
Abstract
Eosinophilic esophagitis (EoE) has been increasingly recognized as a unique clinicopathological entity over the past two decades. In this short time, the mechanisms of a complex disease have begun to emerge. Patient studies suggest that EoE is an immunologic disease related to atopy. At the cellular level, eosinophils, mast cells, and B and T lymphocytes are increased in the esophageal mucosa in a patchy distribution throughout the length of the esophagus. Laboratory investigations have implicated aeroallergens, food allergens, and a unique T helper type 2 cytokine profile. EoE appears to be an antigen-driven hypersensitivity reaction characterized by a mixed IgE-dependent/delayed-type reaction and a distinct cascade of cytokines and growth factors. The causative events that lead to EoE in humans remain unknown.
Similar content being viewed by others

Introduction
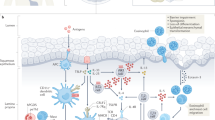
Esophagitis is characterized by inflammatory cells and morphological changes to the esophageal epithelium, including basal zone hyperplasia and elongation of vascular papillae. Esophagitis can be caused by a variety of insults including acid reflux disease and infection, but over the past two decades a new disease termed eosinophilic esophagitis (EoE) has emerged. A clinical review and consensus statement1 defined EoE as ≥15 intraepithelial eosinophils per × 400 high-power field along with the exclusion of other causes of esophagitis. In EoE, eosinophils are recruited to the esophagus and are activated, releasing inflammatory mediators, leading to tissue injury (Figure 1). Clinical manifestations are varied but commonly include dysphagia, food impaction, and retrosternal or epigastric pain.2 The patient population is predominantly male and many patients have concomitant atopic disease.3, 4

Proposed pathophysiology of eosinophilic esophagitis (EoE). Eosinophils are drawn to the esophagus by chemokines. Degranulation of eosinophils is the primary mechanism, which leads to inflammation and clinical manifestations of disease.
EoE appears to be increasing in incidence,5, 6, 7, 8, 9, 10 along with other immunologic diseases.11 The molecular mechanisms of this disease are the subject of much ongoing investigation. Multiple cytokine-driven mechanisms specific to EoE have been identified along with a unique gene expression profile. A comprehensive understanding of the etiology and pathogenesis of EoE will help direct future therapeutic strategies. In this review, we will summarize what is currently known about the molecular mechanisms of this immunologic disease from human and animal studies.
Cell Types Involved in EoE
The eosinophil
The defining histological characteristic of EoE is high numbers of intraepithelial eosinophils. Over time, eosinophils likely evolved as specialized antiparasitic leukocytes. In the western world, eosinophils are most commonly associated with atopic inflammation but are also present during a variety of diseases including autoimmune disorders, inflammatory bowel disease, as well as drug and food sensitivity. Eosinophils derive from CD34+ myeloid precursors in bone marrow and their development is regulated by interleukin (IL)-5.12 IL-5 is also the principle cytokine regulating blood eosinophil number. Eosinophils survive for ∼8–18 h in circulation.13 Survival time in tissue is for ∼2–14 days,14 and can vary depending on the local cytokine milieu.15
In general, eosinophil recruitment from blood to tissue in humans is influenced by a host of proinflammatory and chemoattractant cytokines, secreted by an organ in response to IL-4 and IL-13.12 The eotaxin proteins (eotaxin-1, -2, and -3, also known as CCL (chemokine (C-C motif) ligand)11, CCL24, and CCL26, respectively) and leukotriene B4 are highly potent eosinophil chemoattractants. The three eotaxins are not closely related genetically, but have similar properties with regard to eosinophil recruitment. Eosinophils expressing very late antigen-4 and P-selectin glycoprotein ligand-1 bind to vascular cell adhesion molecule-1 and P-selectin, respectively, and migrate into tissue.16, 17
Chemoattractant cytokines and chemokines draw eosinophils into tissue via integrin-mediated migration.18 Eosinophils express the integrins α4β1, α6β1, αMβ2, αLβ2, αXβ2, αDβ2, and α4β7.18 Activation of eosinophils can result from interaction with cytokines (including IL-3, IL-5, and granulocyte macrophage-colony-stimulating factor), lipid and inflammatory mediators, and from immunogenic antigen exposure, although this may not occur directly.13 Activation leads to release of arachadonic acid metabolites and reactive oxygen species followed by secondary release of cytokines, growth factors, and other protein mediators such as eosinophil-derived neurotoxin, eosinophil peroxidase, eosinophil cationic protein, and major basic protein (MBP).19 These secondary proteins are generally cytotoxic and further increase local reactive oxygen species, in addition to stimulating mast cell degranulation. MBP is the most common mediator and accounts for >50% by mass of the eosinophil secondary granule.20 Many cytokines are produced by eosinophils such as interleukins, interferons, chemokines, and growth factors.21 The primary function of the eosinophil may be to act as an end-effector cell in antiparasite and atopic disease.
Normally, eosinophils are not present in the esophageal mucosa. In patients with EoE, esophageal eosinophil counts fluctuate but remain elevated for long periods.22, 23 Intraepithelial eosinophils can be seen throughout the mucosa or clustered in discrete regions, occasionally forming microabscesses, defined as ≥4 eosinophils in a cluster.1 Eosinophils display signs of activation and degranulation even as they extravasate into the esophageal mucosa.24 The recruitment of eosinophils and subsequent release of granule mediators are likely responsible for the accrued injury to the esophagus in EoE, which in turn leads to symptoms.
