
Abstract
Coxsackievirus B3 (CVB3) and lymphocytic choriomeningitis virus (LCMV) are both neurotropic RNA viruses, which can establish a persistent infection and cause meningitis and encephalitis in the neonatal host. Utilizing our neonatal mouse model of infection, we evaluated the consequences of early viral infection upon the host central nervous system (CNS) by comparing CVB3 and LCMV infection. Both viruses expressed high levels of viral protein in the choroid plexus and subventricular zone (SVZ), a region of neurogenesis. LCMV infected a greater number of cells in the SVZ and targeted both nestin+ (neural progenitor cell marker) and olig2+ (glial progenitor marker) cells at a relatively equal proportion. In contrast, CVB3 preferentially infected nestin+ cells within the SVZ. Microarray analysis revealed differential kinetics and unique host gene expression changes for each infection. MHC class I gene expression, several developmental-related Hox genes, and transthyretin (TTR), a protein secreted in the cerebrospinal fluid by the choroid plexus, were specifically downregulated following CVB3 infection. Also, we identified severe pathology in the choroid plexus of CVB3-infected animals at 48 h post infection accompanied by a decrease in the level of TTR and carbonic anhydrase II. These results demonstrate broader neural progenitor and stem cell (NPSC) tropism for LCMV in the neonatal CNS, whereas CVB3 targeted a more specific subset of NPSCs, stimulated a distinct early immune response, and induced significant acute damage in the choroid plexus.
Similar content being viewed by others


Main
Viral meningitis and encephalitis are serious conditions that can lead to severe morbidity and mortality particularly in the young. Although several enteroviruses and herpes viruses have been found to contribute to neonatal meningitis and encephalitis, the pathology and lasting sequelae of these infections vary substantially and are not fully understood.1, 2, 3 Early symptoms often include fever, irritability, and lack of appetite. However, these infections have been linked to long-term consequences, such as learning disabilities, demyelinating diseases, and behavioral disorders.4, 5, 6, 7, 8 In order to better understand the pathogenesis of neurotropic viral infection in the newborn host, we chose to directly compare lymphocytic choriomeningitis virus (LCMV) and coxsackievirus B3 (CVB3), both of which may cause meningoencephalitis.5, 9 Lymphocytic infiltration of the choroid plexus, or choriomeningitis, may also be an outcome of LCMV infection in the host.
CVB3 and LCMV were utilized for the study not only because of their potent ability to cause neonatal disease in the central nervous system (CNS) but also because of their apparent divergence in stimulating protective T-cell responses.10, 11 Both viruses may be associated with a persistent or chronic infection, although the mechanisms responsible for the establishment of persistence and escape from the immune response may differ substantially between the two viruses.12, 13 Also, CVB3 was previously shown to induce the recruitment of unique nestin+ myeloid cells, which assisted in virus dissemination within the CNS.14 Therefore, we wished to determine whether CVB3 uniquely activated the early host immune response, and whether this activation contributed to distinct aspects of CVB3-mediated neuropathogenesis.
CVB3 is a member of the Picornavirus family and the Enterovirus genus. Enterovirus infections may be responsible for ∼10–15 million symptomatic infections each year in the United States alone.15 LCMV, a rodent-borne Arenavirus often transmitted congenitally, may be considered an emerging cause of neonatal meningitis.3, 16 LCMV is a non-cytolytic, enveloped virus comprised of an ambisense segmented RNA genome inducing a strong cytotoxic T-lymphocyte response in the host.17 In contrast, CVB3, a cytolytic, non-enveloped virus contains a single positive-sense RNA genome, induces high levels of neutralizing antibodies yet generate very weak cytotoxic T-lymphocyte responses in its host.18
Despite considerable inherent dissimilarities between CVB3 and LCMV, including differences in T-cell activation, the presence or absence of a lipid bilayer, and substantial differences in virus replication, both viruses appeared to target neural progenitor and stem cells (NPSCs) in the CNS.19, 20, 21 Our laboratory has previously shown that CVB3 preferentially targets NPSCs in culture and in the neonatal host presumably owing to their undifferentiated status and high proliferative index.20, 22, 23, 24 In addition, we described the recruitment of unique nestin+ myeloid cells into the CNS following CVB3 infection.14 These cells were highly susceptible to infection and assisted in the dissemination of infectious virus into the neurogenic regions of the neonatal CNS at early time points. LCMV has also been shown to infect progenitor cells in neurogenic regions of the CNS, including the subventricular zone, rostral migratory stream, and olfactory bulb.25, 26 The surviving host may exhibit permanent abnormalities in synaptic function, a decrease in GABA-mediated inhibition, and hippocampal neuropathology following LCMV infection.27 Therefore, we wished to inspect progenitor cell tropism of both viruses in greater detail by immunofluorescence microscopy utilizing well-characterized cell markers for neural (nestin+) and glial (olig2+) progenitor cells.
Despite its prevalence in a clinical setting, much remains to be determined regarding CVB3 infection of the neonatal CNS.15 Similarly, while the cellular targets of infection and the subsequent immune response have been extensively studied for LCMV, the virus remains a significant prenatal and postnatal threat and few effective treatments exist.16, 25, 28, 29 We hypothesize that a direct comparison of these two viruses during early infection of the neonatal CNS may help determine previously unidentified targets of infection, illuminate the neonatal host immune response following early infection, and clarify the mechanism of viral pathogenesis. The brains of 1-day-old CVB3 and LCMV-infected mice were analyzed for gene expression changes at 12, 24, and 48 h post infection (PI) utilizing Illumina BeadArray Technology (MouseWG-6 v2 Expression Beadchips) in order to reveal the global neonatal host response for both neurotropic viral infections. In addition, we sought to examine the effects of viral infection upon the choroid plexus by histology, TUNEL assay, and fluorescence microscopy utilizing well-characterized markers of choroid plexus function. We also identified unique immune response genes activated in the neonatal CNS following both LCMV and CVB3 infection, including relative levels of chemokine and MHC class I gene expression. Illumina BeadArray Technology data provided quantitative results of many different gene products upregulated or downregulated at the transcriptional level within the CNS. Moreover, protein levels for key genes were independently verified by immunofluorescence microscopy and found to be localized within distinct regions of the CNS. Hence, both techniques complemented each other and paint a highly accurate view of the consequences of infection upon the neonatal host CNS.
Our results revealed a unique pattern of host immune activation, NPSC tropism, and virus-mediated pathology for CVB3 infection. Hence, although LCMV has been considered the prototypical virus when evaluating the host immune response and identifying mechanisms contributing to the establishment of persistence, each virus may utilize a distinctive strategy to persist within the host.
MATERIALS AND METHODS
Ethics Statement
This study was carried out in strict accordance with the requirements pertaining to animal subjects’ protections within the Public Health Service Policy and USDA Animal Welfare Regulations. Mouse experimentation conformed to the requirements of the San Diego State University Animal Research Committee and the National Institutes of Health. All experimental procedures with mice were approved by the San Diego State University Institutional Animal Care and Use Committee (Animal Protocol Form number 10-05-013F), and all efforts were made to minimize suffering.
