
Abstract
We have established that mucosal immunization can generate high-avidity human immunodeficiency virus (HIV)-specific CD8+ T cells compared with systemic immunization, and interleukin (IL)-13 is detrimental to the functional avidity of these T cells. We have now constructed two unique recombinant HIV-1 vaccines that co-express soluble or membrane-bound forms of the IL-13 receptor α2 (IL-13Rα2), which can “transiently” block IL-13 activity at the vaccination site causing wild-type animals to behave similar to an IL-13 KO animal. Following intranasal/intramuscular prime-boost immunization, these IL-13Rα2-adjuvanted vaccines have shown to induce (i) enhanced HIV-specific CD8+ T cells with higher functional avidity, with broader cytokine/chemokine profiles and greater protective immunity using a surrogate mucosal HIV-1 challenge, and also (ii) excellent multifunctional mucosal CD8+ T-cell responses, in the lung, genito-rectal nodes (GN), and Peyer’s patch (PP). Data revealed that intranasal delivery of these IL-13Rα2-adjuvanted HIV vaccines recruited large numbers of unique antigen-presenting cell subsets to the lung mucosae, ultimately promoting the induction of high-avidity CD8+ T cells. We believe our novel IL-13R cytokine trap vaccine strategy offers great promise for not only HIV-1, but also as a platform technology against range of chronic infections that require strong sustained high-avidity mucosal/systemic immunity for protection.
Similar content being viewed by others


Introduction
It is well established that human immunodeficiency virus (HIV) is a disease of the mucosae, as primary encounter of virus and CD4+ depletion commences respectively in the genito-rectal tract and the gastrointestinal tract.1 As such, induction of mucosal immunity is taking precedence when designing vaccines against HIV-1.2, 3, 4 Furthermore, it is becoming increasingly evident that not only the magnitude of cell-mediated immunity but also the “avidity” or efficacy of the CD8+ T cells induced is important for protection against diseases like HIV-1. It is known that high-avidity cytotoxic T lymphocytes (CTLs) have the ability to recognize low concentrations of antigen and are important in effective pathogen clearance.5, 6 Current findings have demonstrated that despite the fact that mucosal immunization often induces lower magnitude HIV-specific T cells when compared with systemic immunization, mucosal vaccination can nevertheless induce higher avidity CTL with greater protection against HIV challenge.7, 8, 9 Therefore, we have performed studies to understand “how and why” the vaccine delivery routes influence the quality of protective CTL immunity to HIV-1. In previous studies we have found that mucosal immunization generated HIV-specific CD8+ T cells with lower interleukin (IL)-4 and IL-13 expression compared with systemic immunization.8 Furthermore, using animals lacking the IL-13 cytokine (gene knock-out (KO) mice), we have demonstrated that the cytokine IL-13 is critically important in negatively regulating the induction or expansion of effector and memory T cells of heightened avidity.10
Normally IL-13 activity is mediated via two complex receptor systems. IL-13Rα1 binds to IL-13 with low affinity, but when paired with the IL-4Rα it binds with high-affinity forming the functional IL-13 receptor (which is also known as the IL-4 type II receptor) and signals via the STAT6 pathway similar to IL-4.11, 12 The IL-13Rα2 binds to IL-13 with high affinity and it has a membrane bound and a soluble form (Supplementary Schematic Diagram 1 online). The membrane-associated IL-13Rα2 has a short cytoplasmic domain and thought to act as a decoy receptor, sequesting IL-13 preventing binding to IL-4Rα/IL-13Rα1, which is devoid of conventional signaling motifs.12 The soluble IL-13Rα2 receptor is thought to result from either cleavage of the extracellular IL-13 binding domain or in mice from alternate mRNA splicing producing both cell membrane-bound IL-13Rα2 and a secreted IL-13Rα2 lacking the trans-membrane motif.12 The secreted form is found at high levels in both serum and urine in mice. It is clearly established that in allergy and parasitic infection mice regulate IL-13 activity using the soluble IL-13Rα2 decoy receptor.12 However, in humans only the fully transcribed membrane-bound form has been observed with no evidence of an alternatively spliced soluble product.13 It has been found that many human malignancies express membrane-bound IL-13Rα2 with IL-13-mediated cell signaling by an uncharacterized mechanism, possibly involving transforming growth factor-β1 promoter activation,14 which is linked to metastasis. Thus, in cancer studies these molecules have been identified as attractive anti-cancer therapeutics.15
We have now constructed two novel recombinant poxvirus-based HIV-1 vaccines that co-express soluble and membrane-bound forms of IL-13Rα2, together with HIV vaccine antigens that can temporarily inhibit IL-13 activity in vivo at the vaccination site. Using a combined mucosal–systemic prime-boost delivery, here we have investigated (i) the ability of these HIV vaccines to induce robust mucosal/systemic HIV-specific effector and memory T cells with heightened avidity, and enhanced protective immunity, and also (ii) the particular antigen-presenting cell (APC) subsets in the lung mucosae that may govern the induction of these high-avidity CTL.
Results
IL-13 has an important role in HIV-specific CD8+ T-cell avidity and protective immunity
We have previously shown that mucosal immunization generates HIV-specific CTL of higher avidity compared with systemic immunization8 and IL-13 has a critical role in dampening T-cell avidity10 (Supplementary Figure 2 online). In this study, first, we have prime-boost immunized BALB/c and IL-13 KO (H-2d) mice intramuscular (IM)/IM and intranasal (IN)/IN routes (Table 1) with FPV-HIV/VV-HIV, and have evaluated the protective capacity of IL-13 KO mice by transferring 107 splenocytes intravenously from immunized mice into respective naive wild-type BALB/c or native IL-13 KO mice. Then 1 week post transfer, mice were challenged intranasally with 50 plaque-forming unit (PFU) of PR8-KdGag197–205, and body weights were monitored daily thereafter for 10 days (Figure 1a,b). Data clearly indicate that the naive IL-13 KO mice that received the IL-13 KO immune cells were more resistant to challenge compared with wild-type BALB/c control mice that received the BALB/C immune cells (Figure 1a,b). Mice maintaining weight and not succumbing to influenza virus infection are considered as a measure of protective immunity.16 Furthermore, following challenge IL-13 KO mice (mice that received IN/IN or IM/IM IL-13 KO immunized cells) did not display any signs of ruffled fur and looked much healthier than the control BALB/c. At 10 days post challenge, interferon (IFN)-γ ELISpot (Figure 1c) and intracellular cytokine staining (Figure 1d) data revealed that the capacity to produce IFN-γ and/or tumor necrosis factor (TNF)-α by naive IL-13 KO mice that received IL-13 KO immune cells were significantly higher compared with naive BALB/c mice that received BALB/c immune cells (*P=0.0120, **P=0.006, Figure 1c and *P=0.008, **P=0.0009, Figure 1d). Interestingly, these transfer studies also further substantiated that IN/IN immunization is more protective compared with IM/IM pure systemic delivery, as lower weight loss was also observed in mice that received the control vaccine (Figure 1a,b).