Eosinophil recruitment and the eotaxins
A highly conserved gene expression profile, which includes increased eotaxin-3, is seen in EoE patients, independent of other clinical variables such as age, gender, and atopic status.25 This expression profile is not associated with gastroesophageal reflux disease (GERD), which is an interesting finding as esophagitis caused by acid reflux, especially in children, is also characterized by eosinophils.25 Many studies have investigated expression of eotaxin-3 in patients with EoE. Blanchard et al.25 found increased eotaxin-3 (mRNA and protein) in esophageal biopsies of EoE patients and these correlate with eosinophil counts. A correlation (r=0.32) was found by Konikoff et al.26 between eotaxin-3 protein level in peripheral blood and esophageal eosinophilia, but eotaxin-1 and -2 protein levels did not correlate. Bhattacharya et al.27 found increased eotaxin-1, -2, and -3 in EoE. This group did not find a correlation between eotaxin-3 and tissue eosinophilia in EoE. Another study did not find increased eotaxin-3 mRNA in EoE, but no housekeeping gene was used to control for relative sample differences.28 Vascular cell adhesion molecule-1 expression is increased on the esophageal endothelium of patients with EoE and correlates positively (r=0.61) with infiltrating eosinophil number.29 Application of IL-15 to esophageal epithelial cells (which express IL-15Rα) induces eotaxin-1, -2, and -3 expression.30 IL-15 is a cytokine related to IL-2 in its structure and ability to potentiate activated T-cell responses. IL-15Rα-deficient mice were protected from experimental EoE, even while the significant lung eosinophilia associated with this model persisted.30
A single-nucleotide polymorphism in the untranslated region of the eotaxin-3 gene is associated with patients who have EoE.25 In patients taking topical fluticasone for EoE, eotaxin-1 and -3 expression was decreased from pretreatment levels, although only the change in eotaxin-3 reached statistical significance.31
Animal studies have found that intratracheal IL-13 administration resulted in dose- and eotaxin-dependent esophageal eosinophilia,32 although this finding may be indirect as IL-5 knockout mice did not develop experimental EoE after the same treatment.32 IL-15 is also able to induce eotaxin expression in the murine esophagus.30 In an ex vivo model, using isolated rings of esophageal tissue, IL-4 and IL-13 were directly able to induce eotaxin-1, followed by eotaxin-2 expression.33 The transcription factor signal transducer and activator of transcription 6 (STAT6) is activated by IL-4 and IL-13 in these models.32, 33 Eotaxin induction followed by esophageal eosinophilia was also observed in mice exposed to aerosolized insect allergens.34 Eotaxin-deficient animals exposed to the same allergens did not develop experimental EoE.34 No gene homologous to human eotaxin-3 has been found in mice.
C-C chemokine receptor 3 (CCR3)
The cysteine–cysteine motif containing chemokine receptor 3, CCR3, receives signals from multiple T helper type 2 (Th2)-associated chemokines including eotaxin-1, -2, -3, and CCL5 (RANTES (regulated upon activation, normal T-cell expressed, and secreted)).35 In the esophagus, expression of CCR3 is increased27 and correlates positively with esophageal eosinophil number (r=0.71) and biopsy eotaxin-3 message (r=0.68).36 Murine experimental EoE induced by intranasal exposure to either the mold Aspergillus fumigatus or dust mite or cockroach antigen, requires CCR3 to illicit esophageal eosinophilia.25, 34 The chemokine CCL5 (RANTES) is increased on inflammatory cells in biopsies from patients with EoE and may contribute to lymphocyte and eosinophil chemotaxis.37
Eosinophil granule products
In EoE, inflammatory products from eosinophil secondary granules include: eosinophil-derived neurotoxin,38 eosinophil peroxidase,39 eosinophil cationic protein,40 and MBP.41 Eosinophil-derived MBP acts on esophageal epithelial cells in vitro, leading to secretion of fibroblast growth factor 9 (FGF9). FGF9 acts through an autocrine mechanism to induce basal cell hyperplasia.42 Eosinophils in EoE also express transforming growth factor-β1 (TGF-β1), which is known to induce fibrosis.29 This growth factor activates SMAD2/3 signaling in epithelial and lamina propria cells.29 Thus, it appears that eosinophils can contribute to the tissue changes associated with EoE.
Other eosinophil products
Leukotrienes are known to have a role in eosinophil-derived inflammation in other tissues; however, cyclooxygenase-2 expression is downregulated in EoE.43 The concentration of leukotrienes in EoE pediatric patient biopsies is not increased over normal patient biopsies, whereas children with eosinophilic gastroduodenitis did have increased leukotriene levels.44 These findings support a unique mechanism of EoE pathogenesis that is distinct from other gastrointestinal diseases with eosinophilia.
The mast cell
Mast cells are derived from CD34+ progenitors in bone marrow, the same granulocyte precursor cells as eosinophils. Mast cell differentiation is regulated by the surface c-kit receptor (CD117), which is activated by stem cell factor.45 The stem cell factor also induces mast cell chemotaxis and proliferation and increases longevity.45 Mast cells are not easily seen on routine pathology stains and are commonly identified by immunohistochemistry for tryptase.46 Mast cells depend on α4β7 integrin to enter the gastrointestinal mucosa.47 Unlike eosinophils, mast cells mature and undergo mitosis in tissue.
The best-characterized function of mast cells involves their role in immunoglobulin E (IgE)-mediated immediate hypersensitivity, a hallmark of allergic disease. IgE binds to its high-affinity receptor (FcɛRI) on the mast cell surface and can then bind to divalent antigen, leading to activation and degranulation. Many products of mast cell activation, including histamine, cytokines, and eicosanoids, are known to attract eosinophils.48
In the normal esophageal mucosa, mast cells reside at the basement membrane and in the lamina propria.49 Intraepithelial mast cell numbers are increased in EoE.28, 37, 50, 51 When aerosolized ovalbumin was used to induce esophageal eosinophilia in guinea pigs, mast cells preceded eosinophils into the epithelium,52 as they do in other tissues.53 Additionally, in this model, histamine release peaked 2 min after ovalbumin exposure and was followed by sustained C-fiber hyperexcitability.54 In a mouse model of eosinophilic esophagitis, mast cell number was significantly increased after intranasal exposure to cockroach and dust mite allergen.34 IgE is bound to mast cells in EoE,55 and this interaction may be responsible for the increased mast cell degranulation that occurs in EoE.56 As mast cells are present in the esophageal mucosa before the inflammatory response of EoE, and based on the findings from the guinea pig model, it is possible that mast cells are involved in the etiology of EoE. Mast cell counts in EoE correlate with eotaxin-3 level,25 intraepithelial eosinophil count,57 and basal zone hyperplasia57 (r=0.42, 0.37, and 0.67, respectively). Gene array results revealed 301 genes whose expression correlates with esophageal mast cell count.56 Expression of carboxypeptidase A3 has the strongest relation to mast cell number and degranulation activity (number per high-power field and degranulation).56
It should be noted that mast cells and eosinophils are features of reflux-induced esophagitis,58 infectious esophagitis,59 and Barrett's esophagus.60 Therefore, mast cells and eosinophils may be nonspecific features of esophagitis in general, with increased numbers seen in atopic individuals.