Isolation and Production of Recombinant Coxsackievirus and LCMV
The generation of recombinant coxsackievirus expressing GFP (GFP-CVB3) has been described previously.23 Briefly, the CVB3 infectious clone (pH3) (obtained from Dr Kirk Knowlton at University of California at San Diego) was engineered to contain a unique SfiI site that allows the insertion of foreign DNA into the CVB3 genome. Virus stocks were grown on HeLa RW cells maintained in Dulbecco’s modified Eagle’s medium (DMEM, Cellgro) supplemented with 10% fetal bovine serum. Standard plaque assay on HeLa RW cells was used to titer the recombinant virus. Briefly, HeLa RW cells were grown to 70% confluency in a six-well plate, inoculated with 10−2–10−7 dilutions of viral stock, allowed to infect the cells for 1 h, and then overlaid with a 1:1 mix of 2 × DMEM and 1.2% agar for 2 days. Plates were fixed with acetic acid and methanol and stained with crystal violet overnight. Plaque forming units (pfu) were counted the following day. LCMV infections were carried out with Armstrong strain 53b. LCMV stocks were propagated in BHK-21 cells and titers were determined on Vero E6 cells.
Mice and Viral Inoculations
C57BL/6 mice were obtained from Harlan Sprague Dawley. Breeding pairs were checked every day, and 1-day-old pups were infected intracranially with 107 pfu eGFP-CVB3, 5 × 103 pfu LCMV (Armstrong strain), or mock infected with 1 × DMEM, as described previously.14 Alternatively, 3-day-old pups were infected intracranially with 107 pfu eGFP-CVB3 to examine CNS pathology in surviving mice at 30, 75, and 90 days PI.30 The amount of inoculum utilized in the study was chosen to closely equate viral replication kinetics and viral protein expression in the neonatal CNS for each virus. The procedure for intracranial (ic) inoculation of 1-day-old pups has been described previously.20, 23 Young pups were killed by halothane treatment followed by immediate decapitation at 12, 24, and 48 h PI. Older pups (10 days and beyond) were killed by halothane treatment followed by cervical dislocation. The brains were fixed by immersion in 10% neutral-buffered formalin for more than 4 h, paraffin-embedded, and immunostained and/or stained with hematoxylin and eosin.
Immunofluorescence Staining
Paraffin-embedded sections (3–4 μm in thickness) were deparaffinized with xylene followed by rehydration in decreasing percentages of ethanol, PBS, and distilled water. High temperature antigen unmasking was performed for 25 min with citrate-based antigen unmasking solution (Vector Laboratories). For primary antibodies derived from mouse, including mouse anti-LCMV-NP (nucleoprotein, 1:20), Mouse on Mouse Kit was utilized (Vector Laboratories). For GFP (1:100, AB3080; Millipore Corporation; or 1:100, ab13970, Abcam), nestin (1:50, MAB353; Millipore Corporation), and Olig2 (1:100, ab33427; Abcam) antibodies, sections were blocked with 10% normal goat serum for 30 min, primary antibody was incubated overnight at 4 °C, goat anti-rabbit, anti-rat, or anti-mouse antibody conjugated to biotin, bovine anti-chicken FITC (sc2700, Santa Cruz Biotechnology), or goat anti-rabbit Alexa Flour 594 or 488 was applied for 30 min. Finally, sections were incubated with streptavidin-Alexa Fluor 488 or 594 at 1:500 for 30 min. For CCL12/MCP-5 (1:100, anti-MCP-5 (K19)-sc9718; Santa Cruz Biotechnology), Transthyretin (TTR; 1:50, 11891-1-AP; Proteintech Group), Carbonic Anhydrase II (1:50, CAII (G-2): sc-48351; Santa Cruz Biotechnology), and MHC class I (1:50, 5K46: sc-71576; Santa Cruz Biotechnology) antibodies, the Tyramide Signal Amplication Kit number 25 (Invitrogen) was used according to the manufacturer’s protocol. Briefly, sections were quenched with 3% H2O2 for 1 h (unless antibody was generated in mouse), blocked in 1% blocking solution for 1 h (which was used for all subsequent incubations), incubated in primary antibody overnight at 4 °C, goat anti-rabbit or appropriate secondary antibody conjugated to biotin at 1:100 was incubated for 2 h, streptavidin-HRP was incubated at 1:100 for 1 h, and finally sections were incubated with tyramide-594 at 1:100 in amplification buffer for 20 min. In order to ensure antibody specificity, no primary controls were included for each antibody. For detection of DNA/nuclei, sections were overlaid with Vectashield Mounting Medium with DAPI (Vector Laboratories). Sections were observed by fluorescence microscopy (Zeiss Axiovert 200 inverted microscope). Green, red, and blue channel images were merged utilizing AxioVision 4.8 software. Whole-brain images were generated from several × 5 images that were automated and photo-merged using Adobe Photoshop CS2 software.
Quantification of Infected Nestin and Olig2-Positive Progenitor Cells
Fluorescent images ( × 20) of nestin/olig2 colocalized with GFP-CVB3/LCMV-NP-immunostained brain sections were taken using a Zeiss Axiovert 200 inverted microscope. Brain sections of C57BL/6 mice infected with GFP-CVB3 at 24 h PI and LCMV at 48 h PI were used. All three walls of the subventricular zone (SVZ) were imaged using three animals per time point. First, the total number of infected cells per SVZ was counted. Then, the percent of double-positive cells for infection and the given progenitor cell marker was calculated for both GFP-CVB3 and LCMV. Data were analyzed in GraphPad Prism 3.0 software by Student’s t-test and displayed graphically.
ApopTag Staining and Quantification
Cells undergoing apoptosis were identified using the ApopTag Red In Situ Apoptosis Detection Kit (Millipore), as specified by the manufacturer. Briefly, paraffin-embedded sections were deparaffinized and antigen unmasked with proteinase K. Equilibration buffer was added directly onto the specimen for 5 min, and then the samples were incubated in terminal deoxynucleotidyl transferase enzyme in reaction buffer for 1 h at 37 °C. Samples were shaken for 15 s, and then washed in working strength stop/wash buffer for 10 min. Working strength anti-digoxigenin-conjugated rhodamine incubated on the samples at room temperature for 30 min. Sections were washed with 1 × PBS and observed by fluorescence microscopy. ApopTag-positive cells in the choroid plexus were counted, analyzed in GraphPad Prism 3.0 software by ANOVA and displayed graphically.
Microarray Analysis
Total RNA was isolated using the Qiagen RNeasy Kit from homogenized brain samples. Isolated RNA was sent to the BIOGEM Microarray Core at University of California at San Diego and analyzed for quality before microarray analysis was started. Briefly, samples were reverse transcribed to make cDNA and then in vitro transcribed to generate cRNA, which incorporated biotin. As described previously,14 cRNA was hybridized to Illumina Whole Mouse Genome 6 Samples chips, which hybridize ∼45,200 genes. Hybridized chips were incubated with Cy3, washed and read using the Illumina BeadArray Station. Normalized gene expression was used to examine differences and similarities in the following samples: GFP-CVB3, LCMV, and Mock-infected C57BL/6 mice at 12, 24, and 48 h PI. Two biological replicates were analyzed for each treatment and time point. Gene expression levels based on RNA expression in the brain of eGFP-CVB3 or LCMV-infected neonatal mice were compared with mock-infected control mice. The fold change in gene expression levels between GFP-CVB3 and LCMV-infected mice over mock were used to determine how these viral infections altered immune response, developmental, and CNS-specific gene expression in the neonatal host. Data were analyzed using a statistical algorithm developed for high-density oligonucleotide arrays.31
RESULTS
Early Viral Protein Expression for Both LCMV and GFP-CVB3 Localized to the Choroid Plexus and SVZ of the Neonatal CNS
Both LCMV and GFP-CVB3 have been shown previously to target neurogenic regions of the neonatal CNS early after infection.20, 26, 32 However, the progenitor cell type supporting LCMV infection has not been clearly evaluated by immunofluorescence microscopy utilizing informative cell markers. Therefore, we carefully inspected the tropism and virus distribution for LCMV and GFP-CVB3 infection in our neonatal mouse model. In a previous study, we determined the viral growth kinetics for LCMV and GFP-CVB3 in the CNS.14 We observed increasing amounts of LCMV and GFP-CVB3 viral genome via real-time RT-PCR at 12, 24, and 48 h after infection of 1-day-old mice,14 thereby demonstrating the ability of both viruses to successfully replicate in the neonatal CNS. Nonetheless, the early immune response was substantially different for both viruses. Also, the recruitment of novel nestin+ myeloid cells was observed in the choroid plexus at early time points following both wild-type CVB3 and GFP-CVB3 infection. CVB3 infection of NPSCs was observed shortly after nestin+ myeloid cell infiltration into the parenchyma of the CNS. In contrast, LCMV failed to recruit similar myeloid cells into the choroid plexus. Our initial data suggested that CVB3 may activate a novel immune response in the CNS following infection, and that infiltration of nestin+ myeloid cells might represent an intrinsic host CNS repair response. CVB3 induced the recruitment of these myeloid cells by stimulating the expression of a unique set of chemokines including CCL12, which upon infection contributed to virus dissemination within the CNS.14 Therefore, we sought to inspect the global gene expression profile, including the activation of immune response genes, and NPSC tropism of CVB3 in the neonatal CNS in more detail. We utilized LCMV as an informative neurotropic virus to contrast the results for CVB3 infection.
