
Abstract
The goal of this study was to elucidate the functional role of Nox4 during acute kidney injury (AKI). NADPH oxidases are a major source of reactive oxygen species (ROS) in the kidney in normal and pathological conditions. Among NADPH oxidase isoforms, NADPH oxidase4 (Nox4) is highly expressed in the kidney and has an important role in kidney diseases, such as diabetic nephropathy and renal carcinoma. We previously found that Nox4 expression significantly increased in the toxic AKI model. However, its functional role and mechanism of action in AKI are still unknown. We scavenged ROS with apocynin in vitro and in vivo and found it attenuated cisplatin-triggered renal function decline. It also alleviated programmed cell death and renal inflammation, indicating a critical role for ROS in mediating AKI. Nox4 protein and mRNA levels were substantially upregulated by cisplatin in vivo and in vitro. Nox4 knockdown alleviated cisplatin-induced cell death and inflammatory response, while Nox4 overexpression aggravated them. Moreover, N-acetyl-L-cysteine (NAC)-mediated inhibition of ROS suppressed cell injury led by Nox4 overexpression, indicating Nox4-mediated ROS generation may be the key mediator in cisplatin-induced nephrotoxicity. Mechanistically, excessive expression of Nox4 induced programmed cell death, especially RIP-mediated necroptosis. Finally, we tested whether Nox4 is a potential therapeutic target using an AKI mouse model by injecting a lentivirus-packaged Nox4 shRNA plasmid through tail vein. Disruption of Nox4 led to renal function recovery, kidney damage relief and reduced inflammation. We conclude that Nox4 aggravates cisplatin-induced nephrotoxicity by promoting ROS-mediated programmed cell death and inflammation. Thus Nox4 may serve as a potential therapeutic target in the treatment of AKI.
Similar content being viewed by others

Main
Acute kidney injury (AKI) is characterized by sudden renal function decline and is highly correlated with high morbidity and mortality rates.1, 2 Emerging evidence highlights the pathological features of AKI, including excessive inflammation and programmed cell death of tubular epithelial cells.3 AKI can be induced by ischemia reperfusion injury, sepsis insult and nephrotoxics, suchas cisplatin. Drug toxicity accounts for 8–60% of all AKI cases, resulting in a heavy burden to patients and society.4, 5 Cisplatin is a classic chemotherapeutic agent that treats a wide spectrum of tumors. However, its use is limited given adverse effects, including nephrotoxicity, the mechanisms of which are still not clearly understood.6
Recent publications indicate diseases caused by cisplatin, such as ototoxicity, are closely correlated with increased levels of reactive oxygen species (ROS).7 ROS is crucial for maintaining kidney function and structure under physiological and pathological conditions.8, 9 A recent study showed that NAC, an ROS inhibitor, attenuated cisplatin-induced nephrotoxicity in human kidney tubular epithelia cells. However, the detailed mechanism and responsible enzymes are yet to be determined.10 The major enzymatic source of ROS production in cisplatin nephrotoxicity is nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, also known as Noxs.11, 12 The Nox family is the major source of renal ROS where NADPHs transfer electrons across biological membranes.13 The Nox family is composed of seven members, including Nox1 (mainly in colon tissues), Nox2 (primarily expressed in phagocytes), Nox3 (predominantly in the inner ear), Nox4, Nox5 (mainly expressed in lymphoid tissues), Duxo1 and Duxo2 (mainly in the thyroid and bronchus).13 Of note, Nox4 is mainly expressed in the kidney.13, 14 Importantly, recent studies demonstrated that Nox4 is involved in the pathological process of many kidney diseases.8, 15 However, its function varies according to the type of disease; its role in AKI has not been fully elucidated. Given the importance of Nox4 in kidney ROS production and inflammation,16, 17 we hypothesized that Nox4 promotes AKI by initiating ROS-mediated renal injury. We studied the expression and role of Nox4 in cisplatin-induced AKI using in vitro HK2 cell model and in vivo model with cisplatin nephropathy treated with apocynin, as an ROS inhibitor, and lentivirus-packaged Nox4 shRNA plasmid.
Materials and methods
Regents
Cisplatin, NAC and apocynin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Lactacte dehydrogenase (LDH), 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT), Reactive Oxygen Species Assay Kit (ROS) and Annexin V-FITC/PI Apoptosis Detection Kit was obtained from Beyotime (Shanghai, China). Periodic acid Schiff (PAS), Creatinine Assay Kit, BUN Assay Kit and Malondialdehyde (MDA) Assay Kit were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). DMEM, fetal bovine serum (FBS) and other cell culture reagents were obtained from Invitrogen. Antibodies specific to KIM-1, β-actin, RIP1, RIP3, Nox2 and Nox4 were purchased from Santa Cruz Biotechnology. Anti-P-MLKL, anti-cleaved-caspase3 and anti-cleaved-caspase8 were obtained from Cell Signaling Technology (CST, Danvers, MA, USA). IRDye 800-conjugated secondary antibody was purchased from Licor (USA). Lipofectamine 3000 was obtained from SciencBio Technology (Invitrogen, Beijing, China).
Cell Culture
Kidney tubular epithelial cells of human (HK2) and mouse renal tubular epithelial cells (mTECs)) were cultured in DMEM containing 5% FBS under 37 °C and 5% CO2 condition. After starvation in 0.5% FBS for 12 h, cells were treated with or without NAC (50 μM) or apocynin (100 μM) for 12 h and then incubated with 20 μM cisplatin for 24 h. Cells were harvested to detect the level of the following indicators: ROS, kidney injury molecule-1 (KIM-1), necroptosis indexes such as RIP1, RIP3 and phosphor-MLKL protein, apoptosis indexes such as cleaved-caspase3 and inflammatory responses such as TNF-α, IL-1β, IL-6 and mRNA by western blotting analysis, real-time PCR and other methods.