Mast cells in EoE therapy
In a study by Lucendo et al.,50 mast cell number was not statistically decreased in adult EoE patients after 3 months of glucocorticoid therapy (fluticasone propionate, 500 μg, b.i.d), but IgE+ cell count was significantly reduced. In a randomized clinical trial for pediatric EoE, mast cells were significantly decreased by 3 months of fluticasone treatment (880 μg, b.i.d.).61 Treatment of EoE with a mast cell stabilizer (cromoglycic acid, 100 mg q.i.d for 1 month) did not have any effect on eosinophil counts or symptoms in a small pediatric cohort (14 patients).62 Alternatively, biologic therapy using an antibody directed against IL-5 did not significantly decrease mast cell number vs. placebo in adult EoE patients.63
The B lymphocyte
B lymphocytes are bone marrow-derived CD20+ cells that are responsible for antibody production. Naive B cells express IgM isotype antibody and undergo class switching following activation to other isotypes including IgG, which is the most abundant circulating antibody, and IgE, which has a major role in atopic disease.64 Once IgE is secreted from B cells, its primary mechanism of action is to bind to mast cells in tissue, awaiting antigen. The activation of IgE+ B cells is dependent on Th2 cells.65
In EoE, B cells identified by CD20 immunostaining are present in the esophageal mucosa,37, 55 but are not present in higher numbers than in GERD patients.37, 50 Mucosal biopsies from patients with no esophageal pathology do not have intraepithelial B cells.37, 55 Intraepithelial B cell number correlates with mast cells (r=0.74), but not eosinophils.55 B-cell class switch to IgE, as evidenced by ɛ switch circles, may occur in the esophageal mucosa in a subset of EoE patients.55 In experimental murine EoE, induced by intranasal A. fumigatus, the resident B-cell population is approximately doubled.66 In this model, mice deficient in B cells have significantly decreased eosinophil numbers in their bronchoalveolar lavage fluid, but eosinophil numbers in the esophagus remain the same.66 It is likely that B cells have a role in EoE pathogenesis, although the evidence implies that they are not a critical component of the disease development.
Immunoglobulins
IgE has a central part in the pathogenesis of many atopic disorders,13 but its role in eosinophilic gastrointestinal diseases is unclear. Clinically, EoE is sometimes associated with IgE-mediated disorders of the airway or gastrointestinal tract.67 IgE-positive cells are increased in the esophageal mucosa of some patients with EoE,51, 55 but not detectable in esophageal biopsies from normal patients,37 those with GERD,50 or those on fluticasone therapy.50 IgE binds to mast cells in EoE (as demonstrated by double labeling for c-kit and IgE).55 The presence of IgE+c-kit– cells in the esophageal mucosa in EoE implies the presence of IgE-secreting B cells, although evidence for this is indirect.55 Mice sensitized with epicutaneous ovalbumin followed by a single intranasal administration develop EoE and specific IgG1 for ovalbumin and have markedly increased total IgE levels.68 This process is STAT6, IL-4, and IL-13 dependent, but is independent of IL-5.68 Similar induction of specific IgG1 and total IgE can be found in mice exposed to A. fumigatus, and dust mite and cockroach allergens.34, 66 These studies support a role for B cells in EoE that is independent of eosinophils.
A significant number of EoE patients have food and environmental sensitization, and IgE-positive cells in the esophageal mucosa; however, it is unknown how IgE contributes to the disease. Patients with established IgE-mediated food allergy who are then diagnosed with EoE are typically avoiding those foods. Elimination of food antigen with amino acid-based elemental diet is an effective treatment in some individuals. This may have important implications for the pathophysiology of EoE, although elemental diets have effects beyond the removal of antigenic peptides. It is possible that the presence of IgE-positive B cells and mast cells in the esophageal mucosa is incidental. Future studies will be required to fully understand the contribution of IgE to EoE, and to identify potential food or environmental antigens that trigger the inflammatory response in individual patients.
The T lymphocyte
A variety of intraepithelial T cells are found in the normal esophagus and are more numerous in esophagitis. In EoE, CD3+, CD4+, and CD8+ T cells are increased in the esophageal mucosa and can be decreased with corticosteroid treatment.50, 69 Peripheral blood mononuclear cells (which are ∼75% T cells) have been isolated from EoE patients and stimulated with phytohemagglutinin, which caused increased IL-13 production but not IL-5 or interferon-γ production.37 In a subsequent study, some EoE patient peripheral blood mononuclear cells produced IL-5 and IL-13 in response to specific allergens.70 This experimental model differs from subsequent studies (discussed below), which measured cytokine expression from unstimulated peripheral blood mononuclear cells or in specific T-cell subsets. One study of eosinophilic gastrointestinal disease and food allergy found that the Th2 response can be subdivided into an IL-5+ response, associated with the eosinophilic disease, and an IL-5– response, associated with food allergy.71
T helper cells
EoE is a disease associated with a Th2-type inflammatory response.37, 72 Th2 cytokines such as IL-4, IL-5, and IL-13 are expressed by T cells from peripheral blood of EoE patients and are increased in esophageal mucosal biopsies.55, 73 It is interesting to note that Th1-associated cytokines are also part of the inflammatory response in EoE. Th1 cytokines present include tumor necrosis factor-α (expressed by esophageal epithelial cells)37 and interferon-γ (upregulated in peripheral blood T cells,71 increased in esophageal mucosa biopsies,74 and increased following IL-15 treatment of T helper cells30). Increased T helper cells are part of experimental EoE in mice.66 T helper cell-deficient mice do not experience esophageal eosinophilia but do have epithelial basal zone hyperplasia.66 We have recently shown that the esophageal epithelial cell line, HET-1A, is capable of antigen presentation to CD4+ T helper cells.74 T cells clonally multiplied in response to epithelial cells primed with both antigen and interferon-γ.74
IL-4
In general, IL-4 is thought to be responsible for initiating the Th2 response through differentiation of naive T helper cells into Th2 type cells. The initial source of IL-4 in atopic disease remains unclear, but it is produced by T helper cells during the inflammatory response.75 IL-4 mRNA is increased in the mucosa of patients with EoE.55 T helper cells express IL-4 in response to IL-15 treatment.30
