
Abstract
Vascular endothelial cells (ECs) are ideal gene therapy targets as they provide widespread tissue access and are the first contact surfaces following intravenous vector administration. Human recombinant adenovirus serotype 5 (Ad5) is the most frequently used gene transfer system because of its appreciable transgene payload capacity and lack of somatic mutation risk. However, standard Ad5 vectors predominantly transduce liver but not the vasculature following intravenous administration. We recently developed an Ad5 vector with a myeloid cell-binding peptide (MBP) incorporated into the knob-deleted, T4 fibritin chimeric fiber (Ad.MBP). This vector was shown to transduce pulmonary ECs presumably via a vector handoff mechanism. Here we tested the body-wide tropism of the Ad.MBP vector, its myeloid cell necessity, and vector-EC expression dose response. Using comprehensive multi-organ co-immunofluorescence analysis, we discovered that Ad.MBP produced widespread EC transduction in the lung, heart, kidney, skeletal muscle, pancreas, small bowel, and brain. Surprisingly, Ad.MBP retained hepatocyte tropism albeit at a reduced frequency compared with the standard Ad5. While binding specifically to myeloid cells ex vivo, multi-organ Ad.MBP expression was not dependent on circulating monocytes or macrophages. Ad.MBP dose de-escalation maintained full lung-targeting capacity but drastically reduced transgene expression in other organs. Swapping the EC-specific ROBO4 for the CMV promoter/enhancer abrogated hepatocyte expression but also reduced gene expression in other organs. Collectively, our multilevel targeting strategy could enable therapeutic biological production in previously inaccessible organs that pertain to the most debilitating or lethal human diseases.
Similar content being viewed by others
Main
Despite development of targeted small-molecule inhibitors or biologics, both benign and malignant diseases remain challenging or even recalcitrant to control or cure. One problem is that therapies are not tightly targeted to disease and produce systemic complications. Another obstacle is the emergence of disease resistance. Both of these challenges could be addressed by the development of agents that would be systemically administered, home to the disease site, and express high levels of a therapeutic either persistently or under switchable control.2, 3, 4, 5, 6
One option for targeted therapeutic delivery is adenoviral (Ad) vectors. These biologics can be genetically manipulated for disease site homing, infection of relevant target cells, and high-level transgene expression.3, 7, 8, 9 Disease site-specific transductional targeting can be accomplished by genetic manipulation of the viral capsid.6, 9 Transcriptional targeting for expression within specific cell types can be accomplished using DNA enhancer/promoter elements incorporated into the vector transgene.10 Regulated targeted gene expression may be achieved through drug-inducible enhancers/promoters,11 DNA secondary structural elements,12 or microRNA seed sequences placed up or downstream of the transgene open reading frame.13
One portal to disease sites marginally accessed by vectors is vascular endothelium. As many diseases evoke expansion of existent or creation of new blood vessels, endothelial cell (EC) targeted viral vectors could produce therapeutics titrated to disease severity. Moreover, as the vascular ECs are the initial cell layer in contact with intravenously injected therapeutics, first-pass organ targeting can be achieved.5 Several viral vectors have been engineered for vascular EC targeting as potential novel therapeutics for both benign and malignant diseases.14, 15 In oncology, the predominant goal has been vascular ablation. However, the strategy of ‘starving’ tumors has elicited resistance either owing to coordinate upregulation of untargeted factors by malignant cells or dynamic microenvironmental remodeling.2 Other than hypervascular intraocular disease, vessel ablation would be contraindicated for most benign diseases. An alternative to vascular ablation is manipulation of the molecular secretion repertoire of ECs. Angiocrine has been the term assigned to EC production or display of membrane-tethered growth factors, cyto/chemokines, and stem cell nurturing factors.16 Engineering angiocrine functions is a compelling opportunity for vector-based, novel disease therapeutics both for benign and malignant disease.
Here we tested the body-wide cell-type expression of a previously reported vascular EC-targeted Ad5 vector containing a heptapeptide, ‘myeloid-binding peptide’ (MBP). Ad.MBP was engineered for MBP display at the tip of a ‘de-knobbed’ chimeric fiber.5, 17 This vector was shown to bind specifically to myeloid cells ex vivo but predominantly transduced lung vascular endothelium following systemic administration.1 Here we discovered that the Ad.MBP vector affected multi-organ EC-specific expression. Most striking was the finding that hitherto inaccessible organs evidenced EC transgene expression, including the brain, small and large bowel mucosa, kidney glomeruli, medulla, and papilla, skeletal muscle, and cardiac subendocardium and myocardium. The Ad.MBP vector presents an opportunity for targeting the most prominent and vexing human diseases.
MATERIALS AND METHODS
Animals
All mice were of C57BL/6J background and 7–14 weeks of age. Mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA) or through breeding in authors’ animal facility. Experimental procedures involving mice were carried out under protocol nos. 20120029 and 20110035 approved by the Washington University Animal Studies Committee.
Cells and Adenovirus Vectors
Human embryonic kidney HEK293 cells were purchased from Microbix Biosystems (Ontario, Canada). Cells were cultured in DMEM/F12 (Mediatech, Herndon, VA, USA) media containing 10% fetal bovine serum (Summit Biotechnology, Fort Collins, CO, USA), in a humidified atmosphere with 5% CO2 at 37 °C. Replication incompetent E1- and E3-deleted Ad5 vectors were created using a two-plasmid rescue method. Plasmids encoded expression cassettes containing either the cytomegalovirus major immediate-early enhancer/promoter (CMV), or the human roundabout4 (ROBO4) enhancer/promoter, each cloned upstream of enhanced green fluorescent protein (EGFP) followed by the bovine growth hormone polyadenylation signal. These expression cassettes were cloned into a shuttle plasmid (pShuttle, Qbiogene, Carlsbad, CA, USA) to generate the pShuttleCMV-EGFP and pShuttleROBO4-EGFP plasmids, respectively, and inserts were confirmed by using restriction enzyme mapping and partial sequence analysis.
The shuttle plasmids were linearized with Pme I and integrated into the Ad5 genome by homologous recombination with a pAd5 plasmid, encoding the native Ad5 fiber, or a pAdMBP plasmid, encoding an MBP-fiber-fibritin chimera, in the E. coli strain BJ5183. To rescue Ad.MBP.ROBO4, the recombinant viral genome was linearized with Pac I and then transfected into 293F28 cells using SuperFect Transfection Reagent (Qiagen, Chatsworth, CA, USA). 293F28 cells stably express the native Ad5 fiber; thus viruses rescued at this point were mosaic in the sense that the Ad5 virions randomly incorporated a mixture of native Ad5 fibers and MBP-fiber-fibritin chimeras.18 After an additional round of amplification on 293F28 cells, the viruses were amplified in HEK293 cells, which do not express native Ad5 fiber, to obtain virus particles containing only MBP-fiber-fibritin proteins. The Ad.MBP.CMV vector containing the sequence WTLDRGY on a T4 fibritin chimeric fiber knob was created as described previously.1, 17 Recombinant viruses were purified by two rounds of CsCl density ultracentrifugation and dialyzed in storage buffer containing 10 mmol/l HEPES and 1 mmol/l MgCl2, pH 7.8 with 10% glycerol as previously described.19 The viral particle (vp) concentration was determined by absorbance of dissociated virus at A260 nm using a conversion factor of 1.1 × 1012 vp/absorbance unit.