Transfection of Nox4 Small Hairpin RNA and Nox4 Overexpression Plasmid Constructs into HK2 Cells
Knockdown or overexpression of Nox4 was assessed by western blotting analysis after transfection of HK2 cells with human-derived Nox4 shRNA or overexpression plasmid, respectively. All shRNA and plasmid constructs were obtained from GenePharma Co., Ltd. Briefly, cells were seeded in six-well plates and transfected with Nox4 shRNA plasmid and control constructs mixed with Lipo3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Cells were incubated with Opti-MEM at 37 °C and 5% CO2 for 6 h. Then cells were cultured in DMEM containing 5% FBS and selected by neomycin or puromycin. The Nox4 knockdown or overexpression cells were then treated with 20 μM cisplatin for 24 h and consequently harvested for analysis with western blotting, real-time PCR and other experiments.
Determination of ROS Production
The level of intracellular ROS was measured using a fluorescent dye 2’, 7’-dichlorofluorescein diacetate (DCFH-DA). Under the presence of an oxidant such as ROS condition, DCFH was converted to a highly fluorescent 2’, 7’-dichlorofluorescein being detected by fluorescence microscopy. Cells were seeded in six-well culture plates and incubated with 20 μM cisplatin in the presence or absence of Nox inhibitor apocynin. Then cells were treated by subsequent operations according to the instructions.
Western Blotting Analysis
Protein from cultured human tubular epithelial cells and mouse kidney tissue were extracted with RIPA lysis buffer, and concentrations of protein were determined by the BCA Protein Assay Kit (Beyotime, Jiangsu, China). Western blotting analysis was performed as described previously.18, 19 Then membranes were blocked with 5% milk for 2 h in order to break nonspecific binding. Subsequently, membranes were incubated with the primary antibody against KIM-1, β-actin, Nox2, Nox4, RIPK1, RIPK3, Phosphor-MLKL and cleaved-caspase3 overnight at 4 °C, followed by incubation with secondary antibody at room temperature for 2 h. Signals were captured with LiCor/Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE, USA). The intensities of each western blotting band were quantified and analyzed by using the Image J software (NIH, Bethesda, MD, USA).
RNA Extraction and Real-Time PCR Examination
Total RNA was extracted from kidney tissues and cultured human tubular epithelial cells with the use of TRIzol (Invitrogen) according to the manufacturer’s instructions. Single-stranded cDNA was synthesized from total RNA using Real Master Mix (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed as previously described.20 The sequences of primers used in this study, including human and mouse β-actin, KIM-1, TNF-α and IL-1β, were described previously.19 The sequences of other primers used for real-time PCR are listed as follows: human Nox4 (forward, 5′-GGATCACAGAAGGTCCCTAGCAG-3′; reverse, 5′-GCGGCTACATGCACACCTGAGAA-3′); human IL-6 (forward, 5′-CGGGAACGAAAGAGAAGCTCTA-3′; reverse, 5′-GAGCAGCCCCAGGGAGAA-3′); mouse IL-6 (forward, 5′-GAGGATACCACTCCCAACAGACC-3′; reverse, 5′-AAGTGCATCATCGTTGTTCATACA-3′). The ratio for the mRNA of interest index was normalized to β-actin and expressed as the mean±s.e.m.
LDH Assay
Cytotoxicity was detected using LDH assay. HK2 cells were seeded in 96-well plates and then treated in the presence or absence of 20 μM cisplatin for 24 h after incubation with apocynin overnight. A portion was removed from each well as the maximum enzyme activity control wells were added to LDH release reagent. One hour later, 96-well plates were centrifuged for 5 min with a centrifuge of 400 g. Subsequently, 120 μl supernatant of each well was added to a new 96-well plate. Cells were then incubated with 60 μl LDH detection working solution and placed in the dark for 30 min at the room temperature. The optical density (OD) was measured with microplate reader (Multiskan MK3, Thermo, USA) at 490 nm wavelength. The cytotoxicity of each group was calculated according to the manufacturer’s instructions.
Immunofluorescence
HK2 cells were cultured in eight-chamber glass slides in the presence or absence of cisplatin (20 μM) for 24 h after incubation with apocynin overnight. Then the cells were fixed in 2% paraformaldehyde and incubated with antibodies against KIM-1 and TNF-α at 4 °C overnight, followed by incubation with goat anti-rabbit IgG-rhodamine (Bioss Biotechnology, Bei Jing, China) for 1 h. Cells were then counterstained with DAPI and analyzed under fluorescence microscope (Zeissspot; Carl Zeiss MicroImaging GmbH, Gottingen, Germany). The number of positive cells from the randomized 500 nucleated cells was expressed as the percentage of positive cell score.
Flow Cytometric Analysis
The number of apoptotic and necrotic cells was detected by flow cytometric analysis.
The HK2 cells were treated in the presence or absence of 20 μM cisplatin for 24 h after incubation with apocynin overnight. Then HK2 cells were digested with trypsin for 2 min and centrifuged at 1500 r.p.m. for 5 min. According to the manufacturer’s instructions, the density of cells is about 106 cells/ml with the addition of 400 μl Annexin V binding fluid. Subsequently, the cells were stained with 5 μl FITC for 15 min at 4 °C. Cells were re-stained with 10 μl PI for 5 min and lightly placed at 4 °C in the dark. Then the cells were immediately measured with a laser eight-color flow cytometer (FACSVerse, BD, USA). The results were quantified using the FlowJo 7.6 software.
Murine Model of Cisplatin-Induced AKI
Mice were purchased from Laboratory Animal Center of Anhui Medical University. All animal procedures were approved by the Institutional Animal Experimental Ethics Committee. Apocynin (100 mg/kg) was injected intraperitoneally in 8-week-old male mice. One day later, 20 mg/kg cisplatin was injected intraperitoneally and saline were injected as controls. Three days later, mice were killed under anesthesia. Samples of blood and kidney tissues were collected for further analysis.
