Mice. C57BL/6J (SR-BI+/+), B6;129S2-Scarb1tm1Kri/J (SR-BI−/−), and B6.129P2-Apoa1tm1Unc/J (apoA-I−/−) male mice, 7–10 weeks old and weighing 18–22 g, were used; they were from Jackson Laboratories (Bar Harbor, ME) and were bred in-house. ApoA-I−/− mice were backcrossed 10 generations onto a C57/BL/6J background. SR-BI−/− mice were backcrossed >6 generations onto C57BL/6J before use. Experiments conducted using both littermate SR-BI+/+ and commercial SR-BI+/+ (C57BL/6J) controls confirmed very similar responses (Supplementary Figure 2). All experiments were performed in accordance with the Animal Welfare Act and the US Public Health Service Policy on Humane Care and Use of Laboratory Animals after review by the Animal Care and Use Committee of the NIEHS.
In vivo exposures. Exposure to aerosolized LPS (300 μg ml−1, 30 min) was as previously described.19 K. pneumoniae was delivered to the lung by oropharyngeal aspiration (200–2,000 CFU per 50 μl) or intravenously into the retro-orbital venous plexus (7 × 104 CFUs in 200 μl phosphate-buffered saline) during isoflourane anesthesia. In some experiments, corticosterone (10 mg kg−1) or vehicle was injected i.p. 1 h preceding other exposures and daily thereafter, as previously reported.49
BALF collection and analysis. BALF was collected immediately following the killing and cell counts performed as described.19 Protein analysis was performed using the Bradford assay (Bio-Rad, Hercules, CA). Endotoxin levels in BALF were measured with Limulus amebocyte lysate assay (Lonza).
PMN and macrophage harvests and culture. Mature murine bone marrow PMNs were isolated from mouse femurs and tibias by discontinuous Percoll gradient centrifugation as previously reported.19 This preparation was then sorted by a FACSAriaII (Becton Dickinson, Franklin Lakes, NJ) based on live (7AADneg), Ly6G+, and F4/80− to yield a population that was >95% pure with 0% F4/80+ cells (Figure 7a). Peritoneal PMNs were harvested by peritoneal lavage 4 h after i.p. Brewer’s thioglycollate (2 ml, 4%). Peritoneal exudate macrophages were harvested by peritoneal lavage 96 h after i.p. Brewer’s thioglycollate and then plated for 24 h before stimulation. Macrophages were stimulated with media, Pam3CSK4 (Invivogen, San Diego, CA; 100 ng ml−1), Poly I:C (Invivogen, 10 μg ml−1), LPS (List Biologicals, Campbell, CA; 10 ng ml−1), or CpG oligodeoxynucleotide (Invivogen; 10 μg ml−1) for 24 h and cell supernatants harvested for cytokine analysis.
Bactericidal assays. The lung, spleen, and liver were homogenized in phosphate-buffered saline, and serial dilutions were plated on tryptic soy agar for bacterial quantification, as described.19 Blood was collected from the right ventricle and similarly plated. Intracellular killing capacity of PMNs against i.p.-injected K. pneumoniae was quantified as reported.19 In brief, mice received 2 ml of 4% thioglycollate i.p., followed 4 or 96 h later by 1 × 108 CFU K. pneumoniae i.p. Peritoneal leukocytes were collected 30 min later by lavage (HBSS with 100 μg ml−1 gentamicin), washed, and then cells (1 × 106) were incubated (37 °C) for varying durations, followed by lysis (0.1% Triton-X-100) for intracellular CFU quantification by plating. Morphologic analysis of cytospins confirmed that SR-BI+/+ and SR-BI−/− lavages contained equal absolute and relative numbers (≥80% of lavage cells) of PMNs.
Peripheral blood leukocyte typing and enumeration. Blood samples were analyzed using the HEMAVET 1,700 hematology analyzer (Drew Scientific, Dallas, TX). Manual WBC differential counts were reported and smear estimates used for confirmation.
Statistical analysis. Analysis was performed using the GraphPad Prism statistical software (San Diego, CA). Data are represented as mean±s.e.m. Two-tailed Student’s t-test was applied for comparisons of two groups, and analysis of variance for comparisons of more than two groups. Survival was evaluated by log-rank test. For all tests, P<0.05 was considered significant.
Additional methods describing cytokine analysis, RNA isolation and quantitative PCR, flow cytometry, phagocytosis assay, LPS binding and uptake, serum and plasma analysis, generation of bone marrow chimeras, and histopathological evaluation are included in the Supplementary Methods.
References
Waterer, G.W., Rello, J. & Wunderink, R.G. Management of community-acquired pneumonia in adults. Am. J. Respir. Crit. Care Med. 183, 157–164 (2011).
Mizgerd, J.P. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am. J. Respir. Crit. Care Med. 186, 824–829 (2012).