Warfarin and Clodronate Liposome Treatment
Mice were subcutaneously injected with warfarin, 5 mg/kg in peanut oil, 72 and 24 h before virus injection.20 Clodronate- iposomes, 10 μl/g body weight, (ClodronateLiposomes.com, Netherlands) or saline buffer were injected into the tail vein 48 and 24 h before vector injection.21 Twenty-four hours later, peripheral blood was collected by cheek pouch bleeding, and then Ad.MBP was injected.
Virus Injection and Host Organ Harvest
Mice were tail-vein injected with 1 × 1011 or 2 × 1010 particles of virus in 200 μl of saline. Seventy-two hours after virus administration, mice were anesthetized with 2.5% 2, 2, 2-tribromoethanol (Avertin, Sigma-Aldrich, St Louis, MO, USA), perfused via the left ventricle with phosphate-buffered saline (PBS) followed by 10% neutral-buffered formalin. Lung was further inflated and fixed by injecting formalin solution into trachea, closing the trachea by ligature, and then processed as below. Harvested organs were postfixed in formalin at room temperature for 2–4 h and cryopreserved in 30% sucrose in PBS at 4 °C overnight. Treated tissues were embedded in NEG50 (Thermo Fisher Scientific, Waltham, MA, USA) or Tissue-Tek OCT mounting medium (Sakura Torrance, CA, USA), and frozen in a liquid nitrogen pre-chilled, 2-methylbutane-containing glass beaker.
Immunofluorescence Staining
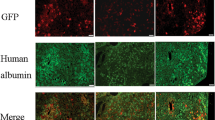
All mouse tissues were cryosectioned at 16 μm. Lung was also cut at 5 μm for determination of transgene microvessel co-localization. Frozen section slides were air-dried for 10 min, washed three times in PBS, blocked with protein block solution (5% donkey serum and 0.1% Triton X-100 in PBS) for 1 h, and incubated at 4 °C overnight in protein block containing primary antibodies, including: rat anti-endomucin 1:1000, rat anti-PDGFRβ 1:200 (nos.14-5851-81 and 14-1402-81, eBioscience, San Diego, CA, USA), Armenian hamster anti-CD31 1:1000, rabbit anti-NG2 chondroitin sulfate proteoglycan 1:100 (nos.MAB1398Z and AB5320, EMD-Millipore, Billerica, MA, USA), rat anti-CD45 1:100 (no.550539, BD Biosciences, San Jose, CA, USA), rat anti-F4/80 1:500 (no.MCA497R, AbD Serotec-BioRad, Raleigh, NC, USA), rabbit anti-GFP 1:400, and chicken anti-GFP 1:400 (nos.A11122 and A10262, Life Technologies, Carlsbad, CA, USA). The two GFP antibodies performed equally well; the chicken anti-GFP antibody was used in the clodronate liposome experiment, and the rabbit antibody was used throughout the rest of the study. On day 2, the slides were washed three times in PBS, incubated with corresponding 1:400 diluted Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), and counterstained with SlowFade Gold Antifade mounting reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies).
Immunofluorescence Microscopy-Based Analysis of Viral Reporter Gene Expression
Immunofluorescence images were collected using an Olympus BX61 microscope equipped with an FVII digital camera (Olympus America, Center Valley, PA, USA). The Extended Focal Imaging (EFI) function was used in collecting all high-magnification micrographs to allow the creation of a single in-focus image from a series of views of the same field at different z-dimensional focal planes at 2 μm intervals. EFI was carried out in a live-processing mode during image acquisition. Camera acquisition time for EGFP immunofluorescence was optimized and set a priori for each organ through independent experiments where the collected data were pooled for statistical analyses. The optimized acquisition time for EGFP immunofluorescence display was 200 msec for the liver, 400 msec for spleen, 300 msec for lung, 300 msec for heart, 300 msec for kidney, 1 s for muscle, 500 msec for pancreas, 1 s for small bowel, 1 s for large bowel, and 500 msec for brain. Wherever a figure contains micrographs collected using a different level of exposure for EGFP, the setting is indicated in the figure legend. Immunofluorescence micrographs were subjected to measurement of both color intensity and color-positive area using the MicroSuite Biological Suite image analysis software Version 5 (Olympus). To determine the EGFP fluorescence intensity, a threshold defining the background green fluorescence color for each pixel was set at 70 while the possible range of intensity values were from 0 representing a complete absence of green color intensity to 255 taken to be of full intensity. A region of interest (ROI) was drawn over the tissue compartment in each image, and positive ID particles in the ROI, defined as containing at least five connected pixels all with above the background color intensity, were identified. The color intensity values from every pixel of positive ID particles were summed and normalized by the tissue ROI area (per μm2). To evaluate the fraction of tissue vascular area expressing EGFP, the endothelial marker-positive area and EGFP-positive area within the tissue ROI were quantified by summing up the areas of all positive ID particles based on the color detection threshold of 70 for both the green and red colors. The percentage ratio of EGFP-positive area to EC-positive area in each organ was calculated for evaluating the vascular EC vector gene expression. Mean and s.d. of data points in each organ derived from experiment mice were plotted.
Quantitative Flow Cytometry
Peripheral blood was collected from mice treated with vehicle or clodronate liposomes, and 50 μl of each sample was spiked with re-fluorescent beads (Invitrogen, CA, USA) as internal standards for absolute counts. Red blood cells were lysed with red blood cell lysis buffer (BioLegend, San Diego, CA, USA), and mononuclear cells (MNCs) were isolated. MNCs were then washed with cold PBS and stained with CD11b-fluorescein isothiocyanate (FITC) and CD45-phycoerythrin (PE) (BD Pharmingen, BD Biosciences, San Jose, CA, USA) for 1 h on ice. Then, cells were washed, re-suspended in PBS, and analyzed by flow cytometry. Forward scatter (FSC) and side scatter (SSC) were used to gate monocytes as high-size (FSC)/low-granulation (SSC) population; moreover, the monocyte population was further characterized as CD11b-positive/CD45-positive. The count of the FSC-high/SSC-low/CD11b-positive/CD45-positive monocyte population was normalized to the count of the fluorescent beads. Results were presented as the percentage of average of vehicle-treated mice.