Blood Measurements
Blood samples collected from mice were used to measure creatinine, blood urea nitrogen (BUN) and MDA levels. Samples of blood were harvested from mice with or without intraperitoneal injection of 20 mg/kg cisplatin after 3 days. The levels of creatinine and BUN in blood samples were measured using the Creatinine and BUN Assay Kit (Nanjing, China) according to the manufacturer’s instructions. The MDA levels were detected using the MDA Assay Kit (Nanjing, China).
PAS Staining and Immunohistochemical Analysis
To evaluate the histological damage, PAS staining was performed with the ‘PAS Kit’ according to the protocol of the manufacturer. Kidney damage in the cortical proximal was scored as the approximate extent of tubules that displayed tubular necrosis, cast formation and tubular dilation as follows: 0=normal; 1=10%; 2=10–25%; 3=26–50%; 4=51–75%; 5=75–95%; 6=>96%. In addition, immunohistochemistry was performed in paraffin sections to examine renal damage and inflammation using a microwave-based antigen retrieval technique. Sections were incubated with rabbit anti-KIM-1 and anti-TNF-α antibody for overnight at 4 °C. After incubation in secondary antibody and chromagen liquid DAB (3, 30-diaminobenzidine tetrahydrochloride), the slides were counterstained with hematoxylin. Non-immune rabbit IgG instead of the primary antibody was used as a negative control. The results were analyzed by Image Pro Plus Analysis System (AxioVision 4, Carl Zeiss, Jena, Germany) as described previously.19
Lentivirus-Mediated Nox4 Knockdown in Mice
Mouse Nox4 shRNA labeled with green fluorescent protein (GFP) were obtained from GenePharm (Shanghai, China). The sequences of murine-derived Nox4 shRNA are listed as follows: serial number 200: 5′-GGCCUAGGAUUGCUUUUAATT-3′, 5′-UUAAACACAAUCCUAGGCCTT-3′; serial number 578: 5′- GCUUCUACCUACGCAAUAATT-3′, 5′-UUAUUGCGUAGGUAGAAGCTT-3′; and serial number 1050: 5′-GACCUGGCCAGUAUAUUAUTT-3′, 5′-AUAAUAUACUGGCCAGGUCTT-3′. The silencing efficiency of Nox4 shRNA was screened on mTEC cells, and then shRNA was packaged with lentivirus for silencing Nox4 in vivo. C57BL/6 mice (18-22 g) were housed at the Animal Experimental Center for one week in order to adapt to the environment. The tails of mice were then wiped with alcohol to expand the tail vein for injection. Mice were slowly injected with 100 μl lentivirus-packaged Nox4 shRNA plasmid with a concentration of 1 × 107 TU/ml through tail vein using a 0.5 ml insulin syringe at the speed of approximately 0.2 μl/min. One week later, mice were intraperitoneally injected with either 20 mg/kg cisplatin or the equal volume of saline for further analysis.
Statistical Analysis
Data are expressed as the mean±s.e.m. One-way analysis of variance followed by the Newman–Keuls post hoc test (Prism 5.0 GraphPad Software, Inc, San Diego, CA, USA) was used to analyze results.
Results
Inhibition of ROS Reduced Cisplatin-Induced Tubular Epithelial Cell Injury by Attenuating Programmed Cell Death and Inflammation In Vitro
We found apocynin, an ROS scavenger, reduced the production of intracellular ROS while suppressing the protein levels of Nox2 and Nox4 in cisplatin-treated HK2 cells (Figures 1a and b). We determined the pharmacological effect of apocynin in cisplatin-treated HK2 cells by western blotting and real-time PCR. Results show that overnight preincubation of apocynin decreased the protein and mRNA levels of KIM-1 in cisplatin-treated HK2 cells (Figures 1c, d and f). Consistently, LDH assay results also show that apocynin prevented cisplatin-induced cell damage (Figure 1e). In addition, apocynin reduced the percentage of cisplatin-induced apoptotic and necrotic cells (Figure 2a). Further, apocynin significantly reduced cisplatin-activated programmed cell death-related signaling pathways, including RIP-mediated necroptosis-associated proteins such as RIP1, RIP3 and P-MLKL, and apoptosis-related cleaved-caspase3 (Figure 2b). Apocynin also downregulated the expression of proinflammatory factors, including TNF-α, IL-1β and IL-6 mRNA; this was further evidenced by immunofluorescence and quantitative data (Figures 1g and h). These results indicate that blocking NADPH oxidases by apocynin prevents cisplatin-induced nephrotoxicity by inhibiting Nox-derived ROS production.

Scavenging ROS by apocynin (Apo) reduced Nox expression, cell injury and inflammatory response in cisplatin (Cis)-treated HK2 cells. (a) DCFH-DA assay in Cis-treated HK2 cells. Treatment of Apo decreased the production of intracellular reactive oxygen species (ROS) in Cis-treated HK2 cells. (b) Western blotting analysis of Noxs in HK2 cells. Treatment of Apo inhibited Cis-induced upregulation of kidney-related NADPH oxidases, including Nox2 and NADPH oxidase 4 (Nox4). (c, d) Western blotting analysis and real-time PCR of kidney injury molecule-1 (KIM-1) in HK2 cells. Treatment of Apo significantly reduced the protein and mRNA levels of KIM-1. (e) Lactate dehydrogenase (LDH) assay. Apo suppressed LDH release in Cis-treated HK2 cells. (f) Immunofluorescence and quantitative analysis of KIM-1. Treatment of Apo decreased KIM-1 protein in Cis-treated HK2 cells. (g) Immunofluorescence and quantitative analysis of TNF-α. Apo decreased the percentage of TNF-α-positive cells in response to Cis. (h) Real-time PCR of inflammatory indexes. Apo reduced mRNA levels of inflammation cytokines, including TNF-α, IL-1β and IL-6, in Cis-treated HK2 cells. Data represent the mean±s.e.m. for 3–4 independent experiments in vitro. *P<0.05, **P<0.01, ***P<0.001 versus normal; ##P<0.01, ###P<0.001 versus Cis-treated group.