(a,b) Protective immunity of interleukin (IL)-13 knock-out mice following PR8-KdGag197–205 challenge. BALB/c wild-type and IL-13 KO (H-2d) mice (n=4 per group) were immunized intramuscularly (IM)/IM or intranasally (IN)/IN with FPV-HIV/VV-HIV as indicated in Table 1 (strategies 1 and 2) (here IM/IM immunization regime was particularly chosen because previously we have shown that IM/IM regime generated the highest tetramer dissociation rate8 (also see Supplementary Figure 2 online) compared with any mucosal immunization regime. Hence, using this regime we could show the increases or decreases in protective immunity much better than a mucosal immunization regime). Four weeks post-booster immunization mice were killed and from each group single-cell spleen suspensions were prepared and 107 splenocytes were intravenously transferred into naive wild-type BALB/c or IL-13 KO mice, respectively. One week following transfer, mice were challenged mucosally (IN) with 50 plaque-forming unit (PFU) influenza virus PR8 expressing KdGag197–205 epitope. Naive wild-type BALB/animals that did not receive any immune cells were also challenged with same dose and used as unimmunized controls, and body weights were monitored for 10 days as described in Methods. Graphs represent (a) originally IN/IN immunized animals, (b) originally IM/IM immunized animals including unimmunized control. The data represent±s.e. obtained with 3–4 mice per group. (c and d) Immune responses at 10 days post PR8-KdGag197–205 challenge in mice that received immune cells. At 10 days post challenge, spleens were harvested and immunity was measured by IFN-γ ELIspot (c), *P=0.0120 (IL-13 KO and BALB/c that received IM/IM immune cells), **P=0.006 (IL-13 KO and BALB/c that received IN/IN immunized cells), ***P=0.006 (BALB/c that received IN/IN or IM/IM immunized cells), and IFN-γ/TNF-α intracellular cytokine staining (d). Data represent mean±s.d. HIV, human immunodeficiency virus; IFN, interferon; TNF, tumor necrosis factor.
HIV-1 vaccines that co-express IL-13Rα2 soluble receptor can induce high-avidity CD8+ T cells
We then tested our novel IL-13Rα2 soluble receptor adjuvanted vaccine as described in Table 1 (strategies 4–7), and assessed the CD8+ T-cell avidity. The IN/IM prime-boost immunization strategy was selected as this strategy was shown to induce robust mucosal and systemic HIV-specific CTL cells that are also of high avidity compared with the IM/IM immunization strategy.8, 16, 17 Our data indicated that FPV-HIV-IL-13RΔ10/VV-HIV-IL-13RΔ10 immunization induced elevated high-avidity KdGag197–205-specific CTL similar to that of IL-13 KO mice (Figure 2a). More interestingly, data indicated that inclusion of the IL-13Rα2 soluble receptor in the prime was essential to generate high-avidity T cells (Figure 2b, gray line). In contrast, if the receptor was only included in the booster immunization, the T-cell avidity was similar to that of the control FPV-HIV/VV-HIV vaccination (Figure 2b, gray dotted line). In these studies, IL-13 KO mice immunized with control IN FPV-HIV/IM VV-HIV, vaccine strategy was considered as the gold standard vaccine (Figure 2a, black dotted line) with the lowest dissociation rate.

Avidity of HIV-specific CD8+ T cells, IL-13Rα2 soluble adjuvanted vaccine compared with control vaccine. BALB/c and interleukin (IL)-13KO mice (n=8 per group) were intranasally/intramuscularly prime-boost immunized with FPV-HIV/VV-HIV, and the recombinant pox virus vaccines co-expressing IL-13Rα2 soluble form. Fourteen days post-booster immunization, percentage of KdGag197–205-positive CD8+ splenocyte loss (dissociation) was measured as described in Methods. (a) The IL-13Rα2 was used in prime and the booster vaccination (gray square), (b) prime only (gray Δ) or booster only (gray dotted). Data represent mean±s.d. These experiments have been repeated three times. HIV, human immunodeficiency virus.
HIV-1 vaccines that co-express IL-13Rα2 soluble receptor can induce enhanced HIV-specific systemic and mucosal effector CD8+ T-cell responses
We had previously found that even though IL-13 KO generated T cells of high avidity the magnitude of KdGag197–205 tetramer reactive cells and the IFN-γ expression by these T cells were similar to that of wild-type control BALB/c mice.10 Therefore, we then tested the magnitude of CD8+ T-cell responses induced by the novel vaccines by evaluating KdGag197–205 tetramer reactive cells and the ability of these CD8+ T cells to produce IFN-γ following KdGag197–205 stimulation. Data indicated that IN FPV-HIV-IL-13RΔ10/IM VV-HIV-IL-13RΔ10 prime-boost immunization (the transient blockage of IL-13 at the vaccination site) induced significantly greater numbers of HIV-specific tetramer reactive systemic (splenic) and mucosal (illiac or GNs, PP, and lung) CD8+ T cells (Figure 3a–d), compared with the control FPV-HIV/VV-HIV prime-boost immunization. Interestingly, when the IL-13Rα2 soluble receptor was delivered only in the prime (IN FPV-HIV-IL-13RΔ10/IM VV-HIV) no such difference in the magnitude of systemic KdGag197–205-specific tetramer reactive cells was observed compared with the control (Figure 3a,d). In contrast, IL-13Rα2 soluble receptor delivered only in the booster vaccination (IN FPV-HIV/IM VV-HIV-IL-13RΔ10) induced a systemic KdGag197–205 tetramer-specific T-cell population similar to that of IN FPV-HIV-IL-13Δ10/IM VV-HIV-IL-13Δ10 prime-boost immunization (Figure 3a,d), although the avidity of the induced T cells was similar to that of the control vaccine strategy (Figure 2b). Interestingly, unlike systemic responses, all three regimes (Table 1, strategies 4–6) showed higher numbers of mucosal KdGag197–205 tetramer reactive CD8+ T cells specifically in the iliac nodes (Figure 3b) compared with the control vaccination regime (Table 1, strategy 3).

Human immunodeficiency virus (HIV)-specific effector CD8+ T-cell responses post interleukin (IL)-13Rα2-adjuvanted vaccination. BALB/c and IL-13 KO mice (n=4–8 per group) were intranasally/intramuscularly prime-boost immunized with FPV-HIV/VV-HIV and/or rFPV or rVV vaccines co-expressing IL-13Rα2 soluble form. Fourteen days post-booster immunization, percentage of KdGag197–205-positive T cells in (a) spleen, (b) iliac nodes (genito-rectal nodes), (c) Peyer’s patches (left, control vaccine; right, FPV-HIV-IL-13RΔ10/VV-HIV-IL-13RΔ10) were evaluated, plots indicate representative fluorescence-activated cell sorting plots. The upper quadrants indicate the percentage of HIV-specific T cells out of the total CD8+ T cells. The graph indicate (d) HIV-specific responses in spleen, error bars represent mean±s.d. These experiments were repeated over three times. FITC, fluorescein isothiocyanate; HIV, human immunodeficiency virus.
The IL-13 receptor used in the prime-boost immunization or only in the booster immunization (Table 1, strategies 5 and 6) induced a larger proportion of CD8+IFN-γ+ T cells (∼14%) compared with the control vaccine strategy (∼4%) (Figure 4a,b). However, when the receptor was only delivered in the IN-priming vaccination, despite the magnitude of CD8+ IFN-γ+ T cells being similar to that of the control vaccination (Figure 4a,b), the induced CD8+ T cells were higher in avidity similar to that of IN FPV-HIV-IL-13RΔ10/IM VV-HIV-IL-13RΔ10 prime-boost immunization strategy (Figure 2). Furthermore, novel vaccines (Table 1, strategies 4–6) generated elevated numbers of IL-2 expressing effector CD8+ T cells in the spleen similar or higher magnitude than that of IL-13 KO mice given the control vaccine (Figure 4c). However, in the mucosal compartment the elevated numbers of IL-2 expressing effector CD8+ T cells was only observed following intranasal delivery of FPV-HIV-IL-13RΔ10, not any other vaccine strategy (Figure 4d).