Statistical Analysis
All data are reported as mean±s.d. Significance of the means between the mouse groups was determined using unpaired Student’s test for each organ with Bonferroni correction for multiple independent comparisons carried out on different organs from the same cohort of mice. Statistical significance was defined as adjusted P<0.05 (GraphPad Prism, San Diego, CA, USA).
RESULTS
MBP Pseudotyping Attenuated Hepatocyte Vector Expression While Producing Widespread Multi-Organ Vascular EC Expression
Our initial work with the Ad.MBP.CMV vector focused on differential liver and lung transduction.1 Here we sought to test the multi-organ biodistribution of Ad.MBP.CMV using semiquantitative tissue section immunofluorescence analysis. As expected, Ad5.CMV-mediated expression was predominantly localized in liver hepatocytes and readily detectable in the spleen reticuloendothelial system and ECs (Supplementary Figure S1A). Vector expression was scarcely found in the lung, heart, kidney, gastrocnemius muscle, pancreas, small bowel, large bowel, and not detectable in any part of the brain (Figure 1b). In contrast, Ad.MBP.CMV produced EC expression throughout the microvasculature of heart, kidney, muscle, pancreas, intestine, and brain (Figure 1a). Vector EC co-localization was confirmed using high-magnification EFI imaging in these organs (Supplementary Figure S2). Surprisingly, robust transgene expression was detectable in ECs within all brain regions, including the cerebrum, cerebellum, hippocampus, and medulla (Supplementary Figure S1B). To quantify vector transgene expression, EGFP fluorescence intensity was summed in a tissue ROI and normalized by the ROI area (per μm2) in each organ. Liver sections from Ad.MBP.CMV-injected mice exhibited a five-fold reduction in the EGFP fluorescence intensity compared with the Ad5.CMV counterparts (Figure 1b). Liver detargeting was associated with two-fold increase in vector expression in splenic reticuloendothelial cells and ECs (Figure 1b). As the lung, heart, kidney, pancreas, small bowel, large bowel, and brain were barely transducible or not at all by the Ad5.CMV, the retargeting enhancement of the Ad.MBP.CMV to these organs was remarkable, ranging from >10-fold increase in the pancreas, small bowel, and large bowel, >100-fold increase in lung and kidney, >1000-fold increase in heart and muscle, and >10 000-fold increase in brain (Figure 1b, red bars vs blue bars for Lu, H, K, M, P, SB, LB, and B).

Incorporation of MBP into Ad5 drastically increased viral gene expression to vascular beds of multiple host organs. (a) Immunofluorescence microscopy analysis of vector EGFP expression in host organs following intravenous injection of 1 × 1011 viral particles (vp) of Ad.MBP.CMV into adult C57BL/6J mice revealed prominent transgene expression in the lung, heart, kidney, gastrocnemius muscle, pancreas, small bowel, large bowel, and brain. Co-staining of tissue sections with an EC-specific endomucin/CD31 cocktail revealed that EGFP expression was restricted to the vasculature. (b) EGFP fluorescence per μm2 of tissue section area (FI, fluorescence intensity) in each organ derived from Ad5.CMV-injected mice (n=4 for all organs) vs that from Ad.MBP.CMV-injected mice (n=10 for liver, spleen, heart, kidney, muscle, small bowel, and brain; n=7 for lung, pancreas, and large bowel). (c) The percentage of vascular EC area expressing EGFP in each organ derived from Ad5.CMV-injected mice (n=4 for all the organs) vs that from Ad.MBP.CMV-injected mice (n=10 for heart, kidney, muscle, small bowel, and brain; n=7 for lung, pancreas, and large bowel). Bar graph is mean±s.d. Asterisk: adjusted P<0.05. Magnification: × 100, Red: endomucin/CD31, Green: EGFP immunofluorescence, Blue: DAPI. Li: liver, S: spleen, Lu: lung, H: heart, K: kidney, M: muscle, P: pancreas, SB: small bowel, LB: large bowel, B: brain.
We next determined the EC expression efficiency of the Ad.MBP.CMV vs Ad5.CMV in multiple organs by quantifying the percentage of total tissue EC area expressing EGFP in each organ. Ad.MBP.CMV targeted >62% of blood vessels in all regions of the brain (B), 21% in lung (Lu), 26% in heart (H), 33% in kidney (K), 38% in muscle (M), 30% in pancreas (P), 16% in small bowel (SB), and 6% in large bowel (LB) (Figure 1c). Other than liver and spleen, pancreas and small bowel were the only organs where Ad5.CMV produced an appreciable, but still rare, vascular EC expression (Figure 1c).
To further test EC-specific expression, immunofluorescence for the pericyte markers, PDGFRβ or proteoglycan nerve-glial antigen 2 (NG2), and the pan-hematopoietic lineage cell marker CD45 or macrophage marker F4/80 were performed. High-magnification revealed that the EGFP-expressing cells were clearly distinct from the PDGFRβ-positive or NG2-positive cells in all the organs (Supplementary Figure S2). Ad.MBP.CMV was expressed in rare CD45-positive hematopoietic cells and F4/80-positive macrophages in the liver and spleen but not in any other sampled organs (Supplementary Figure S3).
Warfarin liver Detargeting Failed to Increase Ad.MBP.CMV Multi-organ EC Expression
Although the Ad.MBP.CMV yielded an impressive level of hepatocyte detargeting, liver remained a substantial transductional and transcriptional target (Figure 2). As the major pathway directing Ad5 hepatocyte sequestration is mediated by coagulation Factor X-viral hexon binding, we tested whether warfarin could affect diminution in the level of Ad.MBP.CMV expression in hepatocytes.22 Warfarin pretreatment diminished the number of EGFP-expressing hepatocytes (Figure 2a). The residual number of vector-expressing hepatocytes was similar to our previous work with Ad5-based vectors with wild-type capsids. This residual hepatocyte vector expression, following warfarin treatment, has been seen by others and likely represents a ‘floor’ for the efficacy of pharmacological blockade.22 However, in contrast to our previous work with vectors with wild-type capsids,23 warfarin failed to enhance EC expression in all other organs (Figures 2a and b). These data suggested that either the Ad.MBP.CMV peripheral vascular EC vector expression was saturated at our 1 × 1011 viral particle dose or that complement opsonization facilitated destruction of the vector dose increment that escaped hepatocyte transduction.24

Warfarin pretreatment reduced Ad.MBP.CMV liver tropism but did not alter gene expression in other host organs. (a) Warfarin, 5 mg/kg, on days −3 and −1 before vector injection diminished hepatocyte expression but did not change transgene expression in the spleen. (b) EGFP fluorescence per μm2 of tissue area in each organ derived from warfarin-treated mice (n=3 for all the organs) normalized as the percentage of the mean value of vehicle-treated or untreated counterparts (n=10 for liver, spleen, heart, kidney, muscle, small bowel, and brain; n=7 for lung) with s.d. Warfarin pretreatment reduced vector liver expression by 68% (Li) but did not lead to a significant change in gene expression in the spleen (S), lung (Lu), heart (H), kidney (K), muscle (M), small bowel (SB), or brain (B). Asterisk indicates adjusted P<0.05. Magnification: × 100, Red: CD31/endomucin, Green: EGFP immunofluorescence, Blue: DAPI.