Scavenging ROS by apocynin (Apo) attenuated programmed cell death in cisplatin (Cis)-treated HK2 cells. (a) Flow cytometry in HK2 cells. Apo decreased the percentage of apoptotic and necrotic HK2 cells in response to Cis. (b) Western blotting analysis of programmed cell death-related molecules. Apo significantly downregulated necroptosis-related indices, including RIPK1, RIPK3 and phosphorylated-mixed lineage kinase domain like (p-MLKL), and apoptosis-related protein such as cleaved-caspase3 (cleaved-C3) in cisplatin-treated HK2 cells. Data represent the mean±s.e.m. for 3–4 independent experiments in vitro. **P<0.01, ***P<0.001 versus normal; ##P<0.01, ###P<0.001 versus cisplatin-treated group. RIP: receptor interacting protein.
Cisplatin-Induced Nephrotoxicity In Vitro was Inhibited after Nox4 Knockdown but Promoted after Nox4 Overexpression
Although apocynin is an ROS scavenger, it is not as specific as Noxs.21 Therefore, we knocked down Nox4 to further study its effect on cisplatin-induced cell injury and the mechanism in both mouse tubular epithelial cell lines and human HK2 cells. Nox4 was silenced by shRNA plasmid (Figure 3a). Western blotting results show that knockdown of Nox4 in mouse tubular epithelial cells reduced cisplatin-induced upregulation of KIM-1 (Figure 3b). This was further confirmed by real-time PCR showing higher KIM-1 mRNA levels (Figure 3c). Also, real-time PCR results show that inflammatory cytokine TNF-α mRNA was inhibited, and the level of IL-1β and IL-6 decreased by 30–50% (Figure 3d). We validated our findings in human HK2 cells by transfecting shRNA plasmid to block Nox4 at the protein level (Figure 4a). Western blotting analysis confirmed that Nox4 knockdown significantly inhibited KIM-1 protein expression in response to cisplatin compared with control group (Figure 4b). The results of real-time PCR also show that Nox4 knockdown significantly decreased the cisplatin-upregulated mRNA level of KIM-1 (Figure 4c). Moreover, Nox4 knockdown reduced the expression of cisplatin-induced inflammatory cytokines, including TNF-α, IL-1β and IL-6 (Figure 4d). These results show that knockdown of Nox4 prevented cisplatin-induced AKI.

Knockdown of Nox4 reduced the level of KIM-1 and inflammatory response in cisplatin (Cis)-treated mouse renal tubular epithelial cells (mTECs). (a) Nox4 knockdown mTEC cells. Results of western blotting show that Nox4 was silenced by transfection of Nox4 shRNA plasmid in mTEC cells. (b, c) Western blotting and real-time PCR of KIM-1. Knockdown of Nox4 significantly reduced the level of KIM-1 protein in Cis-treated mTEC cells, confirmed by real-time PCR at the mRNA level. (d) Real-time PCR of inflammatory indexes. Knockdown of Nox4 decreased inflammatory cytokines, including TNF-α, IL-1β and IL-6 in Cis-treated mTEC cells. Data represent the mean±s.e.m. for 3–4 independent experiments in vitro. *P<0.05, **P<0.01, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus vector control group. Nox4 KD: Nox4 knockdown.

Knockdown of Nox4 reduced programmed cell death and inflammation in cisplatin (Cis)-treated HK2 cells. (a) Nox4 knockdown HK2 cells. (b) Western blotting and quantitative analysis of KIM-1. Knockdown of Nox4 significantly reduced the protein level of KIM-1 in Cis-treated HK2 cells. (c) Real-time PCR of KIM-1. Silencing Nox4 alleviated Cis-induced mRNA level of KIM-1. (d) Real-time PCR of inflammatory indices. Knockdown of Nox4 decreased inflammatory cytokines, including TNF-α, IL-1β and IL-6 in Cis-treated HK2 cells. (e) Western blotting of programmed cell death-related molecules. Knockdown of Nox4 reduced the levels of necroptosis-related proteins such as RIPK1, RIPK3 and P-MLKL and apoptosis-related protein such as cleaved-caspase3 in cisplatin-treated HK2 cells. Data represent the mean±s.e.m. for 3–4 independent experiments in vitro. *P<0.05, **P<0.01, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus vector control group. Nox4 KD: Nox4 knockdown.
To further investigate the role of Nox4 in cisplatin-induced AKI, we established stable Nox4 overexpression HK2 cell lines (Figures 5a and b). We found Nox4 overexpression increased cisplatin-induced KIM-1 protein and mRNA expression compared with the control group (Figures 5c and d). Previously, we reported that inflammatory cytokines have an important role in cisplatin-induced AKI.22, 23 Here we found that Nox4 overexpression increased the levels of cisplatin-induced inflammatory cytokines, including TNF-α, IL-1β and IL-6 mRNA levels, compared with the control group (Figure 5e). Thus these data indicate that Nox4 overexpression accelerates cisplatin-induced AKI.

Overexpression (OE) of Nox4 promoted programmed cell death and inflammation in cisplatin (Cis)-treated HK2 cells. (a, b) Nox4 OE in HK2 cells. (c) Western blotting and quantitative analysis of KIM-1. OE of Nox4 promoted Cis-induced KIM-1 expression in vitro. (d) Real-time PCR of KIM-1. OE of Nox4 significantly upregulated the mRNA level of KIM-1 in Cis-treated HK2 cells. (e) Real-time PCR of inflammatory indices. OE of Nox4 increased TNF-α, IL-1β and IL-6 in Cis-treated HK2 cells. (f) Western blotting of programmed cell death-related molecules. OE of Nox4 increased the levels of necroptosis-related proteins such as RIPK1, RIPK3 and P-MLKL and apoptosis-related protein such as cleaved-caspase3 in cisplatin-treated HK2 cells. Data represent the mean±s.e.m. for 3–4 independent experiments in vitro. *P<0.05, **P<0.01, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus vector control.