(a–f) Cytokine/chemokine expression by effector CD8+ T cells. Mice (n=4–8) were immunized intranasally/intramuscularly, as in Table 1 and at 14 days prime-boost immunization, IFN-γ protein expression in splenic CD8+ T cells (upper right) was measured by intracellular cytokine analysis following AMQMLKETI gag peptide stimulation. (a) Indicates representative fluorescence-activated cell sorting plots, and (b) indicates T-cell responses in spleen. The graphs (c and d) represent the IL-2 ELISpot responses in the spleen and iliac nodes, respectively. The data represent mean+s.d. (e) Pie charts represent the percentage of CD8+, CD8+IFN-γ+, CD8+TNF-α+, and CD8+IFN-γ+TNF-α+ T cells in spleen, iliac nodes, lung, and lung lymph nodes following (i) FPV-HIV-IL-13RΔ10/VV-HIV-IL-13RΔ10 (top two charts) immunization and (ii) the control FPV-HIV/VV-HIV immunization (bottom two charts). In these studies, CD8+ T cells were stimulated with AMQMLKETI gag peptide. (f) To measure cytokine/chemokine expression, 2 × 106 T cells were cultured for 6 h in the presence of AMQMLKETI gag peptide, supernatants were collected and assessed for cytokine/chemokine production using an antibody array as described in Methods (A) FPV-HIV/VV-HIV control (top) and IL-13RΔ10 adjuvanted vaccine (bottom). The positive control intensity is considered as 100%, and negative control as zero. The graph (B) represent the pattern of expression of 20 cytokines, control vaccination FPV-HIV/VV-HIV (left) compared with FPV-HIV-IL-13RΔ10/VV-HIV-IL-13RΔ10 (right) as described in Methods. APC, antigen-presenting cell; FITC, fluorescein isothiocyanate; HIV, human immunodeficiency virus; IFN, interferon; TNF, tumor necrosis factor.
We also evaluated the ability of systemic and mucosal HIV-specific CD8+ T cells to produce IFN-γ and/or TNF-α 14 days post-booster immunization. Data clearly indicated that our novel vaccine strategy can induce elevated systemic and mucosal HIV-specific CD8+TNF-α+ and CD8+IFN-γ+TNF-α+ T-cell numbers compared with the control vaccination (Figure 4e). Interestingly, when the “HIV-specific CD8+ T-cell population” as a whole was further analyzed, although no difference in the relative percentages of splenic CD8+IFN-γ+, CD8+TNF-α+, or CD8+IFN-γ+TNF-α+ were observed between the two groups (Figure 4e, left panel), these populations were much greater in the mucosal compartment; iliac nodes, lung, and lung lymph nodes following IL-13Rα2 soluble receptor adjuvanted vaccination (Figure 4e). Note that extremely low CD8+TNF-α+ T cells number were observed following control vaccination.
HIV-1 vaccines that co-express IL-13Rα2 soluble receptor can induce broader CD8+ T-cell cytokine/chemokine profiles
Following vaccination, the ability of effector T cells to produce an increased number of cytokines and chemokines are known to be a hallmark of protective immunity. We therefore assessed the cytokine/chemokine profiles induced by our novel vaccine strategy at 14 days post-booster vaccination in the spleen. The antibody array data clearly indicated that the co-expression of IL-13Rα2 substantially enhanced the expression profiles of over 20 cytokines and chemokines (Figure 4f), compared with the control vaccine strategy, and IFN-γ expression was outstandingly higher compared with other cytokines (Figure 4f), which correlates with the fluorescence-activated cell sorting (FACS) data (Figure 4f). Also chemokines, CXCL-4, CXCL-5, CCL-5, and CCL-24, expression was greatly enhanced compared with the control (Figure 4f).
HIV-1 vaccines that co-express IL-13Rα2 soluble receptor can induce excellent HIV-specific systemic and mucosal memory CD8+ T-cell responses
We then evaluated the ability of these vaccines to induce strong sustained systemic and mucosal immunity 8 weeks following booster immunization. Results indicated that novel IL-13Rα2 soluble receptor adjuvanted vaccine was able to induce two-fold higher KdGag197–205 tetramer reactive splenic (∼7.4%), PP (∼1.7%), and also lung (∼10% not shown)-specific memory CD8+ T cells compared with the control vaccine (Figure 5a,b). Following KdGag197–205 peptide stimulation, splenic T cells were also able to produce significantly greater CD8+IFN-γ+ single positive and also CD8+IFN-γ+TNF-α+ multifunctional T cells (Figure 5c,d). Furthermore, ELISpot results showed that these cells were able to produce not only IFN-γ but also IL-2 (Figure 5e,f).

Human immunodeficiency virus (HIV)-specific memory T-cell responses post interleukin (IL)-13Rα2-adjuvanted vaccination. BALB/c mice (n=5–8 per group) were intranasally/intramuscularly prime-boost immunized with FPV-HIV/VV-HIV (black bars) or FPV-HIV-IL-13RΔ10/VV-HIV-IL-13RΔ10 (gray bars). Eight weeks post-booster immunization, percentage of KdGag197–205-positive T cells in (a) spleen and (b) Peyer’s patches were evaluated by tetramer staining. Representative FACS plots on the left indicate control vaccine FPV-HIV/VV-HIV, and on the right indicates IL-13Rα2-adjuvanted vaccination FPV-HIV-IL-13RΔ10/VV-HIV-IL-13RΔ10. The upper quadrants indicate the percentage of HIV-specific T cells out of the total CD8+ T cells. (c and d) The percentage of HIV-specific splenic CD8+ T cells that are IFN-γ+ and IFN-γ+TNF-α+ measured by intracellular cytokine analysis (ICS), respectively. (e and f) Splenic IFN-γ and IL-2 responses measured by ELISpot. The error bars represent mean±s.d. These experiments have been repeated over three times. FITC, fluorescein isothiocyanate; IFN, interferon; SFU, spot forming units; TNF, tumor necrosis factor.
HIV-1 IL-13Rα2-based vaccines (soluble or membrane-bound forms) can generate robust protective immunity
We then evaluated the protective efficacy of not only the IL-13Rα2 soluble receptor form of the vaccine (IL-13RΔ10) but also the membrane-bound form of this vaccine (IL-13Rα2m) in a prime-boost modality and compared against the control vaccine. At 8 weeks following booster immunization, mice were challenged with 75 PFU of influenza virus expressing the KdGag197–205 immunodominant epitope, and body weights were monitored daily for 9–10 days. Post challenge IL-13 KO mice that received the control IN FPV-HIV/IM VV-HIV immunization did not lose significant body weight as opposed to the wild-type BALB/c mice that received the same vaccine (Figure 6a). Interestingly, mice that received the IN FPV-HIV-IL-13RΔ10/IM VV-HIV-IL-13RΔ10 or IN FPV-HIV-IL-13Rα2m/IM VV-HIV-IL-13Rα2m-adjuvanted vaccines performed very similar to that of the IL-13 KO mice that received the control vaccines (Figure 6a,b). From day 5 post challenge, the recovery rate of the above three vaccination groups was significantly higher compared with the wild-type BALB/c mice that received the control vaccination (Figure 6, Table). Mice that were prime-boost immunized with the membrane-bound form of the receptor IL-13Rα2m showed less weight loss and better protection compared with the soluble form of the receptor IL-13Rα2Δ10 (Figure 6a,b). The above protective data correlate well with the dissociation rates (Figure 2a) of CD8+ T splenocytes from IL-13 KO mice given the control vaccine or the BALB/c mice that received IN FPV-HIV-IL-13Rα2/IM VV-HIV-IL-13Rα2 receptor adjuvanted vaccines (Table 1, strategies 6 and 7), and also the IFN-γ levels measured in the systemic and mucosal compartments by intracellular cytokine staining (Figure 4b and Supplementary Figure 3A,B online) and KdGag197–205 tetramer staining (Figure 3 and Supplementary Figure 4 online). Post challenge when cytokine production was measured, the mice that were given the novel vaccines showed significantly higher IFN-γ responses compared with the control vaccination strategy (Figure 6c).