In order to more carefully determine sites of LCMV and GFP-CVB3 infection in the neonatal CNS, immunofluorescence microscopy for viral protein was performed (Figure 1). For LMCV, an anti-NP antibody was utilized to track infection at 12, 24, and 48 h PI (Figures 1a, e, and i, respectively; red signal). NP, a viral protein essential for replication, forms a complex with the LCMV genome.33 At higher magnification, increasing amounts of NP were found in the subventricular zone (SVZ) and choroid plexus epithelial cells of LCMV-infected animals over time (Figures 1b, f, and j). Viral protein was also found in the olfactory bulb, retrosplenial cortex, meninges, and regions of the cerebellum by 48 h PI; however, the majority of signal was seen within the choroid plexus and surrounding the lateral ventricle.

Distribution of lymphocytic choriomeningitis virus (LCMV) and coxsackievirus expressing GFP (GFP-CVB3) in the neonatal central nervous system (CNS) at early time points following infection. Sagittal paraffin-embedded brain sections from neonatal mice infected at 1 day post birth and killed at 12, 24, and 48 h post infection (PI) were immunostained with antibodies against LCMV (NP—red signal) or GFP-CVB3 (GFP—green signal). Nuclei were stained with DAPI in blue. A composite of × 5 fluorescent images were arranged to display the whole brain. (a, e, i) An increase in nucleoprotein (NP) protein expression was observed for LCMV in the neonatal CNS at 12, 24, and 48 h PI. (b, f) At higher magnification, early infection was observed within the choroid plexus epithelial cells and SVZ at 12 and 24 h PI. (J) By 48 h PI, higher levels of viral protein expression were seen in the choroid plexus and SVZ. LCMV infection as determined by viral protein expression extended into the cortex, the hippocampus, the meninges, and regions of the cerebellum. (c, g, k) An increase in viral protein expression was observed for GFP-CVB3 in the neonatal CNS at 12, 24, and 48 h PI. (d, h) Similar to LCMV, GFP-CVB3 infection targeted the choroid plexus. The staining pattern was similar to our previously published results whereby we described the recruitment of myeloid cells across the choroid plexus at 12 and 24 h PI, which become targets for CVB3 infection.14 Also, high levels of GFP-CVB3 protein were observed in the SVZ at 24 h PI. (l) After 48 h PI, GFP-CVB3 infection spread into the hippocampus, the retrosplenial cortex, and the olfactory bulb. OLF, olfactory bulb; NC, neocortex; HIP, hippocampus; CER, cerebellum; LV, lateral ventricle; CP, choroid plexus.
In addition, an anti-GFP antibody was used to detect the distribution of GFP-CVB3 infection at 12, 24, and 48 h PI (Figures 1c, g, and k, respectively; green signal) in the CNS. Viral protein expression was observed in the choroid plexus at 12 and 24 h PI at higher magnification (Figures 1d and h). The choroid plexus produces cerebral spinal fluid and regulates the entry of immune cells into the CNS across the tight junctions comprising the choroid plexus epithelial cells.34 GFP-CVB3 protein expression was identical to our previously published data revealing the recruitment of myeloid cells, which become infected as they undergo extravasation through the choroid plexus.14 GFP-CVB3 protein expression was also found in the SVZ at these time points. Previously, we identified infected nestin+ NPSCs in the SVZ following GFP-CVB3 inoculation20, 32 at early time points. By 48 h PI, GFP-CVB3 protein expression was less evident in the choroid plexus and infection spread into the parenchyma of the brain including neurons in the retrosplenial cortex, the rostral migratory stream, olfactory bulb, and hippocampus (Figure 1l).
LCMV Infection Targeted a Greater Number of Cells in the SVZ and Revealed Broader Stem and Progenitor Cell Tropism in Comparison with GFP-CVB3 Infection
We investigated the tropism of each neurotropic virus in greater detail utilizing antibodies against informative stem/progenitor cell markers. The SVZ has been shown to comprise neural progenitor cells, which give rise to glial cells and neuroblasts migrating through the rostral migratory stream.35, 36 Nestin, an intermediate filament implicated in cell motility, was utilized to label NPSCs.37, 38 Olig2, a transcription factor known to have a role in glial cell differentiation was utilized to identify glial progenitor cells.39 Colocalization of viral proteins and stem/progenitor cell markers was evaluated by immunofluorescence microscopy (Figure 2). The peak of SVZ infection as determined by viral protein expression in Figure 1 was used as the optimum time point to inspect LCMV (at 48 h PI) and GFP-CVB3 (at 24 h PI) tropism for stem/progenitor cell populations. Although Olig2 may also label mature oligodendrocytes, we limited the examination to infected Olig2+ cells residing within the SVZ.

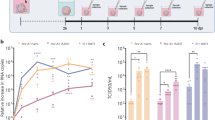
Lymphocytic choriomeningitis virus (LCMV) infection targeted a greater number of cells in the SVZ and revealed broader stem and progenitor cell tropism in comparison with coxsackievirus expressing GFP (GFP-CVB3) infection. Immunofluorescence microscopy for viral protein in combination with nestin and Olig2 immunostaining was performed on sagittal paraffin-embedded brain sections for LCMV at 48 h post infection (PI) and for GFP-CVB3 at 24 h PI. All three walls of the subventricular zone (SVZ) were imaged, using three animals per time point. First, the total number of infected cells per SVZ was counted. Then, the percent of double-positive cells for infection and the given progenitor cell marker was calculated for both GFP-CVB3 and LCMV. (a) LCMV infected a greater number of cells within the SVZ, as compared with GFP-CVB3 (*P<0.05). (b) In addition, LCMV infected a greater percentage of Olig2+ cells in the SVZ, as compared with GFP-CVB3 (*P<0.001). (c) In contrast, GFP-CVB3 infected a greater percentage of nestin+ cells in the SVZ, as compared with LCMV (*P<0.05). Immunofluorescence microscopy revealed the presence of LCMV-infected Olig2+ (d, f, white arrows; h, white arrows) and nestin+ (j, l, white arrow; n, white arrow) cells at 48 h PI. In contrast, GFP-CVB3 infected fewer Olig2+ cells (e, g, white arrow; i, white arrow) and preferentially infected nestin+ cells (k, m; white arrows; o, white arrow) in the SVZ at 24 h PI. In addition, GFP-CVB3-infected cells were observed in the SVZ, which resembled type-B stem cells and failed to express Olig2 (g, notched blue arrow; i, blue arrow). Olig2 or nestin was labeled in red. Virus protein expression for LCMV or GFP-CVB3 was labeled in green. Nuclei/DNA was stained with DAPI (blue) for all images. F, G, L, M at × 63 magnifications. H, I, N, O at × 63 magnification with an additional fourfold digital magnification. D, E, J, K at × 10 magnification. Student’s t-test was utilized to determine statistical significance.