Remmelts, H.H., Meijvis, S.C., Kovaleva, A., Biesma, D.H., Rijkers, G.T. & Heijligenberg, R. Changes in serum cortisol levels during community-acquired pneumonia: the influence of dexamethasone. Respir. Med. 106, 905–908 (2012).
Canton, J., Neculai, D. & Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13, 621–634 (2013).
Qiu, Y. et al. Scavenger receptor A modulates the immune response to pulmonary Cryptococcus neoformans infection. J. Immunol. 191, 238–248 (2013).
Hollifield, M., Bou Ghanem, E., de Villiers, W.J. & Garvy, B.A. Scavenger receptor A dampens induction of inflammation in response to the fungal pathogen Pneumocystis carinii. Infect. Immun. 75, 3999–4005 (2007).
Arredouani, M. et al. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J. Exp. Med. 200, 267–272 (2004).
Acton, S., Rigotti, A., Landschulz, K.T., Xu, S., Hobbs, H.H. & Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520 (1996).
Murphy, A.J. et al. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A-I in in vitro and in vivo models of inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 1333–1341 (2011).
Bocharov, A.V. et al. Targeting of scavenger receptor class B type I by synthetic amphipathic alpha-helical-containing peptides blocks lipopolysaccharide (LPS) uptake and LPS-induced pro-inflammatory cytokine responses in THP-1 monocyte cells. J. Biol. Chem. 279, 36072–36082 (2004).
Cai, L., Ji, A., de Beer, F.C., Tannock, L.R. & van der Westhuyzen, D.R. SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J. Clin. Invest. 118, 364–375 (2008).
Hoekstra, M., van der Sluis, R.J., Van Eck, M. & Van Berkel, T.J. Adrenal-specific scavenger receptor BI deficiency induces glucocorticoid insufficiency and lowers plasma very-low-density and low-density lipoprotein levels in mice. Arterioscler. Thromb. Vasc. Biol. 33, e39–e46 (2013).
McCarthy, J.J. et al. Association of genetic variants in the HDL receptor, SR-B1, with abnormal lipids in women with coronary artery disease. J. Med. Genet. 40, 453–458 (2003).
Vergeer, M. et al. Genetic variant of the scavenger receptor BI in humans. N. Engl. J. Med. 364, 136–145 (2011).
Sahin, F. & Yildiz, P. Distinctive biochemical changes in pulmonary tuberculosis and pneumonia. Arch. Med. Sci. 9, 656–661 (2013).
Deniz, O. et al. Serum HDL-C levels, log (TG/HDL-C) values and serum total cholesterol/HDL-C ratios significantly correlate with radiological extent of disease in patients with community-acquired pneumonia. Clin. Biochem. 39, 287–292 (2006).
Kolleck, I., Sinha, P. & Rustow, B. Vitamin E as an antioxidant of the lung: mechanisms of vitamin E delivery to alveolar type II cells. Am. J. Respir. Crit. Care Med. 166 (12 Pt 2), S62–S66 (2002).
Xu, S., Laccotripe, M., Huang, X., Rigotti, A., Zannis, V.I. & Krieger, M. Apolipoproteins of HDL can directly mediate binding to the scavenger receptor SR-BI, an HDL receptor that mediates selective lipid uptake. J. Lipid Res. 38, 1289–1298 (1997).
Madenspacher, J.H. et al. p53 Integrates host defense and cell fate during bacterial pneumonia. J. Exp. Med. 210, 891–904 (2013).
Mizgerd, J.P. Acute lower respiratory tract infection. N. Engl. J. Med. 358, 716–727 (2008).
Mizgerd, J.P., Peschon, J.J. & Doerschuk, C.M. Roles of tumor necrosis factor receptor signaling during murine Escherichia coli pneumonia. Am. J. Respir. Cell Mol. Biol. 22, 85–91 (2000).
Jeyaseelan, S. et al. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am. J. Respir. Cell Mol. Biol. 32, 531–539 (2005).
Vishnyakova, T.G. et al. Binding and internalization of lipopolysaccharide by Cla-1, a human orthologue of rodent scavenger receptor B1. J. Biol. Chem. 278, 22771–22780 (2003).
Baranova, I.N. et al. Class B scavenger receptor types I and II and CD36 mediate bacterial recognition and proinflammatory signaling induced by Escherichia coli, lipopolysaccharide, and cytosolic chaperonin 60. J. Immunol. 188, 1371–1380 (2012).
Gregory, S.H., Sagnimeni, A.J. & Wing, E.J. Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J. Immunol. 157, 2514–2520 (1996).
Caramori, G. & Adcock, I. Anti-inflammatory mechanisms of glucocorticoids targeting granulocytes. Curr. Drug Targets Inflamm. Allergy 4, 455–463 (2005).
Marik, P.E., Meduri, G.U., Rocco, P.R. & Annane, D. Glucocorticoid treatment in acute lung injury and acute respiratory distress syndrome. Crit. Care Clin. 27, 589–607 (2011).