Protective immunity following PR8-KdGag197–205 challenge. BALB/c and interleukin (IL)-13 KO mice (n=8–14) were intranasally (IN)/intramuscularly prime-boost immunized with FPV-HIV/VV-HIV and/or rFPV or rVV vaccines co-expressing IL-13Rα2 (a) the soluble form–IL-13RΔ10 or (b) the membrane-bound form–IL-13Rα2m. At 6 weeks, post-booster immunization or unimmunized control mice were challenged mucosally (IN) with 75 units influenza virus PR8 expressing KdGag197–205 epitope. Body weights were monitored for 9–10 days, and (c) KdGag197–205-specific IFN-γ T-cell responses in spleen were also measured at 9–10 days following recovery by ELISpot, as described in Methods. The data represent mean±s.e.m., and P-values at 5–10 days are calculated using two-tailed, two sample unequal variance Student’s t-test (see Table). HIV, human immunodeficiency virus; IFN, interferon; SFU, spot forming units; TNF, tumor necrosis factor.
Inhibition of IL-13 at the cell milieu can induce enhanced APC recruitment
Studies have suggested that different dendritic cell (DC) subsets can modulate T-cell avidity.18 As our current study indicated that including the IL-13Rα2 in the priming was critical to induce high-avidity CTL, we then looked at the numbers of APCs (specifically DC subsets) 24-h post rFPV vaccination. The 24-h time point was selected as we have found that following IN rFPV vaccination the peak antigen expression occurs at 12 h in the lung (S Trivedi and C Ranasinghe, personal communication). Following IN immunization, the lung, spleen, and PP were harvested and cells were stained with MHC-II I-Ad, CD45, and CD11c and CD11b as described in the Methods. Compared with the BALB/c mice that received FPV-HIV vaccine, BALB/c mice that received FPV-HIV IL-13RΔ10-adjuvanted vaccine showed 2–3 times higher total APC subsets similar to that of IL-13 KO given FPV-HIV vaccine (Figure 7a). When MHC-II I-Ad+CD45+CD11c+ lung DC subset was further analyzed CD11bhi and CD11blo subsets were detected (Figure 7a). More interestingly, CD11clo and CD11blo DC subsets were greatly enhanced in the lungs of IL-13 KO mice that received the FPV-HIV and BALB/c mice that received the FPV-HIV IL-13RΔ10 (Figure 7a,b). Moreover, four times higher MHC-II I-Ad+CD45+CD11c− APC subset was also detected in these two groups (Figure 7a,b). In contrast, no differences in these APC subsets were detected in distal sites PP or spleen (Figure 7b).

(a) Following intranasal (IN) delivery evaluation of antigen-presenting cell subsets in lung, spleen, and Peyer’s patch (PP). BALB/c and interleukin (IL)-13 KO mice (n=3) were (IN) immunized with FPV-human immunodeficiency virus (HIV) and/or FPV-HIV co-expressing the soluble form–interleukin (IL)-13RΔ10. Twenty-four hours post-immunization lung, spleen, and Peyer’s patch were harvested and cells were stained as described in Methods (due to small sample size, lung and PP were performed as pooled samples). Fluorescence-activated cell sorting plots display cells pre-gated on live cells (R1) followed by doublet discrimination (R2 and R3, not shown) based on forward scatter (FSC) and side scatter (SSC), and gated on CD45+ and MHC-II I-Ad+ population (R4). CD45+ and I-Ad+ population of cells were further analyzed for CD11c expression (R5). R6 and R7 indicate I-Ad+CD45+CD11c+ lung dendritic cell subset expressing various levels of CD11b. R8 indicates the I-Ad+CD45+CD11c− APC subset. (b) Histograms show the surface expression levels of CD11b+ and CD11c+ cells pre-gated on I-Ad+ and CD45+ cell subsets (R4) as defined in panel a. Gray-filled histogram indicates BALB/c mice immunized with FPV-HIV control vaccine, open black and open gray histograms indicate BALB/c mice immunized FPV-HIV IL-13RΔ10 (IL-13Rα2 adjuvanted) vaccine and IL-13 KO mice immunized with FPV-HIV vaccine, respectively.
Discussion
We have now clearly established that following HIV-1 poxvirus prime-boost immunization, both the HIV-specific CTL avidity, protective immunity, and the induction of mucosal immunity are influenced by the route of vaccine delivery and the Th2 cytokine milieu they induce.8 Current findings indicate that our novel HIV-1 IL-13Rα2 cytokine trap vaccines that temporarily inhibit IL-13 activity can induce excellent high-avidity HIV-specific CD8+ T cells with broader cytokine/chemokine profile (i.e., IFN-γ, IL-2, granulocyte/macrophage colony-stimulating factor, CCL-1, CCL-3, CCL-5) with better protective immunity compared to the control IN FPV-HIV/IM VV-HIV vaccination or the previously tested, DNA prime-boost strategies.16 Amazingly these novel vaccines were able to induce a cell milieu (at the vaccination site) that was similar to an IL-13 KO animal. In a vaccine context, several studies have indicated that induction of multifunctional CTL (that express IFN-γ, TNF-α, IL-2) is a hallmark of high-avidity T cells with greater protective immunity.19, 20, 21 Also in recent studies HIV-1 controllers were shown to maintain significantly greater high-avidity CD8+ T cells compared with the non-controllers, suggesting avidity of T cells have a critical role in HIV-1 protective immunity.22, 23 Such studies clearly highlight the importance of developing vaccine strategies that can induce CTL of high avidity, not purely higher numbers of CTL that do not induce protection. More importantly, the IN/IM delivery of the novel vaccines that co-expressed IL-13Rα2 also induce excellent KdGag tetramer reactive CD8+ T cells in the mucosae (see Supplementary Figure 4 online for IL-13Rα2m) that were multifunctional compared with the control vaccine. Data indicated that the IN IL-13Rα2-adjuvanted priming was essential for the induction of these mucosal high-avidity CTL that expressed IL-2. As HIV-1 is first encountered at the genito-rectal tract and the first CD4+ cell depletion occurs in the gut mucosae,1 the induction of greatly elevated sustained high-quality gut-specific CTL, using HIV vaccines that co-expressed IL-13Rα2, is an exciting prospect for a HIV-1 vaccine (Figures 3c and 5b and Supplementary Figure 4 online).