Ad.MBP.CMV Dose Reduction Produced Organ-Specific Non-Linear EC Expression Reduction
To test the sensitivity of each organ vascular bed for Ad.MBP.CMV expression, we challenged mice with injection of 2 × 1010 viral particles. The lower viral dose drastically reduced tissue Ad.MBP.CMV expression in the liver, spleen, pancreas, heart, kidney, muscle, pancreas, small bowel, and brain. Strikingly, the frequency and EC expression level in the lung remained unaffected by vector dose reduction (Figures 3a and b). Comparison of EGFP fluorescence intensity of low- vs high-dose tissue samples revealed that splenic and brain transgene expression was 16 and 31% of the high-dose counterparts (Figures 3c, S and B). These levels of reduction in EGFP expression were within the range of linear response to the viral dose difference (20%). However, the drastically diminished expression in the liver (5% of high-dose level), heart (0.4% of high-dose level), kidney (0.5% of high dose level), muscle (0.1% of high-dose level), pancreas (0.4% of high-dose level), and small bowel (3% of high-dose level) was non-linear. Similar to EC expression frequency analysis, vector dose reduction failed to significantly diminish transgene expression in the lung (91% of high-dose level, P=0.588). These results suggested that Ad.MBP.CMV lung-specific EC expression targeting may be achievable through vector dose fine tuning.

Systemic administration of a low dose of Ad.MBP.CMV into adult mice produced differential and non-linear reduction in gene expression in the host organs. (a) EGFP expression in the host liver, spleen, lung, and brain following intravenous injection of 1 × 1011 or 2 × 1010 vp of Ad.MBP.CMV into adult mice. Lowering vector dosage significantly reduced EGFP expression in vascular ECs of the liver, spleen, and brain, but did not change the expression in the lung. (b) EGFP fluorescence per μm2 of tissue area in each organ derived from the low-dose group (n=6 for each organ). (c) Normalization of the tissue EGFP fluorescence intensity values in panel (b) to the mean value of the high-dose counterparts. The spleen and brain EGFP expression in the low-dose group was 16 and 31% of the high-dose counterparts. However, low virus dose drastically diminished the transgene expression in the heart, kidney, muscle, pancreas, and small bowel (3% of high-dose level). The low dose did not significantly alter the transgene expression in the lung (91% of high-dose level). Asterisk indicates P<0.05. Magnification: × 100, Red: endomucin/CD31, Green: EGFP immunofluorescence, Blue: DAPI. Li: liver, S: spleen, Lu: lung, H: heart, K: kidney, M: muscle, P: pancreas, SB: small bowel, B: brain.
Mononuclear Cell Depletion failed to Diminish Ad.MBP.CMV EC Transgene Expression
The Ad.MBP.CMV acquired a specific and high-affinity binding to myeloid cells ex vivo, compared with the Ad5.1, 17 To test the necessity of mononuclear cells for Ad.MBP.CMV EC transgene expression in intact mice, we administered two doses of intravenous clodronate liposomes and measured the percentage of circulating CD11b-positive cells and the level of multi-organ tissue section EGFP fluorescence (Figures 4a–c). Clodronate treatment depleted circulating CD11b-positive leukocytes by 77% and completely depleted F4/80-positive macrophages in the liver (Kupffer cells) and spleen (Figure 4a and Supplementary Figure S4). In contrast, clodronate barely reduced resident macrophages in the lung, small bowel, heart, and kidney (data not shown). Clodronate increased Ad.MBP.CMV EC lung expression by two-fold but did not significantly alter EC transgene expression level or the EC-specific expression pattern in the liver, spleen, heart, kidney, muscle, pancreas, small bowel, or brain (Figure 4b). The lack of increase in hepatocyte expression was surprising given previous reports on the scavenging function of liver Kupffer cells;25 however, others have also reported a modest, though statistically insignificant level of clodronate-mediated Ad vector liver expression enhancement.26 Most importantly, our data demonstrates that circulating monocytes and macrophages were dispensable for Ad.MBP.CMV organ EC expression. However, the persistence of extrahepatic organ Cd11b and F4/80 cells following clodronate depletion does not rule out the marginated myeloid cell pool as a mechanism for vector-EC handoff.1

Depletion of circulating monocytes and hepatic and splenic macrophages lead to an increased Ad.MBP.CMV gene expression in the lung without a significant change in gene expression in other organs. (a) Representative flow cytometry plots (left panel) quantifying the FSC-high/SSC-low/CD11b-positive/CD45-positive monocyte population in circulation. Relative frequency (right panel) of circulating monocytes from clodronate liposome-treated mice (clod, n=3) vs saline-treated mice (veh, n=3). Intravenous injection of clodronate liposomes depleted circulating CD11b-positive cells by 84%. (b) EGFP fluorescence per μm2 of tissue area in each organ derived from the saline-injected mice (n=7 for liver, spleen, heart, kidney, muscle, pancreas, small bowel, and brain; n=4 for lung) vs clodronate-liposome-injected mice (n=8 for liver, spleen, heart, kidney, pancreas, small bowel, and brain; n=7 for muscle; n=5 for lung). Intravenous clodronate increased Ad.MBP.CMV lung expression by two-fold (Lu) but did not result in a significant change in gene expression in the liver (Li), spleen (S), heart (H), kidney (K), muscle (M), pancreas (P), small bowel (SB), or brain (b). Asterisk indicates adjusted P<0.05.