IL-5
IL-5 has a major role in many eosinophil-related disorders by acting on the bone marrow as an eosinophil differentiation factor and activator. In EoE, mRNA75 and protein expression73 of IL-5 is significantly increased, but plasma IL-5 does not correlate with esophageal eosinophil number.26 Mice that overexpress IL-5 have intense esophageal eosinophilia.77 Eotaxin-deficient mice that overexpress IL-5 can still develop experimental EoE, although to a much lesser extent than isogenic wild-type mice.77 Variable downregulation of IL-5 expression is seen following fluticasone treatment.78
IL-13
IL-13 appears to activate the local tissue inflammatory response in Th2-associated diseases. In the esophagus, IL-13 is increased at the mRNA level in EoE patient biopsies.55, 79 IL-13 decreases esophageal epithelial cell differentiation, a process that may be critical for maintaining the barrier function of the esophageal mucosa.80 Additionally, IL-13 acts through STAT6 on esophageal epithelial cells to upregulate eotaxin-1,81 eotaxin-2,81 and eotaxin-3 production.79, 80 In mice, intratracheal administration of IL-13 caused eosinophil accumulation in a dose-dependent manner,32 whereas human anti-IL-13 (CAT-354) was able to significantly decrease murine experimental EoE.82 IL-13-deficient animals develop EoE in response to ovalbumin but not in response to aerosolized A. fumigatus.68 IL-13 is also expressed in circulating eosinophils in EoE patients.83
Cytotoxic T cells
The increased number of CD8+ cells in the esophageal mucosa in EoE50 is decreased following fluticasone therapy.61, 78 This effect is more marked in nonallergic EoE patients.84 A. fumigatus-induced murine EoE features increased cytotoxic T cells.66 Cytotoxic T cell-deficient mice still develop characteristic eosinophilia and basal zone hyperplasia in this model.66
T regulatory cells
“Regulatory” is a general term for multiple lineages of T cells (e.g., FoxP3+ and CD45RBlow cells) that secrete anti-inflammatory cytokines and function to dampen the immune response. Because of the multiplicity and similarity in surface markers between regulatory and pathogenic T cells, immune inhibitory cells have proven difficult for immunologists to identify and study. Increasingly, an imbalance in T regulatory cells is being implicated in chronic immunologic diseases.85 In mice with A. fumigatus-induced EoE, CD4+CD45RBlow regulatory T helper cells are decreased and express less IL-2 compared with control animals.86 This finding identifies a potential role for T regulatory cell imbalance in EoE. Reconstitution of T cell-deficient mice (Rag2–/–) with a relatively small number (3 × 104) of CD4+ cells causes severe esophageal eosinophilia.87 Disease can be prevented by increasing the number of CD4+ cells (2 × 106) or pretreating animals with CD25+ T regulatory cells.87 Thus, the interaction between lymphocyte subtypes may have a role in the etiology of EoE.
A pediatric study of 10 normal, 8 GERD, and 10 EoE cases revealed increased T regulatory (FoxP3+) cells in both GERD and EoE biopsies, but there was no statistically significant difference in T regulatory cell numbers between GERD and EoE.88 This implies that the T regulatory cell response in the esophagus may be nonspecific. The divergent findings regarding regulatory T-cell alterations in the animal model86 and human disease88 may be because of differences in study design. Each study investigates a different T regulatory cell (CD4+CD45RBlow in the mouse model and FoxP3+ in the human disease). In the animal model, the findings are also based on circulating, tissue, and regional lymph node T-cell populations, whereas only tissue biopsies were examined in the human study.
A subset of T cells, identified by “cutaneous lymphocyte antigen” expression are present in EoE and unchanged by fluticasone treatment.69 The function of cutaneous lymphocyte antigen-positive T cells is unknown, but its expression is associated with homing to the epidermis89 and also with memory function.90
Antigen-presenting cells (APCs)
Dendritic cells and macrophages present antigen to lymphocytes and provide a link between the innate and adaptive immune systems. The number of APCs seen is not a good indicator of their function, as APCs may activate an immune response in tissue or in draining lymph nodes. CD1a+ dendritic cells are present in low numbers in the esophageal mucosa.50 The number of CD1a+ cells is unchanged in normal, untreated EoE, and post-fluticasone therapy adult EoE biopsies.50 In children with EoE, CD1a+ cells are increased in the proximal esophagus but decrease to normal levels after fluticasone treatment. These cells are not increased in the distal esophagus.69
Increased numbers of what are mostly dendritic cells (CD45+CD11c+MHC class II+ cells) and mostly macrophages (CD45+CD11b+MHC class II+ cells) are present in the esophageal mucosa of mice with A. fumigatus-induced EoE.30 It is important to note that the surface markers used to identify the APC subtypes in this study may include additional cell types. Presentation of antigen by professional APCs may be an initiating event in the cytokine cascade that leads to EoE.
We have recently characterized esophageal epithelial cells as potential nonprofessional APCs in EoE.74 Esophageal epithelial cells process and present antigen in vitro, and epithelial MHC class II expression in patient biopsies points to the possibility that antigen presentation has a role in the pathogenesis of EoE.
The epithelial cell
The esophageal epithelium is a nonkeratinized, stratified squamous barrier lining the luminal surface. Regenerative basal cells lie in the layers closest to the basement membrane and mature and replace the superficial cells as they are sloughed off at the luminal surface.49 Basal cells express KI-67 and are commonly identified by the KI-67 immunostaining (a cellular proliferation marker), 5-bromodeoxyuridine incorporation (metabolic activity), or simply by morphological features (high nucleus/cytoplasm ratio).
Basal hyperplasia
Hyperplasia of the basal cells is a characteristic of esophagitis in general. Most studies demonstrate that epithelial cell hyperplasia is more marked in EoE vs. GERD,91 although one study found no difference.43 Basal cell hyperplasia correlates with intraepithelial eosinophil (r=0.68) and mast cell (r=0.71) number.25 EoE-induced basal cell hyperplasia is decreased by fluticasone propionate treatment84 in both the proximal and distal esophagus.61 In mice repeatedly challenged with A. fumigatus or intratracheal IL-13, esophageal eosinophilia and basal hyperplasia occur in a STAT6/IL-5-dependent manner.32, 92, 93 These data indicate that the inflammation of EoE affects the functions of esophageal epithelial cells.