LCMV infected a greater number of cells in the SVZ, as compared with GFP-CVB3 (Figure 2a). In addition, LCMV infected a greater percentage of Olig2+ cells (Figure 2b). In contrast, LCMV infected fewer nestin+ cells (Figure 2c). Although LCMV infected a similar percentage of Olig2+ and nestin+ cells, GFP-CVB3 appeared to preferentially infect nestin+ cells. Representative immunofluorescence images illustrated the colocalization of LCMV NP protein and Olig2 (Figures 2d and f, white arrows; Figure 2h, white arrows) or nestin (Figures 2j and l, white arrow; Figure 2n, white arrow) within the SVZ at 48 h PI. Similarly, representative immunofluorescence images illustrated colocalization for GFP-CVB3 viral protein and Olig2 (Figures 2e and g, white arrow; Figure 2i, white arrow) or nestin (Figures 2k and m, white arrows; Figure 2o, white arrow) within the SVZ at 24 h PI. In addition, GFP-CVB3-infected cells lacking olig2 expression and morphologically similar to a type-B stem cells were observed in the SVZ (Figure 2g, notched blue arrow; Figure 2i, blue arrow).
LCMV Altered the Expression of a Greater Number of Genes at Later Time Points Following Infection, as Compared with GFP-CVB3 Infection
In order to analyze overall changes to the host gene expression profile as a result of the neonatal CNS response to viral infection, microarray analysis was performed following LCMV or GFP-CVB3 infection, and host gene expression levels were compared with mock-infected control mice (Figure 3). In total, LCMV infection altered the expression of 231 genes greater than or equal to a twofold induction, as compared with mock-infected control mice (Figure 3a). Alterations in host gene expression were delayed for LCMV infection; 5 genes were upregulated at 12 h PI. By 24 h PI, 151 genes were upregulated; and by 48 h PI, 135 genes remained upregulated. In total, GFP-CVB3 infection altered the expression of 117 genes greater than or equal to a twofold induction, as compared with mock-infected control mice. Alterations in host gene expression were more rapid following GFP-CVB3 infection, perhaps reflecting the greater inoculum of virus. However, recombinant coxsackieviruses show slightly delayed growth kinetics and reduced morbidity, as compared with wild-type coxsackievirus in our neonatal mouse model. The greater inoculum utilized for GFP-CVB3 was chosen based on previous comparisons of viral growth kinetics and mortality curves between GFP-CVB3 and wild-type CVB3. At 12 h PI, 55 genes were upregulated following GFP-CVB3 infection. By 24 h PI, only 10 genes were upregulated and by 48 h PI, 66 genes were shown to be upregulated.

Lymphocytic choriomeningitis virus (LCMV) infection altered the expression of a greater number of genes at later times following infection, as compared with coxsackievirus expressing GFP (GFP-CVB3) infection. Microarray analysis was performed on RNA isolated from the neonatal brain tissue of mock-infected mice, or mice infected with GFP-CVB3 or LCMV, and collected at 12, 24, and 48 h post infection (PI). (a) The overall number of genes induced (showing more than or equal to twofold increase above mock-infected mice; two animals per time point) following GFP-CVB3 or LCMV infection are shown in total, and for each time point. LCMV altered the expression of 231 genes in total, as compared with 117 genes altered following GFP-CVB3 infection. (b) Venn diagrams depicted the upregulation or downregulation of genes shared or unique for either GFP-CVB3 or LCMV-infected mice for each time point and between time points. No more than seven shared genes were shown to be upregulated or downregulated following GFP-CVB3 or LCMV infection for any time point. (c) Venn diagrams depicted the upregulation or downregulation of genes shared or unique for GFP-CVB3 and LCMV-infected mice at every time point.
The number of genes up- or downregulated following infection were grouped and shown as Venn diagrams for each time point and between time points (Figures 3b and c). The strikingly divergent gene expression profile following LCMV and GFP-CVB3 infection of the neonatal CNS illustrated the significant disparity in host/pathogen interactions for each RNA virus. For example, no more than 7 genes were up- or downregulated in parallel for both viruses at each time point, although the gene expression profile for GFP-CVB3 at 12 h PI showed 20 overlapping genes upgregulated for LCMV at 24 h PI. Similarly, the gene expression profile for GFP-CVB3 at 48 h PI showed 12 overlapping genes upregulated for LCMV at 24 h PI.
Those genes either up- or downregulated by both viruses included the Hox developmental genes, although the significance of these gene expression changes remains unclear. More specifically, Hoxa5 and Hoxb5 were upregulated at 12 h PI for both viruses; also, insulin-degrading enzyme and O-linked N-acetylglucosamine transferase were downregulated by both viruses at 12 h PI. Purkinje cell protein 2 was the only gene upregulated by both viruses at 24 h PI; also, Hoxa5, Hoxb5, Hoxb7, microtubule-associated protein-1B, and pre B-cell leukemia transcription factor-1 were downregulated by both viruses at 24 h PI. By 48 h PI, cDNA sequence BC1179090, S100 calcium-binding protein-A8, S100 calcium-binding protein-A9, and stefin-A1 were upregulated by both viruses; also, claudin-10, ectonucleotide pyrophosphatase/phosphodiesterase-6, G protein-coupled receptor 37-like-1, myelin basic protein (Mbp), myelin-associated glycoprotein, myelin-associated oligodendrocytic basic protein, and tryptophan hydroxylase-2 were downregulated by both viruses at 48 h PI. A complete list of up- and downregulated genes at each time point is shown in Supplementary Table 1.
We performed an analysis of genes modulated following GFP-CVB3 and LCMV infection utilizing the Kegg pathway analysis, a collection of manually drawn pathway maps representing current knowledge of the molecular interactions for a wide spectrum of metabolic, cellular, and organismal processes (http://www.genome.jp/kegg/pathway.html). Perhaps, not surprisingly, both RNA viruses activated many similar pathways following neonatal infection (Table 1; highlighted in light blue). Notable overlapping immunological pathways included genes involved in ‘antigen processing and presentation’, ‘type 1 diabetes mellitus’, and the ‘Toll-like receptor signaling pathway’. However, differences were also observed. For example, LCMV modulated genes involved in the ‘classical complement pathway’ and ‘apoptosis’. In contrast, GFP-CVB3 modulated genes involved in ‘nitrogen metabolism’ and the ‘tight junction’.
Immune Response Gene Expression Varied Temporally and in Magnitude between GFP-CVB3 and LCMV
Although the induction of similar pathways for each virus suggests substantial similarities with regards to the host response, a closer inspection revealed considerable differences in genes modulated within a given pathway (Figure 4). For example, the immune response pathway was activated for both GFP-CVB3 and LCMV infection. However, the kinetics of immune response gene activation was significantly accelerated as soon as 12 h PI following GFP-CVB3 infection. In addition, LCMV activated many interferon (IFN) response genes at 24 h PI, whereas GFP-CVB3 failed to induce high levels of IFN response genes at any time point. Many of the immune response genes induced by GFP-CVB3 and LCMV infection in the neonatal CNS are summarized via the heat map in Figure 4a. GFP-CVB3 induced a narrow set of chemokines at 12 h PI, including CCL12 (outlined with a red box), CCL7, CCL4, CXCL4, CCL2, CXCL10, and CCL9 and CCL4. In contrast, LCMV induced a greater number of chemokines (including the chemokines described above) and high levels of many IFN response genes. Also, relatively high levels of CXCL10, CCL5 CXCL9, OAS1g, and PKR were specifically induced following LCMV infection, although at a later time point (24 h PI).