EC-specific ROBO4 Gene Promoter/Enhancer Ablated the MBP Vector Hepatocyte Expression but Reduced EC Expression in other Organs
We recently engineered an Ad5 vector for transcriptional targeting of ECs using the EC-specific human ROBO4 gene enhancer/promoter fragment.10, 23 To test whether the combination of transcriptional with transductional targeting could produce enhanced multi-organ EC expression, we replaced the CMV enhancer/promoter with the ROBO4 enhancer/promoter. The dual targeted Ad.MBP.ROBO4 vector was administered intravenously, and organs were analyzed for vector transgene expression (Figure 5a). Ad.MBP.ROBO4 abrogated hepatocyte expression and instead EGFP was detectable in a scattered population of liver ECs (Figure 5a). The Ad.MBP.ROBO4 also produced EC transgene expression in the vascular area fraction in the spleen (23%), kidney (23%), lung (10%), muscle (9%), heart (10%), and brain (15%), but produced very low expression in the small bowel and large bowel (1 and 2%, respectively) (Figure 5b, S, Lu, H, K, M, SB, LB, and B). Collectively, the ROBO4 enhancer/promoter produced lower host organ EC expression frequency compared with Ad.MBP.CMV in each organ. However, the undetectable vector transgene expression in hepatocytes highlighted the enhanced EC stringency of the ROBO4 compared with the CMV promoter in the Ad.MBP vector.

Ad.MBP.ROBO4 detargeted hepatocyte expression but reduced levels of vascular EC expression in other host organs. (a) EGFP expression following intravenous injection of 1 × 1011 vp of Ad.MBP.ROBO4 into adult mice. Ad.MBP.ROBO4 yielded punctate vascular EC expression in the liver but showed a reduced targeting efficiency to vascular ECs in the spleen, lung, heart, kidney, muscle, small bowel, and brain. (b) The EGFP-positive vascular area analysis was performed as shown in Figure 1c. Magnification: × 100, Red: endomucin/CD31, Green: EGFP immunofluorescence, Blue: DAPI. Li: liver, S: spleen, Lu: lung, H: heart, K: kidney, M: muscle, SB: small bowel, LB: large bowel, B: brain.
DISCUSSION
Vascular endothelium has been a sought after gene therapy target because of its immediacy to blood-borne therapeutics and its pathophysiological role in a wide range of benign and malignant diseases.2, 5, 15, 27 Despite their accessibility, vascular ECs are poor transduction targets for unmodified Ad5 vectors.14 In addition, systemically administered Ad5 is rapidly opsonized by circulating IgM antibodies and complement components, leading to virus clearance by liver Kupffer cells.8 Ad5 also avidly binds to blood coagulation factor X, which bridges the virus to hepatocytes by interacting with cell surface heparan sulfate proteoglycans.22 Liver Kupffer cell clearance and hepatocyte transduction greatly limit circulating Ad5 vector efficacy. Thus, molecular engineering efforts to achieve Ad vector vascular targeting have focused on diminishing or abrogating liver tropism and opsonization while increasing EC transduction.8, 10 Liver detargeting engaged genetic capsid modification. Virus opsonization diminution has been addressed using chemical shielding of Ad5 capsid proteins.8 One approach to EC transductional targeting has been vector pseudotyping. Ad5 vectors pseudotyped with fibers or fiber knobs from different human, or non-human, serotypes exhibited improved transduction efficiency of cultured human or rodent (rat) ECs.28, 29, 30 Another approach has been addition of ‘angio-adenobodies’ onto the Ad vector capsid. These bispecific biologics shield the native tropism receptors with one antibody site and target EC luminal surface receptors using the other antibody site.6 EC receptors targeted by this mechanism include VEGFR-2, Tie-2, E-selectin, endoglin, and membrane-bound angiotensin converting enzyme.7, 31, 32, 33 EC transduction has also been achieved through capsid fiber knob display of peptide ligands such as the arginine-glycine-asparate (RGD) motif cognate for the angiogenesis-associated integrins αv/β5 and αv/β329, 34 or the DDTRHWG peptide.3 A parallel strategy for EC specificity has been transcriptional targeting using enhancer/promoter elements of endothelial-specific genes, such as VEGFR-2, VEGFR-1, preproendothelin-1, and roundabout-4.10, 23, 35, 36, 37, 38 Transcriptional targeting restricts vector transgene expression to specific EC populations that in most instances are angiogenic and in some cases also hypoxic. However, the transcriptional strategy, when applied alone, does not alter the Kupffer cell sequestration or hepatocyte transduction. Recent efforts have focused on the combination of transductional and transcriptional strategies to achieve enhanced organ or disease-specific EC vector transgene expression.10, 30 Despite progress, systemically administered Ad vectors are still ineffective in gene transfer to some clinically important organs. Dose escalation to achieve appreciable vector expression in marginally accessible organs likely will fail due to dose-limiting adverse effects such as liver toxicity, cytokine storm, or organ imperviousness to the vector.39 Collectively, the limitations of current EC targeting efforts reinforce the need for further vector improvement.
Ad.MBP was previously shown to preserve the myeloid cell-binding specificity of the WTLDRGY MBP peptide ex vivo17 but efficiently and preferentially target gene expression to the lung microvessel ECs in vivo.1 The latter work used single-cell lung suspensions and confirmed that Ad.MBP solely bound to myeloid cells and not to ECs. Co-culture of virus-loaded myeloid cells on an EC monolayer provided indirect evidence supporting a myeloid cell-mediated viral ‘hand-off’ mechanism for potentiating the EC transduction.1 Similar carrier cell hand-off or ‘hitchhiking’ target cell transduction was proposed for other viruses in vivo.40, 41 A central tenet of vector hand-off postulates close contact of virus-carrier cells to the target cells enabling viral penton access with target cell integrins for internalization, bypassing the requirement of an initial attachment step in cell transduction.41 Indeed, the previous lung work revealed that the Ad.MBP virions rapidly bound to the lung following intravenous injection. There the hypothesis was that vector attachment and ‘hand-off’ to lung ECs was mediated by marginated neutrophils.1 Our current data extended these findings and revealed that the MBP peptide possessed a much broader EC-specific tropism in vivo, and many of the permissive recipient organs exhibit a wide range of tissue-specific forms of resident myeloid cells. Our clodronate study provided evidence suggesting that circulating mononuclear cells and tissue-resident macrophages in the liver and spleen were dispensable or redundant for mediating the Ad.MBP EC expression. Nevertheless, it remains to be demonstrated whether the resident myeloid populations in other target organs are essential for the virus EC targeting. Importantly, the existing data on Ad.MBP transduction may also be explained by an alternative mechanism to hand-off, in which a yet to identified MBP cognate cell surface receptor or blood-borne factors also contribute Ad.MBP EC transduction. Of additional interest was the fact that the Ad.MBP vector produced multi-organ vascular expression following warfarin-mediated Factor X depletion. Indeed, previous work demonstrated that Factor X-virus hexon binding ‘shielded’ the vector from peripheral natural antibody-mediated destruction in immunocompetent mice.24 One explanation could be that the fiber modification of the Ad.MBP vector somehow protected the vector in the peripheral circulation. That said, overall our work additionally differs from this paper in that we repeatedly demonstrated a robust warfarin effect in Rag2 knockout complement-deficient mice, whereas warfarin was ineffectual in the reported Rag-deficient background.24 As such, the role of Factor X-complement-mediated systemic vector opsonization will certainly need to be independently tested.