Growth factors
Multiple growth factors known to contribute to epithelial cell proliferation have been linked to the reactive changes that occur in EoE. FGF9 is secreted by esophageal epithelial cells in vitro in response to the eosinophil product MBP.42 FGF9 then acts in an autocrine manner to increase epithelial cell proliferation.42 FGF9 is increased in EoE and correlates with basal hyperplasia (r=0.51). TGF-β1 activates receptor-regulated SMAD2/3 signaling in epithelial cells and may then act to alter epithelial cell gene expression.29 Periostin, a cell adhesion factor, is overexpressed in gene arrays of patients with EoE25 and correlates with eosinophil number (r=0.64).94 Periostin is secreted from primary esophageal epithelial cells when they are treated with IL-13 in a dose-dependent manner, but this production is inhibited by concurrent TGF-β treatment.94
Cytokines
A remarkable similarity exists between gene expression profiles of mucosal biopsies from patients with EoE and primary esophageal epithelial cells treated with IL-13.79 STAT6-dependent eotaxin-3 expression (and protein secretion) is increased in primary esophageal epithelial cell cultures treated with IL-13 and in esophageal squamous carcinoma cell lines (TE-1, -6, -7, and -13) in an IL-13 dose-dependent manner.79 IL-13-induced murine EoE is also enhanced by deletion of IL-13 receptor α2 (IL-13Rα2), implying that cytokine receptor subtypes may alter the expression of EoE.95 The IL-13R is a dimer composed of the IL-4Rα subunit and either the functional IL-13Rα1 or the decoy IL-13Rα2 subunit. IL-13Rα2 is thought to function as a decoy receptor, which thwarts the cytokine's function by preventing STAT6 activation when IL-13 binds. Epithelial differentiation-associated genes such as filaggrin and SPRR3 (small proline rich region protein 3) are downregulated in both EoE and in primary epithelial cell cultures treated with IL-13.80 Biopsies from EoE patients taking glucocorticoids have a gene expression profile similar to normal patients.79 Glucocorticoids act on esophageal epithelial cells to inhibit IL-13 induction of eotaxin-3.96 This process is mediated by glucocorticoid-induced expression of FKBP51 (FK506 binding protein 5).96 Esophageal epithelial cells express tumor necrosis factor-α, but not IL-5.37
Fibrosis and fibroblasts
Fibrosis has been associated with EoE.57 To our knowledge, the role of fibroblasts has not been directly studied in EoE. In pediatric patients, fibrosis, TGF-β1, expression and SMAD2/3 phosphorylation are all increased,41 but can be decreased by corticosteroid treatment.97 Mucin 5AC and TGF-β1 mRNA expression is increased in both A. fumigatus-induced murine EoE and in the human disease.93 In the murine model of EoE, animals treated with A. fumigatus had increased collagen thickness.93
Other cell types
The roles of basophils, neutrophils, and neurons have not, to our knowledge, been investigated in EoE. Neutrophils are rare in EoE,98 but this does not preclude them from having a functional role in disease pathogenesis.
Genetic and Systems Approaches to EoE
Gene arrays and genome-wide association study
Gene array techniques have identified eotaxin-3 polymorphisms25 and cytokine55, 79 and growth factor42, 94 overexpression and gene alterations after therapy79 in EoE. The finding that EoE is associated with a highly conserved gene expression profile25 has allowed for a genome-wide association study. Instead of identifying and investigating individual candidate genes in a population, a genome-wide association study can study the entire gene expression profile of a population. To date, 351 EoE patients and 3,104 control patients have been analyzed.99 EoE patients have various single-nucleotide polymorphisms in a discrete region on the long arm of chromosome 5 (5q22) associated with the TSLP (thymic stromal lymphopioetin) and WDR36 (WD repeat-containing protein 36) genes.99 TSLP is overexpressed in EoE patients, whereas WDR36 is not.99 Genome-wide association studies have identified TSLP gene variation as a common alteration in patients with EoE. The biological function of TSLP overexpression in EoE has yet to be determined.
Biologic therapy
Monoclonal antibodies targeting integral cytokines have clinical benefit in chronic immune disorders,100 and help to elucidate underlying disease mechanisms. Humanized monoclonal antibody directed against IL-5 (mepolizumab) has shown promise in treating various eosinophil-related disorders.101 In a double blind, placebo-controlled trial in adult EoE, mepolizumab did reduce esophageal eosinophilia but not below diagnostic levels, nor did clinical features improve.63 The results of a larger pediatric trial of mepolizumab for EoE have not yet been published.
Alternative biologic therapies for EoE are being investigated (Table 1). A second humanized monoclonal antibody against IL-5, reslizumab, is in trials for treatment of pediatric EoE.102 IL-13 has a central role in EoE pathogenesis and the anti-human IL-13 monoclonal antibody CAT-354 diminished IL-13-induced EoE in mice.82 Additional biologic clinical trials targeting prostaglandin receptor DP2 (OC000459), IgE (omalizumab), and IL-13 (QAX576) are registered. A trial of tumor necrosis factor-α antibody (infliximab) for severe, refractory EoE in three patients did not alter histology or symptoms.103
Conclusions: the Rapidly Evolving Concept of EoE
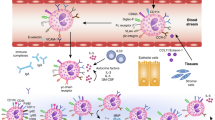
EoE has emerged as a distinct disease entity since the early 1990s, and since then, a large body of research has been generated. Human studies from pediatric and adult populations as well as animal models have begun to elucidate the cellular and molecular mechanisms of this disease. A system of complex immunologic processes is emerging, which feature the interaction of both immune and nonimmune cell types (Figure 2).

The cellular changes to the esophagus that occur with eosinophilic esophagitis (EoE). Marked changes include infiltration of eosinophils and basal zone hyperplasia. Other cellular features of EoE include elongated vascular papillae, B lymphocytes, increased mast cells, and increased T lymphocytes (both CD4+ and CD8+ cells). Dendritic cell number does not change.
Although the inciting factor(s) are unknown, this disorder appears to be triggered most commonly in atopic individuals. This may be related to a milieu of circulating or tissue-derived Th2 cytokines and chemokines that attract and activate eosinophils. Eosinophils themselves produce an array of mediators, including cytokines and other proinflammatory proteins that encourage more eosinophil activation and recruitment. This process is likely responsible for the ongoing tissue damage and subsequent signs and symptoms of this chronic disease.
Despite the current knowledge, many questions remain. What causes EoE? What is the role of food and environmental allergens? Why are we seeing increasing numbers of patients with this disease? Is EoE the same disease in all individuals or are there differences in the mechanisms of disease that lead to phenotypic differences in patients? There are many confounding factors that may have a role including gender, coexisting atopy, and age. Future studies will continue to build upon the work highlighted in this review.
References
Furuta, G.T. et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 133, 1342–1363 (2007).
Odze, R.D. Pathology of eosinophilic esophagitis: what the clinician needs to know. Am. J. Gastroenterol. 104, 485–490 (2009).
Wang, F.Y., Gupta, S.K. & Fitzgerald, J.F. Is there a seasonal variation in the incidence or intensity of allergic eosinophilic esophagitis in newly diagnosed children? J. Clin. Gastroenterol. 41, 451–453 (2007).
Almansa, C. et al. Seasonal distribution in newly diagnosed cases of eosinophilic esophagitis in adults. Am. J. Gastroenterol. 104, 828–833 (2009).
Noel, R.J., Putnam, P.E. & Rothenberg, M.E. Eosinophilic esophagitis. N. Engl. J. Med. 351, 940–941 (2004).