Immune response gene expression varied temporally and in magnitude between coxsackievirus expressing GFP (GFP-CVB3) and lymphocytic choriomeningitis virus (LCMV). (a) Gene expression upregulation (red) or downregulation (blue) as summarized by the heat map illustration showed distinct differences in immune response activation when comparing GFP-CVB3 or LCMV infection in the neonatal central nervous system (CNS) (mean value of two animals per time point). GFP-CVB3 primarily induced a restricted group of chemokines, including CCL12 (highlighted red box) at 12 h post infection (PI). In contrast, LCMV induced the expression of a larger number of chemokines and interferon (IFN) response genes with a greater fold change in gene expression at a later time point (24 h PI). In addition, LCMV induced high levels of MHC class I gene expression at 24 h PI. In contrast, GFP-CVB3 induced only low levels of MHC class I gene expression at a later time point (48 h PI). (b–d) Immunofluorescence microscopy was performed to inspect the expression of MCP-5/CCL12 (red signal) following GFP-CVB3 or LCMV infection at 12, 24, and 48 h PI (representative sample of three animals shown). A higher × 63 magnification (region outlined by a white box) is included below each × 20 image. CCL12 localized to the choroid plexus and was found at high levels in myeloid cells, choroid plexus epithelial cells, and ependymal cells at 12 h PI following GFP-CVB3 infection. However, CCL12 protein expression levels decreased at 24 and 48 h PI. Diffuse and lower expression levels of CCL12 were observed following LCMV infection, although CCL12 levels increased by 24 h PI and remained at high levels at 48 h PI. Little to no CCL12 expression levels were observed for mock-infected control samples at 12 h PI. Detectable levels Of CCL12 were observed in mock-infected control samples by 48 h PI. No primary antibody staining control sections from GFP-CVB3 infected mice were included for 12 h PI. Nuclei/DNA were stained with DAPI (blue signal) for all images.
The magnitude of change for many of the immune response genes was also far greater following LCMV infection compared with GFP-CVB3. For example, CXCL10 showed a 12.5-fold induction in the neonatal CNS following LCMV infection at 24 h PI. Many of the chemokines upregulated following LCMV infection have been shown previously to be induced in the adult CNS.40 In addition, LCMV induced high levels of the MHC genes H2-K1, H2-Q8, and beta-2-microglobulin (B2m) at 24 h PI, whereas the expression of these genes were reduced in magnitude and delayed temporally (48 h PI) following GFP-CVB3 infection. Intriguingly, serum amyloid A3 (Saa3), initially identified as a mediator of metabolic inflammation in adipose tissue,41 was highly upregulated in GFP-CVB3-infected pups at 48 h PI. Recent results indicate that Saa3 may function as an antimicrobial agent in the colon,42 suggesting that the induction of Saa3 may be involved in the host antiviral response against CVB3 in the neonatal CNS.
We wished to inspect the expression of CCL12 in greater detail and over more time points following LCMV and GFP-CVB3 infection. In order to verify the microarray results and inspect the location of CCL12 expression, immunofluorescence microscopy was performed on neonatal brain sections for CCL12 at 12, 24, and 48 h PI (Figures 4b, c, and d, respectively; red signal). Similar to previous studies,14 MCP-5/CCL12 expression localized to infected myeloid cells, choroid plexus epithelial cells, and ependymal cells in GFP-CVB3-infected animals at early time points (12 h PI) following infection. CCL12 expression colocalized with sites of infected myeloid cell infiltration early (12 h PI) after GFP-CVB3 infection. The choroid plexus is known to be a critical entry site for immune cell infiltration into the CNS,43 and CCL12 may be a key chemokine responsible for the unique myeloid cell infiltration observed following GFP-CVB3 infection.14 Expression levels of CCL12 in GFP-CVB3-infected animals rapidly decreased by 24 and 48 h PI. Diffuse and lower expression levels of CCL12 were observed in the neonatal CNS early (12 h PI) following LCMV-infection, although CCL12 levels increased by 24 h PI and remained at high levels at 48 h PI. No CCL12 protein was seen in mock-infected control mice at 12 h PI, although low expression levels were observed in the choroid plexus at 24 and 48 h PI, most likely reflecting a low-level inflammatory response following the ic inoculation procedure. Thus, immunofluorescence microscopy at the protein level served to confirm the microarray results for CCL12 (described in Figure 4a).
MHC Class I Genes were Weakly Expressed in the CNS Following GFP-CVB3 Infection, as Compared with LCMV Infection
High expression levels of the MHC class I genes (H2-K1 and H2-Q8) and B2m was observed via microarray analysis 24 h after LCMV infection. In contrast, the expression profile for these genes was delayed temporally and greatly reduced in magnitude following GFP-CVB3 infection. Immunofluorescence microscopy confirmed the microarray results and aided in determining the location of MHC class I gene expression in the neonatal CNS following both LCMV and GFP-CVB3 infection. An antibody corresponding to the H2-K1 locus and cross-reacting with the H2-Kb haplotype of C57BL/6 mice (Clone 5K46; Santa Cruz Biotechnology) was utilized to evaluate the upregulation of MHC class I gene expression (Figure 5; red signal). MHC class I gene expression was largely absent in the neonatal CNS of GFP-CVB3-infected mice at 12 h (Figure 5a, white arrows) and 24 h PI (Figure 5b, white arrows). At 48 h PI, low levels of MHC class I gene expression was observed in the SVZ (Figure 5c). Moreover, a lack of viral protein colocalization with MHC class I gene expression was evident and MHC class I gene expression in the SVZ was observed only after GFP-CVB3 protein expression was no longer present in the SVZ. These results parallel previous in vitro studies with CVB3 suggesting an inhibition of antigen presentation via the MHC class I pathway44 that may contribute to the evasion of CD8+ T-cell immunity.10

Weak MHC class I expression in the neonatal SVZ following coxsackievirus expressing GFP (GFP-CVB3) infection. Expression levels of MHC class I (red signal) following GFP-CVB3 (GFP- green signal) and LCMV infection (NP—green signal) were evaluated by fluorescence microscopy (representative sample of three animals shown). (a–c) MHC class I expression levels in the neonatal CNS following infection were compared with mock-infected control animals at 12, 24, and 48 h post infection (PI). A higher × 63 magnification (region outlined by a white box) is included below each × 20 image. MHC class I expression was largely absent in brain sections of GFP-CVB3-infected mice at 12 and 24 h PI (white arrows). By 48 h, low levels of MHC class I could be seen in the SVZ. In contrast, detectable levels of MHC class I protein were seen as early as 12 h PI following LCMV infection. Higher levels MHC class I protein were observed at 24 h PI, dropping sharply by 48 h PI. Signal was observed most strongly in the SVZ, but was present in other regions of the brain, including the meninges, olfactory bulb, and cerebellum. MHC class I protein expression was detectable at 12 h PI and 48 h PI in mock-infected brain sections, with little signal observed at 24 h PI. Nuclei/DNA were stained with DAPI (blue signal) for all images.