Wu, Z., Sawamura, T., Kurdowska, A.K., Ji, H.L., Idell, S. & Fu, J. LOX-1 deletion improves neutrophil responses, enhances bacterial clearance, and reduces lung injury in a murine polymicrobial sepsis model. Infect. Immun. 79, 2865–2870 (2011).
Barth, H. et al. Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J. Virol. 82, 3466–3479 (2008).
Schafer, G., Guler, R., Murray, G., Brombacher, F. & Brown, G.D. The role of scavenger receptor B1 in infection with Mycobacterium tuberculosis in a murine model. PLoS ONE 4, e8448 (2009).
Rodrigues, C.D. et al. Host scavenger receptor SR-BI plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe 4, 271–282 (2008).
Baranova, I.N. et al. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J. Immunol. 181, 7147–7156 (2008).
Triantafilou, M. et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol. 9, 2030–2039 (2007).
Yamamoto, K. et al. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 50, 253–262 (2013).
Liu, Y. et al. IL-17A and TNF-alpha exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J. Immunol. 186, 3197–3205 (2011).
Haddad, J.J. Glutathione depletion is associated with augmenting a proinflammatory signal: evidence for an antioxidant/pro-oxidant mechanism regulating cytokines in the alveolar epithelium. Cytokines Cell Mol. Ther. 6, 177–187 (2000).
Hernandez, M.L. et al. Vitamin E, gamma-tocopherol, reduces airway neutrophil recruitment after inhaled endotoxin challenge in rats and in healthy volunteers. Free Radic. Biol. Med. 60, 56–62 (2013).
Carrasco-Marin, E. et al. LIMP-2 links late phagosomal trafficking with the onset of the innate immune response to Listeria monocytogenes: a role in macrophage activation. J. Biol. Chem. 286, 3332–3341 (2011).
Ishiguro, T. et al. Role of macrophage scavenger receptors in response to Listeria monocytogenes infection in mice. Am. J. Pathol. 158, 179–188 (2001).
Higuchi, H., Ito, E., Iwano, H., Oikawa, S. & Nagahata, H. Effects of vitamin E supplementation on cellular alpha-tocopherol concentrations of neutrophils in Holstein calves. Can. J. Vet. Res. 77, 120–125 (2013).
Hall, J.A. et al. Influence of dietary antioxidants and fatty acids on neutrophil mediated bacterial killing and gene expression in healthy Beagles. Vet. Immunol. Immunopathol. 139, 217–228 (2011).
Valacchi, G. et al. Lung vitamin E transport processes are affected by both age and environmental oxidants in mice. Toxicol. Appl. Pharmacol. 222, 227–234 (2007).
Valacchi, G. et al. Cigarette smoke exposure causes changes in Scavenger Receptor B1 level and distribution in lung cells. Int. J. Biochem. Cell Biol. 43, 1065–1070 (2011).
Kimura, T. et al. Induction of scavenger receptor class B type I is critical for simvastatin enhancement of high-density lipoprotein-induced anti-inflammatory actions in endothelial cells. J. Immunol. 181, 7332–7340 (2008).
Zhang, Q., Ma, A.Z., Song, Z.Y., Wang, C. & Fu, X.D. Nifedipine enhances cholesterol efflux in RAW264.7 macrophages. Cardiovasc. Drugs Ther. 27, 425–431 (2013).
Lu, L. et al. Regulations of the key mediators in inflammation and atherosclerosis by aspirin in human macrophages. Lipids Health Dis. 9, 16 (2010).
Gantman, A., Fuhrman, B., Aviram, M. & Hayek, T. High glucose stimulates macrophage SR-BI expression and induces a switch in its activity from cholesterol efflux to cholesterol influx. Biochem. Biophys. Res. Commun. 391, 523–528 (2010).
Vickers, K.C., Palmisano, B.T., Shoucri, B.M., Shamburek, R.D. & Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 13, 423–433 (2011).
Liston, C. & Gan, W.B. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc. Natl Acad. Sci. USA 108, 16074–16079 (2011).
Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences (Z01 ES102005). We thank Drs Donald Cook and Seddon Thomas for manuscript review and Ralph Wilson, Debra King, Ligon Perrow, Maria Sifre, Carl Bortner, and CJ Tucker for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts to interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
About this article

Cite this article
Gowdy, K., Madenspacher, J., Azzam, K. et al. Key role for scavenger receptor B-I in the integrative physiology of host defense during bacterial pneumonia. Mucosal Immunol 8, 559–571 (2015). https://doi.org/10.1038/mi.2014.88
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.88
This article is cited by
-
Scavenger receptor B1 facilitates the endocytosis of Escherichia coli via TLR4 signaling in mammary gland infection
Cell Communication and Signaling (2023)