Straumann, A. & Simon, H.U. Eosinophilic esophagitis: escalating epidemiology? J. Allergy Clin. Immunol. 115, 418–419 (2005).
Cherian, S., Smith, N.M. & Forbes, D.A. Rapidly increasing prevalence of eosinophilic oesophagitis in Western Australia. Arch. Dis. Child 91, 1000–1004 (2006).
Croese, J. et al. Clinical and endoscopic features of eosinophilic esophagitis in adults. Gastrointest. Endosc. 58, 516–522 (2003).
Dalby, K. et al. Eosinophilic oesophagitis in infants and children in the region of southern Denmark: a prospective study of prevalence and clinical presentation. J. Pediatr. Gastroenterol. Nutr. 51 (2010).
Prasad, G.A. et al. Epidemiology of eosinophilic esophagitis over three decades in Olmsted County, Minnesota. Clin. Gastroenterol. Hepatol. 7, 1055–1061 (2009).
Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920 (2002).
Rosenberg, H.F., Phipps, S. & Foster, P.S. Eosinophil trafficking in allergy and asthma. J. Allergy Clin. Immunol. 119, 1303–1310 (2007).
Stone, K.D., Prussin, C. & Metcalfe, D.D. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 125, S73–S80 (2010).
Lim, L.H., Burdick, M.M., Hudson, S.A., Mustafa, F.B., Konstantopoulos, K. & Bochner, B.S. Stimulation of human endothelium with IL-3 induces selective basophil accumulation in vitro. J. Immunol. 176, 5346–5353 (2006).
Simon, H.U., Yousefi, S., Schranz, C., Schapowal, A., Bachert, C. & Blaser, K. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J. Immunol. 158, 3902–3908 (1997).
Okigami, H. et al. Inhibition of eosinophilia in vivo by a small molecule inhibitor of very late antigen (VLA)-4. Eur. J. Pharmacol. 559, 202–209 (2007).
Edwards, B.S., Curry, M.S., Tsuji, H., Brown, D., Larson, R.S. & Sklar, L.A. Expression of P-selectin at low site density promotes selective attachment of eosinophils over neutrophils. J. Immunol. 165, 404–410 (2000).
Barthel, S.R., Johansson, M.W., McNamee, D.M. & Mosher, D.F. Roles of integrin activation in eosinophil function and the eosinophilic inflammation of asthma. J. Leukoc. Biol. 83, 1–12 (2008).
Afshar, K., Vucinic, V. & Sharma, O.P. Eosinophil cell: pray tell us what you do!. Curr. Opin. Pulm. Med. 13, 414–421 (2007).
Plager, D.A., Stuart, S. & Gleich, G.J. Human eosinophil granule major basic protein and its novel homolog. Allergy 53, 33–40 (1998).
Hogan, S.P. et al. Eosinophils: biological properties and role in health and disease. Clin. Exp. Allergy 38, 709–750 (2008).
Spergel, J.M. et al. 14 years of eosinophilic esophagitis: clinical features and prognosis. J. Pediatr. Gastroenterol. Nutr. 48, 30–36 (2009).
Straumann, A. The natural history and complications of eosinophilic esophagitis. Gastrointest. Endosc. Clin. N. Am. 18, 99–118 (2008).
Justinich, C.J., Ricci, A. Jr., Kalafus, D.A., Treem, W.R., Hyams, J.S. & Kreutzer, D.L. Activated eosinophils in esophagitis in children: a transmission electron microscopic study. J. Pediatr. Gastroenterol. Nutr. 25, 194–198 (1997).
Blanchard, C. et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J. Clin. Invest. 116, 536–547 (2006).
Konikoff, M.R. et al. Potential of blood eosinophils, eosinophil-derived neurotoxin, and eotaxin-3 as biomarkers of eosinophilic esophagitis. Clin. Gastroenterol. Hepatol. 4, 1328–1336 (2006).
Bhattacharya, B. et al. Increased expression of eotaxin-3 distinguishes between eosinophilic esophagitis and gastroesophageal reflux disease. Hum. Pathol. 38, 1744–1753 (2007).
Gupta, S.K., Fitzgerald, J.F., Kondratyuk, T. & HogenEsch, H. Cytokine expression in normal and inflamed esophageal mucosa: a study into the pathogenesis of allergic eosinophilic esophagitis 42, 22–26 (2006).
Aceves, S.S., Newbury, R.O., Dohil, R., Bastian, J.F. & Broide, D.H. Esophageal remodeling in pediatric eosinophilic esophagitis. J. Allergy Clin. Immunol. 119, 206–212 (2007).
Zhu, X. et al. Interleukin-15 expression is increased in human eosinophilic esophagitis and mediates pathogenesis in mice. Gastroenterology 139, 182–193 (2010).
Lucendo, A.J., De Rezende, L., Comas, C., Caballero, T. & Bellon, T. Treatment with topical steroids downregulates IL-5, eotaxin-1/CCL11, and eotaxin-3/CCL26 gene expression in eosinophilic esophagitis. Am. J. Gastroenterol. 103, 2184–2193 (2008).
Mishra, A. & Rothenberg, M.E. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 125, 1419–1427 (2003).
Neilsen, C.V. & Bryce, P.J. Interleukin-13 directly promotes oesophagus production of CCL11 and CCL24 and the migration of eosinophils. Clin. Exp. Allergy 40, 427–434 (2010).
Rayapudi, M. et al. Indoor insect allergens are potent inducers of experimental eosinophilic esophagitis in mice. J. Leukoc. Biol. 88, 337–346 (2010).
Erin, E.M., Williams, T.J., Barnes, P.J. & Hansel, T.T. Eotaxin receptor (CCR3) antagonism in asthma and allergic disease. Curr. Drug Targets Inflamm. Allergy 1, 201–214 (2002).
Bullock, J.Z. et al. Interplay of adaptive th2 immunity with eotaxin-3/c-C chemokine receptor 3 in eosinophilic esophagitis. J. Pediatr. Gastroenterol. Nutr. 45, 22–31 (2007).
Straumann, A., Bauer, M., Fischer, B., Blaser, K. & Simon, H.U. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J. Allergy Clin. Immunol. 108, 954–961 (2001).
Kephart, G.M. et al. Marked deposition of eosinophil-derived neurotoxin in adult patients with eosinophilic esophagitis. Am. J. Gastroenterol. 105, 298–307 (2010).
Mueller, S., Neureiter, D., Aigner, T. & Stolte, M. Comparison of histological parameters for the diagnosis of eosinophilic oesophagitis versus gastro-oesophageal reflux disease on oesophageal biopsy material. Histopathology 53, 676–684 (2008).