Several studies also have shown that the vaccine vector combination or order in which immune modulators are delivered (i.e., prime or the booster vaccination) or cell milieu they induce can significantly alter the immune outcomes,17, 24, 25 but the mechanisms governing these differences are currently not well known. In our hands even though several of our previously tested immune modulators showed greatly elevated IFN-γ+ HIV-specific CD8+ T cells, none were able to induce CD8+ T cells of high functional avidity with better protection.26, 27, 28 In great contrast, co-expression of IL-13Rα2 together with HIV antigens delivered in the prime and booster vaccinations was shown to significantly enhance both the avidity and magnitude of HIV-specific CTL that correlated with better protective immunity. More interestingly, the novel IL-13Rα2-adjuvanted vaccines were shown to successfully sequester IL-13 in the cell milieu behaving very similar to an IL-13 KO animal. The inclusion of the IL-13 inhibitor in the priming vaccination was essential for the induction of optimum mucosal and systemic high-avidity CD8+ T cells. In contrast, IL-13 inhibitor delivered only in the booster vaccination, even though induced enhanced IFN-γ+ HIV-specific CD8+ T cells, did not induce high-avidity CTL, (i.e., the avidity observed was similar to that of the control vaccination). This suggests that following vaccination the first antigen encounter, initiating a high-avidity CD8+ T-cell progeny in the mucoase, was central for the maintenance of an effective high-avidity CTL subset. Also the APC data further substantiated that IL-13-depleted cell milieu can chemoattract unique APC subsets into the lung mucosae similar to IL-13 KO mice given the control vaccination, promoting the induction of these high-avidity CTL. Our observations are consistent with Kroger and co-worker studies, where different DC subsets were shown to induce CD8+ T cells of different functional avidities.18 We have also found that unlike other IL-4/IL-13 receptors, IL-4Rα densities on effector CD8+ T cells, especially in IL-13 KO mice, are heavily downregulated at the early stages of vaccination, and IL-4Rα has a significant role in modulating quality/polyfunctionality of CD8+ T cells in a STAT6-dependent manner.29 Interestingly, we have also found that IL-4Rα is upregulated on DC’s following vaccination or viral infection.29 Collectively, our observations suggest that following vaccination CD8+ T-cell avidity is primarily defined at the very early stages of antigen encounter, most likely at the vaccination site, and IL-13 regulation has an important role in T-cell avidity.
The cytokine-specific responsiveness of target cells is mainly regulated via the interaction of cytokines and their receptors.30, 31 Recent studies have shown that IL-13 antibodies can be used to modulate IL-13 activity in humans.32 Similarly, the ability for the IL-15/IL-15R complex to regulate CD8+ T-cell responsiveness33 or use of IL-31/IL-31R complex to reduce type 2 inflammatory responses in the intestinal mucosae34 have been recently documented. Thus, we believe that mimicking these natural in vivo cytokine regulatory mechanisms in a vaccine setting may help design more effective and safer vaccines in the future. We postulate that upregulation of certain Th2 cytokines is a natural mechanism by which hosts have evolved to dampen immune responses to strong pathogens to evade immune exhaustion and tissue damage. While dampening immune response may be useful in a setting of an established chronic or acute viral infection, this could be counterproductive in the setting of many vaccines, as induction of strong sustained immunity is pivotal in establishing protective immunity. Similarly induced upregulation of IL-4 and IL-13 in CD8+ T cells appears to be a mechanism by which particular viruses/pathogens escape the host immune system.35, 36, 37 For example, following chronic HIV-1 infection, IL-4-producing CD8+ T-cell subset with reduced CTL activity has been reported.38 Also some in vitro studies have established that the presence of IL-4 can produce CD8+ T cells of reduced cytolytic activity.39 Inhibition of IL-13 expression by regulatory CD4+ T cells using a monoclonal antibody in the presence of granulocyte/macrophage colony-stimulating factor was shown to increase T-cell activity and enhance protection against viral infection.40 Furthermore, during asthma or nematode infections, where there is an elevated Th2 cytokine, IL-4, and IL-13 bias,41, 42 IL-13 receptors or inhibitors have shown to have an important role in dampening Th2 cytokine production and control of disease progression.43 For example, in mice, IL-13Rα2 has been identified as a powerful inhibitor of IL-13-induced inflammatory remodeling in the lung.44 Also, Morimoto and co-workers have shown that IL-13Rα2 can have an important role in regulating IL-13 following gastrointestinal nematode infection.42 These observations further highlight the importance and uniqueness of our current cytokine trap vaccine strategy.
The HIV vaccines that contained the membrane-bound form (IL-13Rα2m) elicited better protection following mucosal influenza-KdGag197–205 challenge compared with soluble form of the vaccine (IL-13RΔ10). The differences observed with IL-13Rα2m could be a result of (i) membrane-bound form inducing a more localized activity compared with the secreted soluble form and/or (ii) may be the soluble form having the ability to diffuse and/or degrade faster in the milieu. It is also noteworthy that when the receptors were delivered as a single IL-13Rα2 protein together with the control HIV vaccine (IL-13Rα2 not co-expressed) no difference in high-avidity CD8+ T-cell numbers was observed. This is not entirely surprising, as our APC data indicated that localized sustained expression of the IL-13Rα2 at the site of vaccination was pivotal for the optimum depletion of IL-13 in the cell milieu and antigen presentation. However, we believe that in the context of a future HIV-1 vaccine, co-expression of the soluble human IL-13Rα2 form together with HIV antigens may prove much safer and suitable in a clinical setting compared with IL-13Rα2m, which is associated with cancers.45, 46 It is noteworthy that any future human recombinant vaccine, the extracellular IL-13R binding domain will be devoid of the transmembrane and cytoplasmic domains, therefore would be highly unlikely to cause aberrant cell signaling by association with other membrane-bound or cytoplasmic components.
Collectively, our results demonstrate that (i) the novel HIV IL-13Rα2-adjuvanted vaccine strategy not only induced elevated high-avidity HIV-specific CTL with boarder cytokine and chemokine profile but also strong sustained mucosal immunity, which correlated with excellent protective immunity, and (ii) avidity of CD8+ T cells is primarily defined, most likely at the vaccination site. We believe our current findings offer exciting prospects for not only a future HIV-1 vaccine, but also promise for vaccines against many other chronic infections, where high-avidity CD8+ T cells and strong sustained mucosal immunity are required for protection.
Methods
Isolation of soluble and membrane-bound forms of IL-13 receptors. Mouse IL-13Rα2 complementary DNAs were amplified from total RNA using gene-specific primers 5′-AGATCTGAAATGGCTTTTGTGCATATCAGATGCTTGTG-3′ and 5′-GAGCTCTTAACAGAGGGTATCTTCATAAGC-3′ using a One-Step RT-PCR kit (Qiagen, Valencia, CA). Two different length PCR products were isolated; a 1,167-bp product encoding the full-length membrane-bound IL-13Rα2 and a 1,051-bp splice variant encoding the secreted IL-13 receptor, which lacks exon 10 and the trans-membrane domain sequences (sIL-13Rα2, which is named as IL-13RΔ10).47 Note that these IL-13Rα2 complementary DNAs were identical to GenBank entries EF219410 and NM008356, respectively.
Cloning IL-13R into pTK7.5A and pAF09 plasmids. The PCR products were directly ligated into the U-tailed vector pDrive and were transformed into QIAGEN EZ-competent cells (Qiagen). The plasmids were then digested with either BglII and PstI (located in pDrive sequence) or BglII and SacI (treated with Klenow fragment DNA polymerase to make blunt ended). The respective IL-13Rα2 gene fragments were gel-purified and ligated into the BamHI and PstI sites of the FPV vector pAF0948, 49 downstream of the FPV early/late promoter in-frame with the upstream ATG or the BamHI and HincII sites of VV vector pTK7.5A50 immediately downstream of the p7.5 early/late promoter.