In contrast to the results seen for GFP-CVB3, detectable levels of MHC class I protein expression was observed as early as 12 h PI following LCMV infection (Figure 5a). These levels increased by 24 h PI (Figure 5b) and remained high at 48 h PI (Figure 5c). In addition, high levels of LCMV NP expression was observed in regions of the SVZ, which expressed high levels of MHC class I protein. MHC class I gene expression was identified previously on neural progenitor cells.45 However, other cells (downstream progenitor cells, choroid plexus epithelial cells, and infiltrating myeloid cells) may also contribute to MHC class I gene upregulation following LCMV infection. Moderate levels of MHC class I gene expression in LCMV-infected mice were also present in other regions of the brain, including the meninges, olfactory bulb, and cerebellum at every time point analyzed. Detectable levels of MHC class I gene expression was observed in the subventricular zone and choroid plexus of mock-infected animals at 12 and 48 h PI, although little signal was observed at 24 h PI.
The differential immune response in the neonatal CNS following infection with both viruses may reflect differences in viral kinetics and sites of replication at early and at later time points. Also, the restriction in antigen presentation and inhibition of the type I IFN response following CVB3 infection may ultimately contribute to the establishment of persistent infection in the CNS.30 Although Figure 5c revealed a reduction in CVB3 protein expression in the choroid plexus and SVZ at 48 h PI, these results more likely reflect the restricted tropism for CVB3 and the migration of CVB3-infected progenitor cells away from the SVZ and into the olfactory bulb, hippocampus, and retrosplenial cortex (shown in Figure 1k).20 Low amounts of MHC class I protein expression was also observed in mock-infected mice, most likely reflecting limited levels of protein expression or, alternatively, a low-level inflammatory response following the ic inoculation procedure.
Similar Developmental and CNS-Specific Gene Expression Changes Following GFP-CVB3 or LCMV Infection
We recently described CNS developmental defects in mice surviving infection with GFP-CVB3.46 To analyze developmental and CNS-specific gene expression changes following GFP-CVB3 or LCMV infection, microarray results were summarized via heat map shown in Figure 6a. The gene expression profile for both GFP-CVB3 and LCMV was similar at 12 and 24 h PI. Hox gene expression was upregulated following infection of GFP-CVB3 and LCMV at 12 h PI. However, by 24 and 48 h PI, GFP-CVB3 infection downregulated several Hox genes, including Hoxa5 (−5.8- and −2.9-fold, respectively), Hoxb5 (−5.0- and −2.7-fold, respectively), and Hoxb7 (−2.6- and −1.4-fold, respectively). In contrast, LCMV induced these Hox genes (+5.5-, +4.9-, and +2.1-fold, respectively) at 48 h PI, subsequent to a precipitous decline of Hox gene expression at 24 h PI. Both GFP-CVB3 and LCMV downregulated expression levels of Mbp (−5.2- and −5.7-fold, respectively) at 48 h PI. Also, LCMV specifically downregulated glial fibrillary acidic protein (GFAP; −3.1-fold) at 48 h PI, whereas GFP-CVB3 specifically reduced the expression of TTR (−3.3-fold; Figure 6a, red box) at 48 h PI.

Similar developmental and central nervous system (CNS)-specific gene expression changes following coxsackievirus expressing GFP (GFP-CVB3) and lymphocytic choriomeningitis virus (LCMV) infection. (a) Gene expression upregulation (red) or downregulation (blue) as summarized by the heat map illustration showed similarities in developmental and CNS-specific gene expression levels when comparing GFP-CVB3 or LCMV infection in the neonatal CNS. The gene expression profile was relatively similar between the two viruses; however, GFP-CVB3 continuously downregulated more genes from 24 to 48 h post infection (PI), including Hoxa5, Hoxb5, Hoxb7 and transthyretin (TTR; red box), as compared with LCMV. (b–d) Immunofluorescence microscopy was performed to inspect the expression of TTR (red signal) following GFP-CVB3 or LCMV infection at 12, 24, and 48 h PI (representative sample of three animals shown). A higher × 63 magnification (region outlined by a white box) is included below each × 20 image. A decrease in TTR following GFP-CVB3 infection was observed in the choroid plexus (white arrows) at 12, 24, and 48 h PI. Further, no colocalization occurred between TTR and GFP-CVB3-infected myeloid cells at any time point. In contrast, high levels of TTR were seen at every time point in brain sections of LCMV-infected mice or in mock-infected control mice. Nuclei/DNA were stained with DAPI (blue signal) for all images.
TTR protein produced by the choroid plexus accounts for ∼25% of the protein secreted in the CSF. The reduction of TTR expression following GFP-CVB3 infection was of particular interest as we previously observed choroid plexus pathology at 48 h PI.14 Further, hydrocephalus was previously seen in a significant number of mice surviving GFP-CVB3 infection.32 The reduction of TTR expression may reflect virus-mediated pathology in the choroid plexus following GFP-CVB3 infection. By immunofluorescence microscopy, TTR protein was expressed at high levels in the choroid plexus of LCMV or mock-infected mice at every time point following infection (Figures 6b, c, and d). In contrast, GFP-CVB3-infected animals showed reduced levels of TTR protein in the choroid plexus at 12, 24, and 48 h PI. The apparent lack of TTR gene expression downregulation following GFP-CVB3 infection as determined by microarray analysis at 12 and 24 h PI might reflect regional reductions of TTR protein specifically within the choroid plexus in the lateral ventricle, as compared with the third and fourth ventricles.
Downregulation of CAII, a Marker of the Choroid Plexus Functional Activity, Following GFP-CVB3 Infection
Although reduced TTR expression suggested choroid plexus dysfunction following GFP-CVB3 infection, we examined a functional maker that would clearly show a reduced capacity of the choroid plexus to produce CSF. By microarray analysis, we inspected gene expression levels for CAII that catalyzes the reversible hydration of carbon dioxide and comprises one of the key enzymes responsible for the secretion of cerebrospinal fluid. Microarray results for CAII were summarized via heat map shown in Figure 7a. Immunofluorescence microscopy using an antibody against CAII was also performed (Figures 7b, c, and d; red signal). CAII expression was observed in choroid plexus epithelial cells of mock-infected mice at 12, 24, and 48 h PI. LCMV-infected mice also expressed high levels of CAII at every time point analyzed, including in cells expressing high levels of LCMV NP at 48 h PI. Immunofluorescence staining of CAII and LCMV NP was carried out on serial sections owing to the technical inability of both antibodies to work together (see Materials and Methods). Although high levels of CAII were observed in the choroid plexus and in nearby infected myeloid cells at early time points (12 and 24 h PI), CAII protein expression levels fell dramatically in eGFP-CVB3-infected mice at 48 h PI (Figure 7d, white arrows).

Downregulation of carbonic anhydrase II (CAII), a functional marker of the choroid plexus, following coxsackievirus expressing GFP (GFP-CVB3) infection. (a) Gene expression as summarized by the heat map illustration (upregulation—red; downregulation—blue) showed similar minimal reductions in gene expression levels for two isoforms of carbonic anhydrase when comparing GFP-CVB3 or lymphocytic choriomeningitis virus (LCMV) infection in the neonatal central nervous system (CNS). Expression levels of CAII (red signal) following GFP-CVB3 (GFP—green signal) and LCMV infection (NP—green signal) were also evaluated by fluorescence microscopy (representative sample of three animals shown). (b–d) CAII expression levels in the neonatal CNS following infection were compared with mock-infected control animals at 12, 24, and 48 h post infection (PI). A higher × 63 magnification (region outlined by a white box) is included below each × 20 image. CAII protein expression following GFP-CVB3 infection was observed within infected myeloid cells and choroid plexus epithelial cells at 12 and 24 h PI. CAII protein signal greatly decreased specifically within the choroid plexus at 48 h PI (white arrows). Relatively high levels of CAII were seen at every time point in the choroid plexus of LCMV-infected mice or in mock-infected control mice. Nuclei/DNA were stained with DAPI (blue signal) for all images.