Ooi, C.Y., Day, A.S., Jackson, R., Bohane, T.D., Tobias, V. & Lemberg, D.A. Eosinophilic esophagitis in children with celiac disease. J. Gastroenterol. Hepatol. 23, 1144–1148 (2008).
Aceves, S.S. & Ackerman, S.J. Relationships between eosinophilic inflammation, tissue remodeling, and fibrosis in eosinophilic esophagitis. Immunol. Allergy Clin. North Am. 29, 197–211 (2009).
Mulder, D.J. et al. FGF9-induced proliferative response to eosinophilic inflammation in oesophagitis. Gut 58, 166–173 (2009).
Lewis, C.J., Lamb, C.A., Kanakala, V., Pritchard, S., Armstrong, G.R. & Attwood, S.E. Is the etiology of eosinophilic esophagitis in adults a response to allergy or reflux injury? Study of cellular proliferation markers. Dis. Esophagus 22, 249–255 (2009).
Gupta, S.K. et al. Cysteinyl leukotriene levels in esophageal mucosal biopsies of children with eosinophilic inflammation: are they all the same? Am. J. Gastroenterol. 101, 1125–1128 (2006).
Okayama, Y. & Kawakami, T. Development, migration, and survival of mast cells. Immunol. Res. 34, 97–115 (2006).
Caughey, G.H. Mast cell tryptases and chymases in inflammation and host defense. Immunol. Rev. 217, 141–154 (2007).
Hallgren, J. & Gurish, M.F. Pathways of murine mast cell development and trafficking: tracking the roots and routes of the mast cell. Immunol. Rev. 217, 8–18 (2007).
Minai-Fleminger, Y. & Levi-Schaffer, F. Mast cells and eosinophils: the two key effector cells in allergic inflammation. Inflamm. Res. 58, 631–638 (2009).
Collins, M.H. Histopathologic features of eosinophilic esophagitis. Gastrointest. Endosc. Clin. N. Am. 18, 59–71 (2008).
Lucendo, A.J. et al. Immunophenotypic characterization and quantification of the epithelial inflammatory infiltrate in eosinophilic esophagitis through stereology: an analysis of the cellular mechanisms of the disease and the immunologic capacity of the esophagus. Am. J. Surg. Pathol. 31, 598–606 (2007).
Kirsch, R., Bokhary, R., Marcon, M.A. & Cutz, E. Activated mucosal mast cells differentiate eosinophilic (allergic) esophagitis from gastroesophageal reflux disease. J. Pediatr. Gastroenterol. Nutr. 44, 20–26 (2007).
Yu, S., Stahl, E., Li, Q. & Ouyang, A. Antigen inhalation induces mast cells and eosinophils infiltration in the guinea pig esophageal epithelium involving histamine-mediated pathway. Life Sci. 82, 324–330 (2008).
Kung, T.T. et al. Mast cells modulate allergic pulmonary eosinophilia in mice. Am. J. Respir. Cell Mol. Biol. 12, 404–409 (1995).
Yu, S., Kollarik, M., Ouyang, A., Myers, A.C. & Undem, B.J. Mast cell-mediated long-lasting increases in excitability of vagal C fibers in guinea pig esophagus. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G850–856 (2007).
Vicario, M. et al. Local B cells and IgE production in the oesophageal mucosa in eosinophilic oesophagitis. Gut 59, 12–20 (2010).
Abonia, J.P. et al. Involvement of mast cells in eosinophilic esophagitis. J. Allergy Clin. Immunol. 126, 140–149 (2010).
Chehade, M., Sampson, H.A., Morotti, R.A. & Magid, M.S. Esophageal subepithelial fibrosis in children with eosinophilic esophagitis. J. Pediatr. Gastroenterol. Nutr. 45, 319–328 (2007).
Morganstern, J.A., Wang, M.Y. & Wershil, B.K. Direct evidence of mast cell participation in acute acid-induced esophageal inflammation in mice. J. Pediatr. Gastroenterol. Nutr. 46, 134–138 (2008).
Muller-Stover, I., Richter, J. & Haussinger, D. Infection with gnathostoma spinigerum as a cause of eosinophilic oesophagitis. Dtsch. Med. Wochenschr 129, 1973–1975 (2004).
Wolfsen, H.C., Hemminger, L.L. & Achem, S.R. Eosinophilic esophagitis and Barrett's esophagus with dysplasia. Clin. Gastroenterol. Hepatol. 5, A18 (2007).
Konikoff, M.R. et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology 131, 1381–1391 (2006).
Liacouras, C.A. et al. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin. Gastroenterol. Hepatol. 3, 1198–1206 (2005).
Straumann, A. et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut 59, 21–30 (2010).
Schroeder, H.W., Jr. & Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 125, S41–S52 (2010).
Cambier, J.C. & Getahun, A. B cell activation versus anergy; the antigen receptor as a molecular switch. Immunol. Lett. 128, 6–7 (2010).
Mishra, A., Schlotman, J., Wang, M. & Rothenberg, M.E. Critical role for adaptive T cell immunity in experimental eosinophilic esophagitis in mice. J. Leukoc. Biol. 81, 916–924 (2007).
Simon, D., Marti, H., Heer, P., Simon, H.U., Braathen, L.R. & Straumann, A. Eosinophilic esophagitis is frequently associated with IgE-mediated allergic airway diseases. J. Allergy Clin. Immunol. 115, 1090–1092 (2005).
Akei, H.S., Mishra, A., Blanchard, C. & Rothenberg, M.E. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology 129, 985–994 (2005).
Teitelbaum, J.E. et al. Eosinophilic esophagitis in children: immunopathological analysis and response to fluticasone propionate. Gastroenterology 122, 1216–1225 (2002).
Yamazaki, K. et al. Allergen-specific in vitro cytokine production in adult patients with eosinophilic esophagitis. Dig. Dis. Sci. 51, 1934–1941 (2006).
Prussin, C., Lee, J. & Foster, B. Eosinophilic gastrointestinal disease and peanut allergy are alternatively associated with IL-5+ and IL-5- Th2 responses. J. Allergy Clin. Immunol. 124, 1326–1332 (2009).
Bullock, J.Z. et al. Interplay of adaptive th2 immunity with eotaxin-3/c-C chemokine receptor 3 in eosinophilic esophagitis. J. Pediatr. Gastroenterol. Nutr. 45, 22–31 (2007).
Straumann, A., Bauer, M., Fischer, B., Blaser, K. & Simon, H.U. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J. Allergy Clin. Immunol. 108, 954–961 (2001).