Construction of FPV-HIV and VV-HIV vaccines co-expressing IL-13 receptor. Recombinant viruses co-expressing the HIV gag/pol(mut) antigen and mouse IL-13Rα2 (membrane-bound or soluble forms) were constructed using parent viruses FPV-HIV 086 and VV-HIV 336.51 rFPV were constructed by infecting chicken embryo skin cell cultures with FPV-HIV 086 (MOI 0.05) followed by transfection with pAF09-IL-13Rmα2 or IL-13Δ10 using Lipofectamine 2000 (Invitrogen). rFPV were selected by passage of viruses on chicken embryo skin cells in the minimal essential medium containing MX-HAT (2.5 μg/ml mycophenolic acid, 250 μg/ml xanthine, 100 μg/ml hypoxanthine, 0.4 μg/ml aminopterine, and 30 mg/ml thymidine) to select for viruses expressing the gpt (xanthine guanine phosphoribosyl transferase) gene. Plaques containing recombinant viruses were identified using an agar overlay (1% agar in minimal essential medium) containing X-gal (200 μg/ml) to detect co-expression of the lacZ gene, three to four plaque purification rounds were performed under selection media and recombinants were confirmed by PCR.
Similarly, rVV were constructed by infecting H143B TK-cells with VV-336 (multiplicity of infection 0.05) and transfection with pTK7.5A- IL-13Rmα2 or IL-13RΔ10. Recombinant viruses were selected using minimal essential medium containing 100 μM hypoxanthin, 0.4 μM aminopterin, 16 μM thymidine (HAT) supplement to select for viruses expressing the Herpes Simplex Virus TK gene contained in the vector. Viruses were plaque purified under selection and purity confirmed similar to rFPV.
Western blotting. Expression of the membrane-bound (IL-13Rmα2) and secreted (IL-13RΔ10) forms of the recombinant viruses were confirmed by western blotting of infected cells and filtered culture media, respectively, using rat monoclonal anti-mouse IL-13Rα2 antibody (R&D Systems, Minneapolis, MN) (Supplementary Figure 1 online).
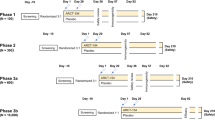
Immunization of mice. Pathogen-free 6- to 7-week-old female BALB/c (H-2d) mice were obtained from the Animal Breeding Establishment, John Curtin School of Medical Research (JCSMR). All animals were maintained and used in accordance with the approved Australian National University (ANU) animal experimentation ethics committee guidelines. Mice were prime-boost immunized with 1 × 107 PFU rFPV followed by 1 × 107 PFU rVV-expressing HIV-1 antigens and/or IL-13R, as described in Table 1 (we have also evaluated the different doses of our novel vaccines and 1 × 107 PFU was found to be the optimal dose–see Supplementary Figure 5 online), under mild methoxyfluorane anesthesia 2 weeks apart using IM/IM-purely systemic, IN/IN-purely mucosal, or IN/IM-combined mucosal–systemic routes of vaccination (Table 1, strategies 1–3). Immediately prior to delivery, rFPV and rVV were diluted in phosphate-buffered saline and sonicated 20–30 s to obtain an homogeneous viral suspension, IN rFPV was given in a final volume of 20–25 μl and IM rFPV or rVV were delivered, 50 μl per quadriceps. To evaluate protective immunity at 6 weeks after the final vaccine booster, mice were challenged intranasally with 50 or 75 PFUs of influenza virus PR8 expressing the KdGag197–205 epitope of HIV in the neuraminidase stalk, as described previously.16 This was constructed using reverse genetic technology.52, 53 Body weight was monitored for 9–10 days after challenge.
Preparation of mucosal and systemic lymphocytes. To measure systemic and mucosal T-cell responses, mice were euthanized at different time intervals (2 and 8 weeks) post-boost immunization, and 10 days post influenza-KdGag197–205 challenge; the spleen, GNs or iliac lymph nodes, lung, and PP were removed and cell suspensions prepared in complete (5%) RPMI. Briefly, the spleen cells were dissociated through a cell strainer and were treated with red blood cell lysis buffer, similarly GN and PP were prepared without the red cell lysis as described previously.16, 17 Lung samples were first cut into small pieces and digested in 2 ml of complete RPMI buffer containing 2 mg/ml collagenase (Sigma-Aldrich, St Louis, MO), 2.4 mg/ml dispase (Gibco, Auckland, NZ), and 5 Units/ml DNAse (Calbiochem, La Jolla, CA) at 37 °C for 1 h with gentle vortexing. Sample was then filtered through a cell strainer, rinsed with complete RPMI, red cells lysed and filtered through sterile gauze to remove debris as described previously.16, 28 The single-cell suspensions were then kept at 4 °C for minimum of 4–6 h for recovery prior to performing assays, as we have observed that digestion can downregulate the expression of some surface markers.
Tetramer staining and dissociation assays. Allophycocyanin-conjugated KdGag197–205 tetramers were synthesized at the Bio-Molecular Resource Facility at The John Curtin School of Medical Research (BRF/JCSMR), ANU. Splenocytes or mucosal lymphocytes (2—5 × 106) were stained with anti-CD8-FITCa antibody (BD PharMigen, San Diego, CA) and allophycocyanin-conjugated KdGag197–205 tetramer at room temperature and analyzed as described previously16, 17 (note that all appropriate controls have been performed and we have found that the background tetramer counts in naive mice are between 0.05 and 0.5% in spleen, 0.2 and 0.8% in lung, 0.02 and 0.1% in PP and GN).
Similarly, following KdGag197–205 tetramer staining the dissociation assays were performed as described previously.16 Plates were configured to assess five time points per sample (0–60 min). Anti-H-2Kd competitive binding antibody (50 μg/ml; BD PharMigen) was added to each well to prevent dissociated tetramer from re-binding, and plates were incubated at 37°C, 5% CO2. At each time point, cells were transferred into ice-cold FACS buffer to stop the reaction, washed and resuspended in 100 μl of FACS buffer containing 0.5% paraformaldehyde. A total of 100,000 events were acquired on a FACs Calibur flow cytometer (Becton-Dickinson, San Diego, CA)) and analyzed using Cell Quest Pro software (BD Biosciences, San Diego, CA).
IFN-γ and IL-2 ELISpot assay. IFN-γ or IL-2 HIV-specific T-cell responses were measured by IFN-γ or IL-2 capture ELISpot assay as described previously.8, 16 Briefly, 2 × 105 spleen or GN cells were added to 96-well Millipore polyvinylidene difluoride plates (Millipore, Billerica, MA) coated with 5 μg/ml of mouse anti-IFN-γ or IL-2 capture antibodies (BD PharMigen), and stimulated for 12 or 22 h, respectively, for IL-2 or IFN-γ ELISpot, in the presence of H-2Kd immunodominant CD8+ T-cell epitope, Gag197–205 (AMQMLKETI) (synthesized at the Bio-Molecular Resource Facility at JCSMR). ConA-stimulated cells (Sigma) were used as positive controls and unstimulated cells as negative controls. For both ELISpot assays, all steps were carried out exactly as described previously.16, 17 Plotted data are expressed as spot forming units (SFU) per 106 T cells and represent mean values±s.d. Unstimulated cell counts were subtracted from each stimulated value before plotting the data. All instances of the background SFU counts were extremely low.
Intracellular cytokine analysis. IFN-γ and TNF-α producing HIV-specific CD8 T cells were analyzed as described in Ranasinghe et al.16, 17 Briefly, 2 × 106 lymphocytes were stimulated with AMQMLKETI peptide at 37°C for 16 h, and further incubated with Brefeldin A (e-Biosciences, San Diego, CA) for 4 h. Cells were surface-stained with CD8-allophycocyanin (BD PharMigen) then fixed and permeabilized prior to intracellular staining with IFN-γ-FITC and TNF-α-PE (BD PharMigen). Total 100,000 gated events per sample were collected using FACS Calibur flow cytometer (Becton-Dickinson), and results were analyzed using Cell Quest Pro software. Prior to plotting the graphs, the unstimulated background values were subtracted from the data.