NAC, an ROS Inhibitor, Alleviated Nox4 Overexpression-Promoted Kidney Injury in Cisplatin-Treated HK2 Cells
According to our findings, Nox4 promotes cisplatin-induced AKI. We found that NAC treatment suppressed the KIM-1 protein levels that are promoted by Nox4 overexpression in the cisplatin-treated group (Figure 6a). This was confirmed by real-time PCR showing reduced mRNA level of KIM-1 after NAC treatment (Figure 6b). Moreover, Nox4-induced upregulation of inflammatory cytokines TNF-α, IL-1β and IL-6 mRNA were suppressed after NAC treatment (Figure 6c). These results indicate that Nox4 promotes cisplatin-induced AKI by generating ROS.

N-acetyl-L-cysteine (NAC) suppressed programmed cell death and inflammation promoted by Nox4 overexpression in cisplatin (Cis)-treated cells. (a, b) Western blotting and real-time PCR of KIM-1. NAC suppressed the level of KIM-1 protein promoted by Nox4 overexpression in Cis-treated cells, which was further confirmed at the mRNA level. (c) Real-time PCR of inflammatory indices. NAC suppressed Nox4-upregulated mRNA level of TNF-α, IL-1β and IL-6 in Cis-treated HK2 cells. (d) Western blotting of programmed cell death-related molecules. NAC suppressed Nox4-upregulated RIPK1, RIPK3, P-MLKL and cleaved-caspase3 in Cis-treated cells. Data represent the mean±s.e.m. for 3–4 independent experiments in vitro. *P<0.05, **P<0.01, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus Nox4-EV group; $P<0.05, $$P<0.01, $$$P<0.001 versus cisplatin-treated group.
Nox4 Accelerated Cisplatin-Induced Kidney Cell Injury by Initiating ROS-Mediated Necroptosis and Apoptosis
Interfering with RIP-mediated necroptosis prevents cisplatin-induced AKI.24 In addition, renal tubular epithelial cells are victims of apoptosis in AKI caused by cisplatin.25 Here we scavenged ROS with apocynin and then knocked down or overexpressed Nox4 in HK2 cells to observe the effects on RIPK-mediated necroptosis and apoptosis induced by cisplatin. We found that apocynin downregulated necroptosis-related proteins, including RIPK1, RIPK3 and P-MLKL. It also suppressed apoptosis-related protein cleaved-caspase3 in cisplatin-treated HK2 cells (Figure 2b). In Nox4 knockdown HK2 cells, the activation of RIP1/RIP3/MLKL axis and cleavage of caspase3 were inhibited; whereas Nox4 overexpression had the opposite effect (Figures 4e and 5f). Moreover, Nox4 overexpression-promoted kidney injury was largely inhibited by NAC treatment, indicating Nox4 induced kidney damage via ROS generation (Figure 10d). Taken together, these results show that Nox4 accelerates cisplatin-induced AKI by enhancing ROS-mediated necroptosis and apoptosis.
Intraperitoneal Injection of Apocynin Prevented Cisplatin-Induced AKI In Vivo
We determined the functional role of ROS after intraperitoneal injection of apocynin in a toxic AKI model. We found that apocynin suppressed cisplatin-induced upregulation of ROS-mediated proteins, such as MDA, in mouse serum (Figure 7a). Western blotting and quantitative data show that intraperitoneal injection of apocynin (100 mg/kg) significantly suppressed cisplatin-induced Nox4 protein expression in vivo (Figure 7b). PAS staining shows that apocynin attenuated the degree of renal injury (Figure 7c), consistent with the findings that apocynin downregulated creatinine (Figure 7d) and BUN (Figure 7e) 3 days after intraperitoneal injection of cisplatin (20 mg/kg). In addition, apocynin attenuated cisplatin-induced KIM-1 mRNA level compared with cisplatin-treated mice (Figure 7f). Consistently, apocynin also decreased the level of KIM-1 protein in cisplatin-induced AKI mice model (Figure 7g), which was further confirmed by IHC of KIM-1 (Figure 7h). We also observed the effect of apocynin on the inflammatory response triggered by cisplatin in mice. Immunohistochemistry and quantitative analysis show apocynin suppressed TNF-α protein level in cisplatin-induced AKI mouse model (Figure 8a). Real-time PCR results also show that apocynin significantly downregulated mRNA levels of TNF-α, IL-1β and IL-6 compared with the cisplatin-induced AKI group (Figure 8b). Consistently, western blotting results show that apocynin significantly reduced the protein levels of RIPK1, RIPK3, P-MLKL and cleaved-caspase3 in cisplatin nephropathy (Figure 8c). Taken together, our findings suggest that apocynin prevents renal damage and inflammation in response to cisplatin by scavenging ROS in vivo.

Apocynin (Apo) prevented cisplatin (Cis)-induced renal injury, supported renal function and decreased KIM-1 and reactive oxygen species (ROS) in vivo. (a) Malondialdehyde (MDA) assay in mice. Apo significantly reduced the level of ROS in Cis nephropathy. (b) Western blotting analysis of Nox4. Intraperitoneal injection of Apo significantly reduced the Cis-upregulated Nox4 protein level. (c) Periodic acid-Schiff (PAS) staining. Apo prevented Cis-induced renal injury in vivo. (d, e) Renal function. Results of serum creatinine and BUN showed that Apo suppressed the decline of renal function caused by Cis. (f) Real-time PCR of KIM-1. Apo reduced the mRNA level of KIM-1 in Cis nephropathy. (g and h) Western blotting analysis and immunohistochemistry (IHC) of KIM-1. Apo reduced the protein level of KIM-1 protein in Cis nephropathy, which was further confirmed by IHC. Data represent the mean±s.e.m. for 6–8 independent experiments in vivo. *P<0.05, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus cisplatin-treated group. Noxs: NADPH oxidases.