Multi-organ expression analysis also enabled us to expand on previous observations and discover the exquisite lung tropism of our Ad.MBP vector. Viral particle dose reduction essentially eliminated gene transfer to most organs while maintaining robust lung expression. This apparent pulmonary vascular avidity suggests that the Ad.MBP vector could be an ideal vehicle for treatment of pulmonary diseases, particularly those initiated by single gene mutations. Over the past two decades, several gene therapy trials have been accomplished for alpha-1 antitrypsin deficiency (AATD). AATD is caused by a mutation in the alpha-1 antitrypsin (AAT) gene that produces a misfolded, nonfunctional protease inhibitor.42 Affected individuals develop emphysema due to pulmonary tissue destruction and cirrhosis from hepatocyte AAT protein aggregation. Gene therapy has sought to restore lung AAT levels via elevation of circulating wild-type enzyme and inhibition of hepatocyte mutant protein accumulation.43 Recently, a combinatorial approach using microRNA (miR) targeting the AAT mutant protein and a transgene encoding a miR-resistant wild-type protein was highly efficacious in a mouse genetic model.44, 45 The dual hepatocyte and lung EC expression profile of our Ad.MBP vector suggests that it could be an ideal vehicle for AATD therapy with a similar miR/wild-type bicistronic transgene. Likewise, cystic fibrosis (CF) is a lung disease caused by disrupted chloride channel function particularly affecting the airway epithelium owing to mutations in the gene cystic fibrosis transmembrane conductance regulator (CFTR) gene. So far, 25 viral and non-viral gene therapy clinical trials for CF have been conducted or are in progress aiming at direct gene transfer to the airway epithelium.46 Some the studies evidenced partial correction of the chloride transport defect, but clinical benefits have not been equivocal.46 Although the primary CF defect is abnormal epithelial mucous production, marked neutrophil recruitment, neutrophil elastase production, and overall inflammatory hypersensitivity accompany the disease and lead to bronchial wall destruction (bronchiectasis). Here, the perialveolar and peribronchial Ad.MBP vascular EC expression could be a platform for secretion of vector transgenes inhibiting neutrophil elastase or inflammatory mediator expression.47 In addition, the unique multi-organ vascular expression of the Ad.MBP vector, in particular within the intestinal and pancreatic vasculature, presents a tantalizing opportunity to treat nearly all of the major organ systems affected by CF. Again, however, the epithelial-centric nature of CF will require a creative choice of transgenes to achieve multi-site efficacy.
Although Ad.MBP has many conceivable applications in other organs, its widespread expression in cardiac and brain vasculature is particularly exciting. In the heart, gene therapy has focused on ischemic disease.48 Although the immediate cause of cardiac ischemia is coronary artery atherosclerosis, myocardial remodeling is the principal mechanism for development of chronic congestive heart failure.49 Restoration of blood flow has been approached using gene therapy as a surgical adjuvant or as a primary treatment.4, 50 So far, more than a dozen of trials using first-generation Ad5-based vectors have been or are being carried out, to deliver angiogenic factors such as the VEGF family members, FGF-4, HGF, or angiogenic regulator HIF1α to ischemic myocardium via the intracoronary or intramuscular route. Although a few Phase I and II studies resulted in encouraging end points, such as statistically significant increase in regional myocardial perfusion51, 52 or improved exercise time and angina,53, 54, 55 several phase III trials yielded overall disappointing results.56, 57 The reasons for the variegated therapeutic outcomes have not been fully understood, but vector types, therapeutic genes, routes of administration, and the presence of anti-Ad5-neutralizing antibodies may have accounted for the observed outcomes.4 Our Ad.MBP vector could solve the dual challenge of coronary perfusion and myocardial remodeling. Coronary perfusion could be increased using Ad.MBP vector armed with constitutively active hypoxia-inducible factors (HIFs).38 The advantage of HIFs is that these transcription factors upregulate a collection, rather than a single angiogenic factor, and induce a leakage-resistant hypervascularity.58, 59 The widespread myocardial vascular distribution of Ad.MBP presents the opportunity to capitalize on EC angiocrine functions such that ECs are transformed into local sources of HIF-mediated angiogenic factors to both preserve marginal zone myocardial viability and potentially augment arteriogenesis. Similarly, Ad.MBP vectors containing polycistronic transgenes encoding the same molecules apparently secreted by myocardial stem cells or endothelial stem cells could possibly effect restorative rather than pathological myocardial remodeling by inducing expansion and myocardial differentiation of perivascular-resident cardiac stem cells.60, 61
In contrast to the goal of improvement of vascularity through transient angiogenic gene expression for heart gene therapy, brain gene therapy strives to achieve long-term expression in neurological disorders such as Alzheimer’s, amyotrophic lateral sclerosis (ALS), or brain cancer.62, 63, 64 Benign neurological diseases have been approached using intraventricular vector injection. Although intraventricular vector injection has produced some efficacy preclinically, this strategy predominantly targets the brain surface rather than the brain parenchyma. Recent studies have highlighted the potential for local production of secreted antibodies targeting mutant SOD in amyotrophic lateral sclerosis and tau protein in Alzheimer’s.65, 66 Here again, the ability of our Ad.MBP vector to target >62% of blood vessel beds in all regions of the brain offers the intriguing potential for treating the multifocal intraparenchymal mechanisms for both diseases. In brain cancer, glioblastoma (GBM) in particular, gene therapy has predominantly been restricted to vector injections into the surgical bed.67 This approach ultimately fails due to the invasiveness of brain cancer wherein host vessel co-option leads to widespread permeation. Moreover, GBM is notoriously resistant to either conventional chemo-irradiation or molecularly targeted therapies. This resistance is based in part on GBM stem cells that have been shown to reside in perivascular niches throughout the brain.68 Collectively, the remarkable tropism of the Ad.MBP vector for brain vascular ECs offers the possibility for targeting perivascular GBM stem cells by angiocrine-mediated secretion of cytotoxics or molecules blocking signaling pathways maintaining this therapy-resistant cell population.69, 70 Indeed, we have preclinical studies underway to test these hypotheses.
In summary, our Ad.MBP vector enables unprecedented multi-organ vascular access. This vector can be used to harness ECs for production of a variety of therapeutic molecules for a diverse collection of benign and malignant diseases. Although promiscuous vascular vector expression might be detrimental, its multi-organ tropism may in fact be uniquely beneficial. In cases wherein greater disease specificity is required, the inherent EC vector tropism enables swapping in enhancer/promoters tailored to the altered microenvironment created by each disease in each organ. Clearly, considerable preclinical modeling, functional testing, and possibly additional genetic engineering will need to be done to validate the Ad.MBP vector’s efficacy and possibly increase its expression duration. However, its potential to treat multiple diseases in previously inaccessible organs is compelling.