Cytokine antibody arrays. Splenocytes (2 × 106) were cultured in complete RPMI without IL-2 for 16 h in the presence of H-2Kd binding Gag197–205 peptide as described previously.10, 16 Supernatants were collected and cytokine antibody arrays were performed, the signals were detected using chemiluminescence according to the manufacturer’s instructions (Ray Biotech, Norcross, GA). Protein expression signal intensities were calculated as a percentage absorbance as described previously.10, 16
Evaluation of APCs using FACS. BALB/c and IL-13 KO mice were immunized with control FPV-HIV, and a second group of BALB/c mice were immunized with IL-13RΔ10-adjuvanted vaccine. Twenty-four hours post vaccination, mice were euthanized and the lung, spleen, and PP were collected and single-cell suspensions were prepared as described above. From each sample, 4 × 106 cells were aliquoted, and first cells were incubated with Fc block antibody (anti-mouse CD16/CD32 Fc Block, BD Biosciences) for 20 min at 4°C and then cells were surface stained with allophycocyanin-conjugated MHC-II I-Ad (e-Biosciences), CD45 FITC (30-F11 clone, Biolegend, San Diego, CA), CD11b PE (Biolegend), CD11c PerCp-Cy5.5 (HL3 clone, BD Biosciences), biotin-conjugated F4/80 (BM8 clone, e-Biosciences) followed by streptavidin Brilliant violet 421 (Biolegend). Cells were fixed and total 100,000 gated events per sample were collected using Fortessa flow cytometer (Becton-Dickinson), and results were analyzed using Cell Quest Pro software.
Statistical analysis of data. Standard deviation (s.d.) or standard error of the mean (s.e.m.) was calculated and P-values were determined using a two-tailed, two sample equal variance or unequal variance Student’s t-test. The P-values less than 0.05 were considered significant. Except where stated experiments have been repeated for minimum three times.
References
Veazey, R.S. et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280, 427–431 (1998).
Shacklett, B.L., Shacklett, B.L., Critchfield, J.W., Ferre, A.L. & Hayes, T.L. Mucosal immunity to HIV: a review of recent literature. Mucosal T-cell responses to HIV: responding at the front lines. Curr Opin HIV AIDS 3, 541–547 (2008).
Ahlers, J.D. & Belyakov, I.M. Strategies for optimizing targeting and delivery of mucosal HIV vaccines. Eur J Immunol 39, 2657–2669 (2009).
Corbett, M. et al. Aerosol immunization with NYVAC and MVA vectored vaccines is safe, simple, and immunogenic. Proc Natl Acad Sci USA 105, 2046–2051 (2008).
Alexander-Miller, M.A., Leggatt, G.R., Sarin, A. & Berzofsky, J.A. Role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. J Exp Med 184, 485–492 (1996).
Alexander-Miller, M.A. High-avidity CD8+ T cells: optimal soldiers in the war against viruses and tumors. Immunol Res 31, 13–24 (2005).
Belyakov, I.M. et al. Impact of vaccine-induced mucosal high-avidity CD8+ CTLs in delay of AIDS viral dissemination from mucosa. Blood 107, 3258–3264 (2006).
Ranasinghe, C. et al. Mucosal HIV-1 pox virus prime-boost immunization Induces high-avidity CD8+ T cells with regime-dependent cytokine/granzyme B profiles. J Immunol 178, 2370–2379 (2007).
Kent, S.J. et al. Mucosally-administered human-simian immunodeficiency virus DNA and fowlpoxvirus-based recombinant vaccines reduce acute phase viral replication in macaques following vaginal challenge with CCR5-tropic SHIV(SF162P3). Vaccine 23, 5009–5021 (2005).
Ranasinghe, C. & Ramshaw, I.A. Immunisation route-dependent expression of IL-4/IL-13 can modulate HIV-specific CD8(+) CTL avidity. Eur J Immunol 39, 1819–1830 (2009).
Takeda, K., Kamanaka, M., Tanaka, T., Kishimoto, T. & Akira, S. Impaired IL-13-mediated functions of macrophages in STAT6-deficient mice. J Immunol 157, 3220–3222 (1996).
Tabata, Y. & Khurana Hershey, G.K. IL-13 receptor isoforms: breaking through the complexity. Curr Allergy Asthma Rep 7, 338–345 (2007).
Chen, W. et al. IL-13R alpha 2 membrane and soluble isoforms differ in humans and mice. J Immunol 183, 7870–7876 (2009).
Fichtner-Feigl, S., Strober, W., Kawakami, K., Puri, R.K. & Kitani, A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med 12, 99–106 (2006).
Fujisawa, T., Joshi, B.H. & Puri., R.K. IL-13 regulates cancer invasion and metastasis through IL-13Ralpha2 via ERK/AP-1 pathway in mouse model of human ovarian cancer. Int J Cancer 131, 344–356 (2012).
Ranasinghe, C. et al. A comparative analysis of HIV-specific mucosal/systemic T cell immunity and avidity following rDNA/rFPV and poxvirus-poxvirus prime boost immunisations. Vaccine 29, 3008–3020 (2011).
Ranasinghe, C. et al. Evaluation of fowlpox-vaccinia virus prime-boost vaccine strategies for high-level mucosal and systemic immunity against HIV-1. Vaccine 24, 5881–5895 (2006).
Kroger, C.J., Amoah, S. & Alexander-Miller, M.A. Cutting edge: dendritic cells prime a high avidity CTL response independent of the level of presented antigen. J Immunol 180, 5784–5788 (2008).
Almeida, J.R. et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 204, 2473–2485 (2007).
Harari, A. et al. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J Exp Med 205, 63–77 (2008).
Burgers, W.A. et al. Broad, high-magnitude and multifunctional CD4+ and CD8+ T-cell responses elicited by a DNA and modified vaccinia Ankara vaccine containing human immunodeficiency virus type 1 subtype C genes in baboons. J Gen Virol 90, 468–480 (2009).
Mothe, B. et al. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One 7, e29717 (2012).
Berger, C.T. et al. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J Virol 85, 9334–9345 (2011).
Gherardi, M.M., Ramirez, J.C. & Esteban, M. Interleukin-12 (IL-12) enhancement of the cellular immune response against human immunodeficiency virus type 1 env antigen in a DNA prime/vaccinia virus boost vaccine regimen is time and dose dependent: suppressive effects of IL-12 boost are mediated by nitric oxide. J Virol 74, 6278–6286 (2000).
Dale, C.J. et al. Evaluation in macaques of HIV-1 DNA vaccines containing primate CpG motifs and fowlpoxvirus vaccines co-expressing IFNgamma or IL-12. Vaccine 23, 188–197 (2004).
Harrison, J.M. et al. 4-1BBL coexpression enhances HIV-specific CD8 T cell memory in a poxvirus prime-boost vaccine. Vaccine 24, 6867–6874 (2006).
Day, S.L., Ramshaw, I.A., Ramsay, A.J. & Ranasinghe, C. Differential effects of the type I interferons alpha4, beta, and epsilon on antiviral activity and vaccine efficacy. J Immunol 180, 7158–7166 (2008).
Xi, Y., Day, S.L., Jackson, R.J. & Ranasinghe, C. Role of novel type I interferon epsilon (IFN-ɛ) in viral infection and mucosal immunity. Mucosal Immunol 5, 610–622 (2012).