Apocynin (Apo) decreased renal inflammation and programmed cell death in cisplatin (Cis) nephropathy. (a) IHC and quantitative analysis of TNF-α in mice. Apo significantly reduced Cis-induced TNF-α protein expression in vivo. (b) Real-time PCR of inflammatory indices in mice. Apo largely decreased the mRNA levels of inflammatory cytokines, including TNF-α, IL-1β and IL-6. (c) Western blotting analysis of programmed cell death-related molecules. Intraperitoneal injection of Apo significantly reduced Cis-induced upregulation of necroptosis-related proteins such as RIPK1, RIPK3 and P-MLKL while decreasing apoptosis-related protein, such as cleaved-caspase3. Data represent the mean±s.e.m. for 6–8 independent experiments in vivo. *P<0.05, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus cisplatin-treated group. A full color version of this figure is available at the Laboratory Investigation journal online.
Lentivirus-Mediated Knockdown of Nox4 Protected Against Cisplatin-Induced AKI In Vivo
Immunohistochemistry and quantitative data show Nox4 significantly upregulated in cisplatin nephropathy (Figure 9a). This was further confirmed by real-time PCR and western blotting in vivo (Figures 9b and c). We determined whether Nox4-targeted therapy could prevent cisplatin-induced kidney injury in vivo. We found GFP signal in the kidneys transfected with Nox4 shRNA and control vector plasmid (Supplementary Figure 1). Accordingly, Nox4 was reduced in Nox4 shRNA-transfected kidney (Figure 9d). We found that disruption of Nox4 relieved cisplatin-induced kidney damage as shown by PAS staining and quantitative data (Figure 9e). Moreover, results of serum creatinine and BUN assays show that deficiency of Nox4 protected against the loss of renal function in response to cisplatin (20 mg/kg) (Figures 9f and g). This was further confirmed by real-time PCR, western blotting and immunohistochemistry measuring the mRNA and protein levels of KIM-1 (Figures 10a–c). In addition, disruption of Nox4 attenuated cisplatin-induced renal inflammation by downregulating the TNF-α protein and mRNA levels (Figures 10d and e). IL-1β and IL-6 mRNA levels were also reduced after Nox4 knockdown in cisplatin-treated kidney (Figure 10e). Importantly, we found that loss of Nox4 prevented the activation of RIPK1/RIPK3/MLKL and cleavage of caspase3 (Figure 10f). These results further confirm that knockdown of Nox4 protects cisplatin-induced AKI by preventing programmed cell death and inflammation. In this setting, we propose that Nox4 is a potential therapeutic target for cisplatin-induced AKI.

Lentivirus-mediated knockdown of Nox4 protected against renal injury in cisplatin nephropathy. (a) Immunohistochemistry and quantitative analysis of Nox4 in mice. (b) Real-time PCR of Nox4 in mice. Nox4 protein was increased in cisplatin nephropathy, which was further confirmed by real-time PCR for mRNA. (c) Western blotting and quantitative analysis of Nox4 in mice. Nox4 was significantly upregulated in a time-dependent manner in response to cisplatin. (d) Nox4 knockdown in mice. Western blotting analysis showed a substantial knockdown of Nox4 protein in the kidney. (e) PAS staining. Silencing Nox4 attenuated cisplatin-induced kidney damage in vivo. (f, g) Detection of renal function. Results of serum creatinine and BUN show that knockdown of Nox4 protected against the loss of renal function in cisplatin nephropathy. Data represent the mean±s.e.m. for 6–8 independent experiments in vivo. *P<0.05, **P<0.01, ***P<0.001 versus normal. ##P<0.01 versus vector control mice.

Deficiency of Nox4 reduced kidney damage, inflammation and programmed cell death in cisplatin nephropathy. (a–c) Real-time PCR, western blotting and immunohistochemistry of KIM-1 in mice. Lentivirus-mediated knockdown of Nox4 reduced the protein level of KIM-1 in cisplatin nephropathy, further confirmed by real-time PCR and immunohistochemistry. (d) IHC and quantitative analysis of TNF-α. Disruption of Nox4 from mice largely suppressed the protein level of TNF-α in cisplatin nephropathy. (e) Real-time PCR of inflammatory cytokines. Deficiency of Nox4 reduced cisplatin-upregulated mRNA level TNF-α, IL-1β and IL-6 in mice. (f) Western blotting of programmed cell death-related molecules. Knockdown of Nox4 suppressed the levels of necroptosis-related proteins, such as RIPK1, RIPK3 and P-MLKL, and apoptosis-related protein, such as cleaved-caspase3 in cisplatin nephropathy. Data are mean±s.e.m. for 6–8 mice. **P<0.01, ***P<0.001 versus normal; #P<0.05, ##P<0.01, ###P<0.001 versus vector control mice.
Discussion
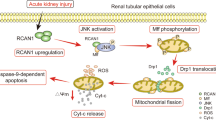
In the current study, we blocked general ROS and Nox4-mediated ROS production and found that Nox4 promoted cisplatin-induced nephrotoxicity in vivo and in vitro. Our findings show that Nox4 accelerated AKI, which is possibly correlated with its regulative role in renal inflammation and programmed cell death.