References
Alberti MO, Deshane JS, Chaplin DD et al. A myeloid cell-binding adenovirus efficiently targets gene transfer to the lung and escapes liver tropism. Gene Therapy 2013;20:733–741.
Dong Z, Nor JE . Transcriptional targeting of tumor endothelial cells for gene therapy. Adv Drug Deliv Rev 2009;61:542–553.
Nicol CG, Denby L, Lopez-Franco O et al. Use of in vivo phage display to engineer novel adenoviruses for targeted delivery to the cardiac vasculature. FEBS Lett 2009;583:2100–2107.
Bradshaw AC, Baker AH . Gene therapy for cardiovascular disease: perspectives and potential. Vasc Pharmacol 2013;58:174–181.
Muro S, Koval M, Muzykantov V . Endothelial endocytic pathways: gates for vascular drug delivery. Curr Vasc Pharmacol 2004;2:281–299.
Glasgow JN, Everts M, Curiel DT . Transductional targeting of adenovirus vectors for gene therapy. Cancer Gene Ther 2006;13:830–844.
Haisma HJ, Kamps GK, Bouma A et al. Selective targeting of adenovirus to alphavbeta3 integrins, VEGFR2 and Tie2 endothelial receptors by angio-adenobodies. Int J Pharmaceut 2010;391:155–161.
Duffy MR, Parker AL, Bradshaw AC et al. Manipulation of adenovirus interactions with host factors for gene therapy applications. Nanomedicine (Lond) 2012;7:271–288.
Beatty MS, Curiel DT . Chapter two—adenovirus strategies for tissue-specific targeting. Adv Cancer Res 2012;115:39–67.
Kaliberov SA, Kaliberova LN, Hong LuZ et al. Retargeting of gene expression using endothelium specific hexon modified adenoviral vector. Virology 2013;447:312–325.
Magnenat L, Schwimmer LJ, Barbas CF 3rd . Drug-inducible and simultaneous regulation of endogenous genes by single-chain nuclear receptor-based zinc-finger transcription factor gene switches. Gene Therapy 2008;15:1223–1232.
Bochman ML, Paeschke K, Zakian VA . DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet 2012;13:770–780.
van Rooij E . The art of microRNA research. Circ Res 2011;108:219–234.
Baker AH, Kritz A, Work LM et al. Cell-selective viral gene delivery vectors for the vasculature. Exp Physiol 2005;90:27–31.
Lindemann D, Schnittler H . Genetic manipulation of endothelial cells by viral vectors. Thromb Haemost 2009;102:1135–1143.
Franses JW, Edelman ER . The evolution of endothelial regulatory paradigms in cancer biology and vascular repair. Cancer Res 2011;71:7339–7344.
Alberti MO, Roth JC, Ismail M et al. Derivation of a myeloid cell-binding adenovirus for gene therapy of inflammation. PLoS One 2012;7:e37812.
Belousova N, Korokhov N, Krendelshchikova V et al. Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol 2003;77:11367–11377.
He TC, Zhou S, da Costa LT et al. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 1998;95:2509–2514.
Short JJ, Rivera AA, Wu H et al. Substitution of adenovirus serotype 3 hexon onto a serotype 5 oncolytic adenovirus reduces factor X binding, decreases liver tropism, and improves antitumor efficacy. Mol Cancer Ther 2010;9:2536–2544.
van Rooijen N, Hendrikx E . Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol 2010;605:189–203.
Waddington SN, McVey JH, Bhella D et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008;132:397–409.
Lu ZH, Kaliberov S, Sohn RE et al. Transcriptional targeting of primary and metastatic tumor neovasculature by an adenoviral type 5 roundabout4 vector in mice. PLoS One 2013;8:e83933.
Xu Z, Qiu Q, Tian J et al. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat Med 2013;19:452–457.
Wolff G, Worgall S, van Rooijen N et al. Crystal RG. Enhancement of in vivo adenovirus-mediated gene transfer and expression by prior depletion of tissue macrophages in the target organ. J Virol 1997;71:624–629.
Bradshaw AC, Coughlan L, Miller AM et al. Biodistribution and inflammatory profiles of novel penton and hexon double-mutant serotype 5 adenoviruses. J Control Release 2012;164:394–402.
Aird WC . Endothelial Biomedicine. Cambridge University Press: Cambridge, UK; New York, NY, USA, 2007.
Shinozaki K, Suominen E, Carrick F et al. Efficient infection of tumor endothelial cells by a capsid-modified adenovirus. Gene Therapy 2006;13:52–59.
Preuss MA, Glasgow JN, Everts M et al. Enhanced gene delivery to human primary endothelial cells using tropism-modified adenovirus vectors. Open Gene Ther J 2008;1:7–11.
White KM, Alba R, Parker AL et al. Assessment of a novel, capsid-modified adenovirus with an improved vascular gene transfer profile. J Cardiothorac Surg 2013;8:183.
Reynolds PN, Zinn KR, Gavrilyuk VD et al. A targetable, injectable adenoviral vector for selective gene delivery to pulmonary endothelium in vivo. Mol Ther 2000;2:562–578.
Nettelbeck DM, Miller DW, Jerome V et al. Targeting of adenovirus to endothelial cells by a bispecific single-chain diabody directed against the adenovirus fiber knob domain and human endoglin (CD105). Mol Ther 2001;3:882–891.
Bachtarzi H, Stevenson M, Subr V et al. E-selectin is a viable route of infection for polymer-coated adenovirus retargeting in TNF-alpha-activated human umbilical vein endothelial cells. J Drug Target 2011;19:690–700.
Nicklin SA, Von Seggern DJ, Work LM et al. Ablating adenovirus type 5 fiber-CAR binding and HI loop insertion of the SIGYPLP peptide generate an endothelial cell-selective adenovirus. Mol Ther 2001;4:534–542.
Song W, Sun Q, Dong Z et al. Antiangiogenic gene therapy: disruption of neovascular networks mediated by inducible caspase-9 delivered with a transcriptionally targeted adenoviral vector. Gene Therapy 2005;12:320–329.
Greenberger S, Shaish A, Varda-Bloom N et al. Transcription-controlled gene therapy against tumor angiogenesis. J Clin Invest 2004;113:1017–1024.
Reynolds PN, Nicklin SA, Kaliberova L et al. Combined transductional and transcriptional targeting improves the specificity of transgene expression in vivo. Nat Biotechnol 2001;19:838–842.