The Choroid Plexus of GFP-CVB3-Infected Animals Displayed Severe Pathology
We inspected choroid plexus pathology following infection in H&E-stained sections (Figure 8). In addition, the level of apoptosis in the choroid plexus was determined by ApopTag staining. The morphology of the choroid plexus following LCMV infection and the functional integrity of the choroid plexus epithelial cells (Figure 8a) appeared similar to that of mock-infected control mice (Figure 8g). In contrast, the presence of red blood cells within the lateral ventricle indicated possible hemorrhaging of the choroid plexus following GFP-CVB3 infection (Figure 8d, blue arrows). Also, representative ApopTag-stained images showed an increase in apoptosis within the choroid plexus of GFP-CVB3-infected mice (Figures 8e and f, white arrows), as compared with LCMV-infected (Figures 8b and c) or mock-infected animals (Figures 8h and i). These results were quantified using ImageJ software to reveal a significantly higher degree of apoptosis in GFP-CVB3-infected mice, as compared with LCMV-infected or mock-infected animals (Figure 8j; *P<0.05). Separately, we also observed an increase in apoptosis within the SVZ suggesting that CVB3 infection may result in the reduction of NPSCs within the neonatal CNS.46 No difference in the levels of apoptosis was observed between LCMV and mock-infected control mice (P>0.05).

The choroid plexus displayed severe pathology following coxsackievirus expressing GFP (GFP-CVB3) infection. ApopTag and hematoxylin and eosin (H&E) staining were performed on sagittal brain sections from mock-infected mice or mice infected with lymphocytic choriomeningitis virus (LCMV) or GFP-CVB3 (representative sample of three animals shown). (a, d, g) At 48 h post infection (PI), no alterations in morphology were observed in the choroid plexus following LCMV infection. In contrast, H&E staining revealed disruption of the choroid plexus with a reduction in size and some nearby hemorrhaging following GFP-CVB3 infection. The blood-cerebral spinal fluid barrier appeared to be compromised, as shown by red blood cells found in the lateral ventricle (d, blue arrows). (b, c, e, f, h, i) Representative images of ApopTag staining (red signal) are shown from brain sections of mock-infected mice or mice infected with either LCMV or GFP-CVB3. (c, f, i) Higher magnification of fluorescence images revealed greater levels of ApopTag staining following GFP-CVB3 infection (white arrows). (j) At 48 h PI, significantly greater levels of apoptosis were observed in the choroid plexus of GFP-CVB3-infected animals, as compared with LCMV or mock-infected animals using ANOVA with Tukey’s post hoc analysis (*P<0.05). No difference in levels of apoptosis was observed between LCMV and mock-infected animals. Nuclei/DNA were stained with DAPI (blue signal). Some autofluorescing red blood cells were observed in the sections (yellow signal). Images are shown at × 20 magnification (a, b, d, e, g, h) or digitally magnified by an additional ∼5-fold (c, f, i).
Recovery of CAII Expression in the Choroid Plexus at Later Time Points Following GFP-CVB3 Infection
We inspected the expression of choroid plexus functional markers by immunofluorescence microscopy and morphology changes by H&E staining following recovery from acute GFP-CVB3 infection (day 30 and 90 PI; Figure 9). Mock-infected mice expressed high levels of CAII protein (Figures 9a and e) and the choroid plexus appeared normal by H&E staining at day 30 and 90 PI (Figures 9b and f, respectively). At later time points following GFP-CVB3 infection (day 30 and day 90 PI), high levels of CAII protein were observed once again in the choroid plexus (Figures 9c and g, respectively; white arrows). In addition, by H&E staining, the choroid plexus appeared normal with few signs of inflammation (Figures 9d and h) at these later time points following infection.

Recovery of carbonic anhydrase II (CAII) expression in the choroid plexus at late time points following coxsackievirus expressing GFP (GFP-CVB3) infection. Expression levels of CAII in the choroid plexus were evaluated by fluorescence microscopy in mock-infected mice, and in mice following GFP-CVB3 infection at 30 and 90 days post infection (PI) (representative sample of three animals shown). in addition, the morphology of the choroid plexus was determined by hematoxylin and eosin (H&E) staining. (a, e) High levels of CAII were seen in the choroid plexus of mock-infected control mice at 30 and 90 days post inoculation. (b, f) Normal choroid plexus morphology was seen in mock-infected control mice by H&E staining. (c, g) In contrast to the reduced levels of CAII seen at 48 h PI, levels of CAII in GFP-CVB3-infected mice recovered to levels seen in mock-infected control mice by day 30 and 90 PI, (white arrows). (d, h) In addition, the choroid plexus appeared normal by H&E staining in GFP-CVB3-infected mice by day 30 and 90 PI. The levels of CAII and transthyretin (TTR) were evaluated in a GFP-CVB3-infected mouse suffering from hydrocephalus at 75 days PI. (i, j) Relatively normal levels of CAII and TTR protein were observed in the choroid plexus despite clear signs of hydrocephalus in the infected mouse. (k) In addition, the choroid plexus appeared relatively normal by H&E staining, although the lateral ventricle appeared enlarged. Nuclei/DNA were stained with DAPI (blue signal) for immunostained images, and all images are shown at × 20 magnification.
High levels of Functional Markers and Few Signs of Pathology in the Choroid Plexus of Mice Suffering from GFP-CVB3-Induced Hydrocephalus
Previously, we described the high incidence of hydrocephalus in mice surviving GFP-CVB3 infection characterized by an enlarged cranium, increased ic pressure, and an abnormal accumulation of cerebral spinal fluid (CSF) in the ventricles.32 Therefore, we determined whether a previous infection might permanently alter choroid plexus function and/or structure in mice exhibiting signs of hydrocephalus. The choroid plexus of a GFP-CVB3-infected mouse suffering from hydrocephalus expressed moderate levels of CAII and TTR (Figures 9i and j, respectively; white arrows). Also, the structure of the choroid plexus appeared normal by H&E staining, although an enlarged lateral ventricle was readily apparent in the section (Figure 9k). These results suggest that GFP-CVB3-mediated hydrocephalus may be the result of CSF blockage of outflow from the ventricles. Alternatively, choroid plexus recovery following GFP-CVB3 infection may result in dysregulated or compensatory overproduction of CSF in the ventricles.