Wijesundara, D.K., Tscharke, D.C., Jackson, R.J. & Ranasinghe, C. Reduced interleukin-4 receptor α expression on CD8+ T cells correlates with high quality anti-viral immunity. PLoS One (in press).
Khodoun, M. et al. Differences in expression, affinity, and function of soluble (s)IL-4Ralpha and sIL-13Ralpha2 suggest opposite effects on allergic responses. J Immunol 179, 6429–6438 (2007).
de Weerd, N.A. & Nguyen, T. The interferons and their receptors-distribution and regulation. Immunol Cell Biol 1038, 9 (2012).
Kasaian, M.T. et al. IL-13 antibodies influence IL-13 clearance in humans by modulating scavenger activity of IL-13Ralpha2. J Immunol 187, 561–569 (2011).
Stoklasek, T.A., Colpitts, S.L., Smilowitz, H.M. & Lefrancois, L. MHC class I and TCR avidity control the CD8 T cell response to IL-15/IL-15Ralpha complex. J Immunol 185, 6857–6865 (2010).
Perrigoue, J.G., Zaph, C., Guild, K., Du, Y. & Artis, D. IL-31-IL-31R interactions limit the magnitude of Th2 cytokine-dependent immunity and inflammation following intestinal helminth infection. J Immunol 182, 6088–6094 (2009).
Costa, D.L. et al. BALB/c mice infected with antimony treatment refractory isolate of Leishmania braziliensis present severe lesions due to IL-4 production. PLoS One 5, e965 (2011).
Liu, L. et al. Vaccinia virus induces strong immunoregulatory cytokine production in healthy human epidermal keratinocytes: a novel strategy for immune evasion. J Virol 79, 7363–7370 (2005).
Imrie, A. et al. Differential functional avidity of dengue virus-specific T-cell clones for variant peptides representing heterologous and previously encountered serotypes. J Virol 81, 10081–10091 (2007).
Maggi, E. et al. Th2-like CD8+ T cells showing B cell helper function and reduced cytolytic activity in human immunodeficiency virus type 1 infection. J Exp Med 180, 489–495 (1994).
Kienzle, N., Baz, A. & Kelso, A. Profiling the CD8low phenotype, an alternative career choice for CD8 T cells during primary differentiation. Immunol Cell Biol 82, 75–83 (2004).
Ahlers, J.D. et al. A push-pull approach to maximize vaccine efficacy: abrogating suppression with an IL-13 inhibitor while augmenting help with granulocyte/macrophage colony-stimulating factor and CD40L. Proc Natl Acad Sci USA 99, 13020–13025 (2002).
Hahn, C. et al. Inhibition of the IL-4/IL-13 receptor system prevents allergic sensitization without affecting established allergy in a mouse model for allergic asthma. J Allergy Clin Immunol 111, 1361–1369 (2003).
Morimoto, M. et al. IL-13 receptor alpha2 regulates the immune and functional response to Nippostrongylus brasiliensis infection. J Immunol 183, 1934–1939 (2009).
Bree, A. et al. IL-13 blockade reduces lung inflammation after Ascaris suum challenge in cynomolgus monkeys. J Allergy Clin Immunol 119, 1251–1257 (2007).
Zheng, T. et al. IL-13 receptor alpha2 selectively inhibits IL-13-induced responses in the murine lung. J Immunol 180, 522–529 (2008).
Takenouchi, M. et al. Epigenetic modulation enhances the therapeutic effect of anti-IL-13R(alpha)2 antibody in human mesothelioma xenografts. Clin Cancer Res 17, 2819–2829 (2011).
Joshi, B.H., Leland, P., Calvo, A., Green, J.E. & Puri, R.K. Human adrenomedullin up-regulates interleukin-13 receptor alpha2 chain in prostate cancer in vitro and in vivo: a novel approach to sensitize prostate cancer to anticancer therapy. Cancer Res 68, 9311–9317 (2008).
Tabata, Y. et al. Allergy-driven alternative splicing of IL-13 receptor alpha2 yields distinct membrane and soluble forms. J Immunol 177, 7905–7912 (2006).
Boyle, D.B. & Coupar, B.E. Construction of recombinant fowlpox viruses as vectors for poultry vaccines. Virus Res 10, 343–356 (1988).
Heine, H.G. & Boyle, D.B. Infectious bursal disease virus structural protein VP2 expressed by a fowlpox virus recombinant confers protection against disease in chickens. Arch Virol 131, 277–292 (1993).
Coupar, B.E., Andrew, M.E. & Boyle, D.B. A general method for the construction of recombinant vaccinia viruses expressing multiple foreign genes. Gene 68, 1–10 (1988).
Coupar, B.E.H. et al. Fowlpox virus vaccines for HIV and SIV clinical and pre-clinical trials. Vaccine 24, 1378–1388 (2006).
Cukalac, T. et al. Narrowed TCR diversity for immunised mice challenged with recombinant influenza A-HIV Env(311-320) virus. Vaccine 27, 6755–6761 (2009).
Sexton, A. et al. Evaluation of recombinant influenza virus-simian immunodeficiency virus vaccines in macaques. J Virol 83, 7619–7628 (2009).
Acknowledgements
We thank Dr David Boyle, CSIRO Animal Health Laboratories, for providing the parent HIV vaccines; Kerong Zhang and Kerry McAndrew at the BRF/JCSMR ANU for synthesizing the HIV-specific peptides and tetramers; Annette Buchanan, Lisa Pavlinovic, Megan Glidden, Sherry Tu, and Jill Medveczky for their technical assistance with various aspects of the project; and Professor Ian Ramshaw for some early discussions. This work was supported by the Australian National Health and Medical Research Council project grant award 525431 (C.R.), Australian Centre for Hepatitis and HIV Virology EOI 2010 grant (C.R.), and Bill and Melinda Gates Foundation GCE Phase I grant OPP1015149 (C.R.).
Author contributions
C.R. conceived the study, designed all the immunological experiments, data analysis, and wrote the manuscript. S.T. conducted all the APC studies. J.S. designed and constructed the influenza-HIV challenge virus and critical evaluation of the manuscript. R.J.J. designed and constructed all the IL-13Rα2 co-expression (adjuvanted) vaccines, also prepared the Supplementary Figure 1 online and critical evaluation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article
Cite this article
Ranasinghe, C., Trivedi, S., Stambas, J. et al. Unique IL-13Rα2-based HIV-1 vaccine strategy to enhance mucosal immunity, CD8+ T-cell avidity and protective immunity. Mucosal Immunol 6, 1068–1080 (2013). https://doi.org/10.1038/mi.2013.1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2013.1
This article is cited by
-
STAT3 determines IL-4 signalling outcomes in naïve T cells
Scientific Reports (2021)
-
Unique IL-13Rα2/STAT3 mediated IL-13 regulation detected in lung conventional dendritic cells, 24 h post viral vector vaccination
Scientific Reports (2020)
-
Mucosal IL-4R antagonist HIV vaccination with SOSIP-gp140 booster can induce high-quality cytotoxic CD4+/CD8+ T cells and humoral responses in macaques
Scientific Reports (2020)
-
Mucosal and systemic SIV-specific cytotoxic CD4+ T cell hierarchy in protection following intranasal/intramuscular recombinant pox-viral vaccination of pigtail macaques
Scientific Reports (2019)
-
Vaccination route can significantly alter the innate lymphoid cell subsets: a feedback between IL-13 and IFN-γ
npj Vaccines (2018)