Tal R, Shaish A, Rofe K et al. Endothelial-targeted gene transfer of hypoxia-inducible factor-1alpha augments ischemic neovascularization following systemic administration. Mol Ther 2008;16:1927–1936.
Zaiss AK, Machado HB, Herschman HR . The influence of innate and pre-existing immunity on adenovirus therapy. J Cell Biochem 2009;108:778–790.
Cole C, Qiao J, Kottke T et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med 2005;11:1073–1081.
Roth JC, Curiel DT, Pereboeva L . Cell vehicle targeting strategies. Gene Therapy 2008;15:716–729.
Mueller C, Flotte TR . Gene-based therapy for alpha-1 antitrypsin deficiency. COPD 2013;10 (Suppl 1):44–49.
Flotte TR, Mueller C . Gene therapy for alpha-1 antitrypsin deficiency. Hum Mol Genet 2011;20:R87–R92.
Mueller C, Tang Q, Gruntman A et al. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther 2012;20:590–600.
Li C, Xiao P, Gray SJ et al. Combination therapy utilizing shRNA knockdown and an optimized resistant transgene for rescue of diseases caused by misfolded proteins. Proc Natl Acad Sci USA 2011;108:14258–14263.
Oakland M, Sinn PL, McCray PB Jr. . Advances in cell and gene-based therapies for cystic fibrosis lung disease. Mol Ther 2012;20:1108–1115.
Kelly E, Greene CM, McElvaney NG . Targeting neutrophil elastase in cystic fibrosis. Expert Opin Ther Targets 2008;12:145–157.
Tang T, Hammond HK . Gene transfer for congestive heart failure: update 2013. Transl Res 2013;161:313–320.
van Berlo JH, Maillet M, Molkentin JD . Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest 2013;123:37–45.
Kaminsky SM, Rosengart TK, Rosenberg J et al. Gene therapy to stimulate angiogenesis to treat diffuse coronary artery disease. Hum Gene Ther 2013;24:948–963.
Hedman M, Hartikainen J, Syvanne M et al. Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation 2003;107:2677–2683.
Hedman M, Muona K, Hedman A et al. Eight-year safety follow-up of coronary artery disease patients after local intracoronary VEGF gene transfer. Gene Therapy 2009;16:629–634.
Rosengart TK, Lee LY, Patel SR et al. Six-month assessment of a phase I trial of angiogenic gene therapy for the treatment of coronary artery disease using direct intramyocardial administration of an adenovirus vector expressing the VEGF121 cDNA. Ann Surg 1999;230:466–470 discussion 470–472.
Rosengart TK, Bishawi MM, Halbreiner MS et al. Long-term follow-up assessment of a phase 1 trial of angiogenic gene therapy using direct intramyocardial administration of an adenoviral vector expressing the VEGF121 cDNA for the treatment of diffuse coronary artery disease. Hum Gene Ther 2013;24:203–208.
Stewart DJ, Hilton JD, Arnold JM et al. Angiogenic gene therapy in patients with nonrevascularizable ischemic heart disease: a phase 2 randomized, controlled trial of AdVEGF(121) (AdVEGF121) versus maximum medical treatment. Gene Therapy 2006;13:1503–1511.
Henry TD, Grines CL, Watkins MW et al. Effects of Ad5FGF-4 in patients with angina: an analysis of pooled data from the AGENT-3 and AGENT-4 trials. J Am Coll Cardiol 2007;50:1038–1046.
Kastrup J, Jorgensen E, Fuchs S et al. A randomised, double-blind, placebo-controlled, multicentre study of the safety and efficacy of BIOBYPASS (AdGVVEGF121.10NH) gene therapy in patients with refractory advanced coronary artery disease: the NOVA trial. EuroIntervention 2011;6:813–818.
Oladipupo SS, Hu S, Santeford AC et al. Conditional HIF-1 induction produces multistage neovascularization with stage-specific sensitivity to VEGFR inhibitors and myeloid cell independence. Blood 2011;117:4142–4153.
Elson DA, Thurston G, Huang LE et al. Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev 2001;15:2520–2532.
Kamdar F, Jameel MN, Score P et al. Cellular therapy promotes endogenous stem cell repair. Can J Physiol Pharmacol 2012;90:1335–1344.
Ou DB, Zeng D, Jin Y et al. The long-term differentiation of embryonic stem cells into cardiomyocytes: an indirect co-culture model. PLoS One 2013;8:e55233.
Coune PG, Schneider BL, Aebischer P . Parkinson's disease: gene therapies. Cold Spring Harb Perspect Med 2012;2:a009431.
Ramaswamy S, Kordower JH . Gene therapy for Huntington's disease. Neurobiol Dis 2012;48:243–254.
Assi H, Candolfi M, Baker G et al. Gene therapy for brain tumors: basic developments and clinical implementation. Neurosci Lett 2012;527:71–77.
Patel P, Kriz J, Gravel M et al. Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther 2014;22:498–510.
Yanamandra K, Kfoury N, Jiang H et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013;80:402–414.
Tobias A, Ahmed A, Moon KS et al. The art of gene therapy for glioma: a review of the challenging road to the bedside. J Neurol Neurosurg Psychiatry 2013;84:213–222.
Gilbertson RJ, Rich JN . Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 2007;7:733–736.
Galan-Moya EM, Le Guelte A, Lima Fernandes E et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep 2011;12:470–476.
Zhu TS, Costello MA, Talsma CE et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res 2011;71:6061–6072.
Acknowledgements
We thank Andrew Thomas for providing technical assistance.
Sources and funding: This work was funded by National Institutes of Health (NIH) Grant numbers R01CA159959 to JMA and R01CA154697 to DTC.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
An adenovirus serotype 5 vector with a myeloid cell-binding peptide (MBP) incorporated into the fiber knob (Ad.MBP) produces widespread endothelial cell expression in lung, heart, kidney, skeletal muscle, pancreas, small bowel, and brain. While binding specifically to myeloid cells ex vivo, multi-organ Ad.MBP vascular expression is independent of circulating monocytes or tissue macrophages. This vector could enable production of biologic therapeutics in previously inaccessible organs.
Rights and permissions
About this article

Cite this article
Lu, Z., Kaliberov, S., Zhang, J. et al. The myeloid-binding peptide adenoviral vector enables multi-organ vascular endothelial gene targeting. Lab Invest 94, 881–892 (2014). https://doi.org/10.1038/labinvest.2014.78
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2014.78
This article is cited by
-
Untangling the Intricacies of Infection, Thrombosis, Vaccination, and Antiphospholipid Antibodies for COVID-19
SN Comprehensive Clinical Medicine (2021)
-
ADAR1 Suppresses the Activation of Cytosolic RNA-Sensing Signaling Pathways to Protect the Liver from Ischemia/Reperfusion Injury
Scientific Reports (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}