Nox4 is a major renal isoform of the Nox family; it is highly expressed in the kidney and associated with pathological processes of different kidney diseases, including streptozotocin-induced diabetic nephropathy,26, 27 renal tumorigenesis,28 UUO nephropathy and 5/6 nephrectomy remnant kidney model.29, 30 However, its functional role is still unclear. One group showed Nox4 deficiency reduced the level of hypoxia-inducible factor-1α, thereby promoting TGF-β-induced apoptosis and interstitial fibrosis in obstructive kidney diseases.28 In contrast, other groups showed that disruption of Nox4 reduced glomerulosclerosis and inflammatory cytokines in long-term diabetic nephropathy.26, 27 Clearly, this is not consistent with the previous study indicating that loss of Nox4 increases albuminuria but does not alter matrix deposition in diabetic mice.30 This evidence suggests that the function of Nox4 is highly varied depending on the type and stage of disease.
To date, the functional role of Nox4 in AKI, especially cisplatin-induced AKI, is not well understood. To further explore this critical issue, we blocked the generation of ROS in cisplatin-treated tubular epithelial cells by preincubating with apocynin. Our results demonstrate that Nox-derived ROS has a predominant role in mediating cisplatin-mediated kidney damage. Moreover, we found that cisplatin significantly induced Nox4 in tubular epithelial cells and injured kidneys. After blocking this enzyme in tubular epithelial cells from mouse and human origin, the expression of KIM1, a key index for kidney injury, was significantly reduced. This is in line with our in vivo results showing that lentivirus-mediated knockdown of Nox4 attenuated the deterioration of renal function, tubular injury and inflammation. Moreover, we also found that scavenging Nox4 overexpression-induced ROS generation by NAC abrogated its detrimental role in kidney injury.
We further explored the mechanisms of how Nox4 knockdown protects against renal injury and inflammation. Our in vitro study results show that loss of Nox4-derived ROS substantially downregulated programmed cell death, which was consistent with the findings collected from cisplatin-treated tubular epithelial cells where the ROS was scavenged by apocynin. In addition to limiting apoptosis, Nox4 deficiency significantly blocked RIPK-mediated programmed cell death. Necroptosis is regulated by a death-signaling pathway and a recently identified cell death pattern that has a key role in AKI.24 Necroptosis is initiated in response to a variety of stimuli, including TNF-α, FasL, TRAIL, Toll-like receptors and cisplatin.24, 31 Typically, the major components for this signaling pathway include receptor-interacting proteins 1 and 3. MLKL, phosphorylated on Thr357 and Ser358, is recruited and binds RIPK1/RIPK3, forming RIPK1/RIPK3/MLKL complex that is regarded as the predominant mediator for necroptosis.32, 33 Pharmacological inhibition of RIPK1, knockdown of RIPK3 or MLKL protein prevents kidney injury caused by cisplatin.24 Emerging evidence shows that necroptosis, instead of apoptosis, triggers severe inflammatory response by destroying cell membranes, facilitating the release of endogenous pro-inflammatory molecules known as damage-associated molecular patterns.34 All of these lines of evidence indicate a key role for RIPK-mediated necroptosis in cisplatin-induced AKI. Excessive ROS production results in tissue injury, but the detailed mechanisms are not understood.35 Our current study provides novel evidence showing that inhibition of Nox4-induced ROS suppresses necroptosis and apoptosis of tubular epithelial cells, which alleviates cisplatin-induced kidney injury and inflammation.
Finally, we tested whether Nox4 is a potential therapeutic target using a mouse model of cisplatin nephropathy by injecting lentivirus-mediated Nox4 shRNA plasmid through tail vein. Our results show that Nox4 inhibition prevented the deterioration of renal function while relieving kidney damage and inflammation. This is consistent with our recent findings showing that protocatechuic aldehyde attenuated cisplatin-induced AKI by suppressing Nox4-mediated oxidative stress and renal inflammation.36 Consistently, Nox4 loss in the kidney attenuated cisplatin-induced cell necroptosis and apoptosis.
Of note, a recent study showed Nox4 knockdown deteriorated renal function and accelerated tubular apoptosis in acute ischemic reperfusion kidney injury by suppressing hydrogen peroxide, a key regulator of the pro-survival pathway, in NRF2-dependent mechanisms.9 We offer two possible explanations for the discrepancy in this report and our findings. First, Nox4 induced hydrogen peroxide to initiate cell survival in response to stimuli at a very early stage of disease, but Nox4 knockout abrogated this protective property. Our data indicate that Nox4 is pathogenic in the progression of kidney disease, because overactivation of Nox4 results in excessive production of ROS and destroys renal cells while enhancing inflammation. In this setting, rebalancing, instead of totally shutting down, the level of Nox4 seems to be protective in the treatment of kidney diseases. Second, in different types of kidney disease, the functional role of Nox4 may vary. Taken together, the role of Nox4 may be disease type and state dependent, which needs to be carefully evaluated.
In conclusion, we show that cisplatin induces kidney injury, programmed cell death and inflammation via Nox4-correlated mechanisms. Nox4 knockdown in vivo effectively attenuated cisplatin-induced kidney damage and inflammation, indicating that it may serve as a therapeutic target in treatment of AKI.
References
Zeng X, McMahon GM, Brunelli SM et al. Incidence, outcomes, and comparisons across definitions of AKI in hospitalized individuals. Clin J Am Soc Nephrol 2014;9:12–20.
Yang L, Xing G, Wang L et al. Acute kidney injury in China: a cross-sectional survey. Lancet 2015;386:1465–1471.
Mulay SR, Linkermann A, Anders HJ . Necroinflammation in kidney disease. J Am Soc Nephrol 2016;27:27–39.
Pavkovic M, Vaidya VS . MicroRNAs and drug-induced kidney injury. Pharmacol Ther 2016;163:48–57.
Kandasamy K, Chuah JK, Su R et al. Prediction of drug-induced nephrotoxicity and injury mechanisms with human induced pluripotent stem cell-derived cells and machine learning methods. Sci Rep 2015;5:12337.
Galgamuwa R, Hardy K, Dahlstrom JE et al. Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties. J Am Soc Nephrol 2016;27:3331–3344.