Mulder, D.J., Pooni, A., Mak, N., Hurlbut, D.J., Basta, S. & Justinich, C.J. Antigen presentation and MHC class II expression by human esophageal epithelial cells: Role in eosinophilic esophagitis. Am. J. Pathol in press (2010).
Paul, W.E. & Zhu, J. How are T(H)2-type immune responses initiated and amplified? Nat. Rev. Immunol. 10, 225–235 (2010).
Gupta, S.K., Fitzgerald, J.F., Kondratyuk, T. & HogenEsch, H. Cytokine expression in normal and inflamed esophageal mucosa: a study into the pathogenesis of allergic eosinophilic esophagitis. J. Pediatr. Gastroenterol. Nutr. 42, 22–26 (2006).
Mishra, A., Hogan, S.P., Brandt, E.B. & Rothenberg, M.E. IL-5 promotes eosinophil trafficking to the esophagus. J. Immunol. 168, 2464–2469 (2002).
Lucendo, A.J., De Rezende, L., Comas, C., Caballero, T. & Bellon, T. Treatment with topical steroids downregulates IL-5, eotaxin-1/CCL11, and eotaxin-3/CCL26 gene expression in eosinophilic esophagitis. Am. J. Gastroenterol. 103, 2184–2193 (2008).
Blanchard, C. et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J. Allergy Clin. Immunol. 120, 1292–1300 (2007).
Blanchard, C. et al. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J. Immunol. 184, 4033–4041 (2010).
Neilsen, C.V. & Bryce, P.J. Interleukin-13 directly promotes oesophagus production of CCL11 and CCL24 and the migration of eosinophils. Clin. Exp. Allergy 40, 427–434 (2010).
Blanchard, C., Mishra, A., Saito-Akei, H., Monk, P., Anderson, I. & Rothenberg, M.E. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354). Clin. Exp. Allergy 35, 1096–1103 (2005).
Schmid-Grendelmeier, P. et al. Eosinophils express functional IL-13 in eosinophilic inflammatory diseases. J. Immunol. 169, 1021–1027 (2002).
Noel, R.J. et al. Clinical and immunopathologic effects of swallowed fluticasone for eosinophilic esophagitis. Clin. Gastroenterol. Hepatol. 2, 568–575 (2004).
Maloy, K.J. & Powrie, F. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2, 816–822 (2001).
Zhu, X., Wang, M., Crump, C.H. & Mishra, A. An imbalance of esophageal effector and regulatory T cell subsets in experimental eosinophilic esophagitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G550–G558 (2009).
Milner, J.D., Ward, J.M., Keane-Myers, A. & Paul, W.E. Lymphopenic mice reconstituted with limited repertoire T cells develop severe, multiorgan, Th2-associated inflammatory disease. Proc. Natl. Acad. Sci. USA 104, 576–581 (2007).
Tantibhaedhyangkul, U., Tatevian, N., Gilger, M.A., Major, A.M. & Davis, C.M. Increased esophageal regulatory T cells and eosinophil characteristics in children with eosinophilic esophagitis and gastroesophageal reflux disease. Ann. Clin. Lab. Sci. 39, 99–107 (2009).
Fuhlbrigge, R.C., Kieffer, J.D., Armerding, D. & Kupper, T.S. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature 389, 978–981 (1997).
Clark, R.A. et al. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. 176, 4431–4439 (2006).
Steiner, S.J., Kernek, K.M. & Fitzgerald, J.F. Severity of basal cell hyperplasia differs in reflux versus eosinophilic esophagitis. J. Pediatr. Gastroenterol. Nutr. 42, 506–509 (2006).
Mishra, A., Hogan, S.P., Brandt, E.B. & Rothenberg, M.E. An etiological role for aeroallergens and eosinophils in experimental esophagitis. J. Clin. Invest. 107, 83–90 (2001).
Mishra, A. et al. Esophageal remodeling develops as a consequence of tissue specific IL-5-induced eosinophilia. Gastroenterology 134, 204–214 (2008).
Blanchard, C. et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses 1, 289–296 (2008).
Zuo, L. et al. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13R alpha2-inhibited pathway. J. Immunol. 185, 660–669 (2010).
Caldwell, J.M. et al. Glucocorticoid-regulated genes in eosinophilic esophagitis: a role for FKBP51. J. Allergy Clin. Immunol. 125, 879–888 (2010).
Aceves, S.S. et al. Resolution of remodeling in eosinophilic esophagitis correlates with epithelial response to topical corticosteroids. Allergy 65, 109–116 (2010).
Basseri, B. et al. Redefining the role of lymphocytes in gastroesophageal reflux disease and eosinophilic esophagitis. Dis. Esophagus. 23, 368–376 (2010).
Rothenberg, M.E. et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat. Genet. 42, 289–291 (2010).
Ardizzone, S. & Bianchi Porro, G. Biologic therapy for inflammatory bowel disease. Drugs 65, 2253–2286 (2005).
Walsh, G.M. Mepolizumab and eosinophil-mediated disease. Curr. Med. Chem. 16, 4774–4778 (2009).
Walsh, G.M. Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions. Curr. Opin. Mol. Ther. 11, 329–336 (2009).
Straumann, A., Bussmann, C., Conus, S., Beglinger, C. & Simon, H.U. Anti-TNF-alpha (infliximab) therapy for severe adult eosinophilic esophagitis. J. Allergy Clin. Immunol. 122, 425–427 (2008).
Acknowledgements
We acknowledge support from Kingston General Hospital and the Queen's University Gastrointestinal Diseases Research Unit.
This study was supported by Physicians’ Services Incorporated (PSI) grant PAED-237-09.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Mulder, D., Justinich, C. Understanding eosinophilic esophagitis: the cellular and molecular mechanisms of an emerging disease. Mucosal Immunol 4, 139–147 (2011). https://doi.org/10.1038/mi.2010.88
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.88
This article is cited by
-
A multi-omic analysis reveals the esophageal dysbiosis as the predominant trait of eosinophilic esophagitis
Journal of Translational Medicine (2023)
-
An Overview of the Diagnosis and Management of Eosinophilic Esophagitis
Clinical and Translational Gastroenterology (2016)
-
Diagnosis and treatment of benign inflammatory esophageal diseases
European Surgery (2015)
-
Involvement of the iNKT Cell Pathway Is Associated With Early-Onset Eosinophilic Esophagitis and Response to Allergen Avoidance Therapy
American Journal of Gastroenterology (2014)
-
Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis
Mucosal Immunology (2014)