Kaur T, Borse V, Sheth S et al. Adenosine A1 receptor protects against cisplatin ototoxicity by suppressing the NOX3/STAT1 inflammatory pathway in the cochlea. J Neurosci 2016;36:3962–3977.
Holterman CE, Read NC, Kennedy CR . Nox and renal disease. Clin Sci (Lond) 2015;128:465–481.
Nlandu-Khodo S, Dissard R, Hasler U et al. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci Rep 2016;6:38598.
Chen Q, Peng H, Dong L et al. Activation of the NRF2-ARE signalling pathway by the Lentinula edodes polysaccharose LNT alleviates ROS-mediated cisplatin nephrotoxicity. Int Immunopharmacol 2016;36:1–8.
Wang Y, Luo X, Pan H et al. Pharmacological inhibition of NADPH oxidase protects against cisplatin induced nephrotoxicity in mice by two step mechanism. Food Chem Toxicol 2015;83:251–260.
Schreck C, O'Connor PM . NAD(P)H oxidase and renal epithelial ion transport. Am J Physiol Regul Integr Comp Physiol 2011;300:R1023–R1029.
Sedeek M, Nasrallah R, Touyz RM et al. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 2013;24:1512–1518.
Geiszt M, Kopp JB, Varnai P et al. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA 2000;97:8010–8014.
Gorin Y, Block K . Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med 2013;61:130–142.
Sanchez-Gonzalez PD, Lopez-Hernandez FJ, Lopez-Novoa JM et al. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit Rev Toxicol 2011;41:803–821.
Kaushal GP, Shah SV . Autophagy in acute kidney injury. Kidney Int 2016;89:779–791.
Meng XM, Ren GL, Gao L et al. Anti-fibrotic effect of wogonin in renal tubular epithelial cells via Smad3-dependent mechanisms. Eur J Pharmacol 2016;789:134–143.
Meng XM, Huang XR, Xiao J et al. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int 2012;81:266–279.
Meng XM, Huang XR, Xiao J et al. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J Pathol 2012;227:175–188.
Ray R, Murdoch CE, Wang M et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 2011;31:1368–1376.
Liang H, Zhang Z, He L et al. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 2016;7:31652–31662.
Nozaki Y, Kinoshita K, Yano T et al. Signaling through the interleukin-18 receptor alpha attenuates inflammation in cisplatin-induced acute kidney injury. Kidney Int 2012;82:892–902.
Xu Y, Ma H, Shao J et al. A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol 2015;26:2647–2658.
Benedetti G, Fredriksson L, Herpers B et al. TNF-alpha-mediated NF-kappaB survival signaling impairment by cisplatin enhances JNK activation allowing synergistic apoptosis of renal proximal tubular cells. Biochem Pharmacol 2013;85:274–286.
Jha JC, Gray SP, Barit D et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 2014;25:1237–1254.
Jha JC, Thallas-Bonke V, Banal C et al. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 2016;59:379–389.
Gregg JL, Turner RM 2nd, Chang G et al. NADPH oxidase NOX4 supports renal tumorigenesis by promoting the expression and nuclear accumulation of HIF2alpha. Cancer Res 2014;74:3501–3511.
Jung KJ, Min KJ, Park JW et al. Carnosic acid attenuates unilateral ureteral obstruction-induced kidney fibrosis via inhibition of Akt-mediated Nox4 expression. Free Radic Biol Med 2016;97:50–57.
Babelova A, Avaniadi D, Jung O et al. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 2012;53:842–853.
LaRocca TJ, Stivison EA, Mal-Sarkar T et al. CD59 signaling and membrane pores drive Syk-dependent erythrocyte necroptosis. Cell Death Dis 2015;6:e1773.
Nogusa S, Thapa RJ, Dillon CP et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 2016;20:13–24.
Newton K . RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol 2015;25:347–353.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191–195.
Faria A, Persaud SJ . Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential. Pharmacol Ther 2017;172:50–62.
Gao L, Wu WF, Dong L et al. Protocatechuic aldehyde attenuates cisplatin-induced acute kidney injury by suppressing Nox-mediated oxidative stress and renal inflammation. Front Pharmacol 2016;7:479.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 81300580, 81570623) and by Science and Technological Fund of Anhui Province for Outstanding Youth of China (Grant number: 1608085J07). We thank Dr Hui Yao Lan (The Chinese University of Hong Kong) for providing mouse tubular epithelial cell lines (mTECs). We thank Dr Austin Cape at ASJ Editors for careful reading and feedback.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
This study reveals that Nox4 is significantly induced in cisplatin-treated tubular epithelial cells and in a mouse model of acute kidney injury. Nox4 aggravates cisplatin-induced nephrotoxicity possibly by promoting reactive oxygen species-mediated programmed cell death and inflammation. These results also indicate that Nox4 may serve as a potential therapeutic target in the treatment of kidney injury.
Supplementary information
Rights and permissions
About this article

Cite this article
Meng, XM., Ren, GL., Gao, L. et al. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab Invest 98, 63–78 (2018). https://doi.org/10.1038/labinvest.2017.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.120
This article is cited by
-
The Protective Effects of Vitamin B Complex on Diclofenac Sodium-Induced Nephrotoxicity: The Role of NOX4/RhoA/ROCK
Inflammation (2024)
-
Cytosine–phosphate–guanine oligodeoxynucleotides alleviate radiation-induced kidney injury in cervical cancer by inhibiting DNA damage and oxidative stress through blockade of PARP1/XRCC1 axis
Journal of Translational Medicine (2023)
-
Cisplatin nephrotoxicity: new insights and therapeutic implications
Nature Reviews Nephrology (2023)
-
NOX4 blockade suppresses titanium nanoparticle-induced bone destruction via activation of the Nrf2 signaling pathway
Journal of Nanobiotechnology (2022)
-
Role of TRPC6 in kidney damage after acute ischemic kidney injury
Scientific Reports (2022)

{kind=link}