DISCUSSION
Viral infection of the CNS may cause serious clinical disease particularly in newborn infants. Enhanced susceptibility and disease following viral infection in the young may be a consequence of virus-mediated alterations in development. In addition, an inefficient antiviral response, perhaps reflecting the immaturity of the neonatal host immune response, may contribute to increased pathology in newborn infants. Enteroviruses are significant human pathogens contributing to the great majority of clinical cases of aseptic meningitis and encephalitis in the United States. Few studies have determined the target cells for enteroviral infection in the developing CNS. We previously established a neonatal mouse model to study both CVB3 and LCMV infection of the CNS,14 and persistent CVB3 infection.30 We utilized a recombinant CVB3 expressing GFP (GFP-CVB3) to track virus spread in the neonatal CNS and compared these results with that of LCMV (Armstrong strain). GFP-CVB3 can infect NPSCs and is highly pathogenic in the neonatal CNS of 1-day-old pups.32 Nonetheless, 3-day-old pups survive acute infection and establish a persistent infection characterized by chronic immunopathology and CNS developmental defects.30, 32, 46 In addition, early GFP-CVB3 infection induced the recruitment of a novel population of infected myeloid cells expressing nestin into the neonatal CNS.14 The induction of a family of chemokines that included CCL12 was observed as early as 12 h PI.14 Our recent studies suggested that NPSCs and neurogenic regions of the CNS may support persistent CVB3 infection,22, 24 and infected mice surviving infection may suffer a chronic depletion of neural stem cells.46
Here, we directly compared neural stem cell tropism for both CVB3 and LCMV. We utilized 1-day-old or 3-day-old pups to study either acute or persistent GFP-CVB3 infection, respectively. In addition, nestin (neural) and Olig2 (glial) markers were used to label both neural and glial progenitor cell populations in the neonatal CNS, respectively. Viral protein expression for both GFP-CVB3 and LCMV localized to the choroid plexus and neurogenic regions near the lateral ventricle of the neonatal CNS. We also observed a gradual increase in viral protein expression accompanied by an increase in gene expression for many immune response and other host genes via microarray analysis following LCMV infection. Despite the temporal dissimilarities between the two viruses, both viruses exhibited tropism for the subventricular zone (SVZ) and NPSC populations within the neonatal CNS. We determined that LCMV infected both nestin+ and olig2+ progenitor cells. In contrast, coxsackievirus preferentially infected nestin+ progenitor cells. Supporting our results, LCMV has been shown to infect both neurons and astrocytes in the CNS. LCMV infection in our study led to decreased expression of GFAP, an astrocytic cell marker, at 48 h PI. In contrast, GFP-CVB3 has been previously shown to exhibit restricted tropism for neurons at later time points.32 Initial infection of particular subsets of neural progenitor cells most likely reflects the final target cell for each virus.
Double immunofluorescence staining of nestin and Olig2 revealed an overlap of both markers in the neurogenic regions of the neonatal CNS (data not shown) as described in previous studies.47 We estimate that ∼25–50% of Olig2+ cells may express detectable levels of nestin. Therefore, the total percent of neural or glial progenitor cells infected for GFP-CVB3 and LCMV (∼90 and 77%, respectively) may actually be lower, suggesting that additional cell types in the SVZ may become infected. These additional cell types might include downstream progenitor cells or immature neurons and glial cells. Thus, future studies inspecting viral tropism in more detail using additional markers along the pathway of cell lineage commitment might be informative.
Early immune activation in the host is critical in predicting the quality of the protective immune response following each viral infection. For example, although T-cell exhaustion may occur in the host following LCMV infection12; in contrast, CVB3 may hinder the activation of CD8+ T cells by inhibiting antigen presentation.10 Many previous studies have characterized LCMV as a prototypical RNA virus infection in the mammalian host. As a consequence of this premise, the rules governing immune activation, immunoevasion, and viral persistence following LCMV infection have been used as a blue print to project all RNA virus/host interactions. However, early immune activation following CVB3 infection appeared to be unique; hence, LCMV might not be considered a general model for RNA virus infection and immune activation in the neonatal CNS. We suggest that each RNA virus family may uniquely induce the host immune response and establish a persistent infection.
CVB3 is considered a highly cytopathic virus that can directly harm NPSCs.22 In contrast, the non-cytolytic nature of LCMV readily evokes a potential mechanism of persistence within target cells. Our study may be of value in recognizing and predicting gene expression signature patterns of immune activation and disease following RNA virus infection in the neonatal CNS. Most likely, the early host response may be critical in predicting the quality of the subsequent humoral and T-cell immune response. Therefore, we directly compared host gene expression changes at an early time point following infection of these two distinct viruses. The broader tropism of LCMV may reflect the relatively greater effect on gene expression in the neonatal CNS for this virus. Symptoms of LCMV infection in mice vary based on the timing of infection.48
Although LCMV altered the expression of a greater number of host genes as compared with GFP-CVB3, both viruses profoundly affected host chemokine and immune response genes at the early time points analyzed. The substantial induction of MHC class I gene expression by LCMV may account for the high numbers of cytotoxic T lymphocytes typically observed following LCMV infection.28, 49 In GFP-CVB3-infected mice, MHC class I gene expression was either absent or observed at very low levels. These results parallel previous findings suggesting the potent ability of CVB3 to decrease MHC class I gene expression by shutting down host cell translation and inhibiting surface expression of these key immune activation molecules.10, 44 Hence, these divergent neurotropic RNA viruses may differentially evade the host immune response utilizing unique strategies. Nonetheless, limited expression of MHC class I was observed at later time points following GFP-CVB3 infection at a time when virus protein expression waned in the choroid plexus.
NPSCs have been previously shown to respond to IFN50, 51 and express immune activation molecules including MHC antigens.45 Also, the immune system has been shown to modulate neurogenesis.52 Although infected NPSCs may partake in the early immune response following each viral infection, additional cells in the CNS may also contribute. For example, infected myeloid cells were found to express high levels of CCL12 early following GFP-CVB3 infection.14 Infected choroid plexus epithelial cells may also contribute to the early activation of the immune response following infection.
The mechanism of CVB3-mediated hydrocephalus remains largely unknown. As the choroid plexus is an early target for CVB3 infection and the choroid plexus is responsible for CSF production, we considered the possibility that the associated virus-mediated pathology in this critical organ may have a mechanistic role in the subsequent hydrocephalus seen in mice. For this reason, TTR and CAII were utilized as surrogate markers to evaluate choroid plexus function. A dramatic early reduction in both TTR and CAII was observed in the choroid plexus following GFP-CVB3 infection. Signs of choroid plexus pathology and apoptosis were readily apparent following GFP-CVB3 infection. Nevertheless, recovery of the choroid plexus was observed at later time points. Microarray analysis identified reduced TTR levels, most likely reflecting GFP-CVB3-induced damage in the choroid plexus following infection. TTR is synthesized in the choroid plexus and acts as a carrier for thyroid hormone thyroxine and retinol. Intriguingly, altered TTR levels have been shown to have a role in depression and schizophrenia.53, 54 Thus, a marked decrease in TTR expression following GFP-CVB3 infection may provide a functional link between previous viral infection and these neurological disorders.55, 56
We further analyzed choroid plexus function following GFP-CVB3 infection by inspecting the expression levels of CAII. CAII was shown to be greatly decreased 48 h following GFP-CVB infection, indicating potential dysfunction in the production of CSF in these mice. GFP-CVB3-infected myeloid cells near the choroid plexus also expressed high levels of CAII following GFP-CVB3 infection. The cytosolic CAII isoform is the most commonly expressed isoform for carbonic anhydrases and may be expressed within myeloid cell populations.57, 58 In contrast, a lack of choroid plexus pathology was observed following LCMV infection at every time point. We hypothesize that the recovery of the choroid plexus at later time points may contribute to a compensatory increase and overproduction of CSF in the choroid plexus, although a reduction in CSF resorption may also have a role. Hence, our animal model may provide insight and a model system to inspect the mechanism of communicating hydrocephalus associated with infection.
Despite the large number of genes induced at early time points following either GFP-CVB3 or LCMV infection, surprisingly, very few overlapping genes were induced in both infection models, perhaps suggesting the relative unique host response for each virus. In addition, an unexpected paucity of genes appeared to be altered in the host after GFP-CVB3 at 24 h PI (10 genes). We suggest that the earliest time point (12 h PI) may point to an active host response upon initially entry of the virus. However, upon establishment of infection at 24 h PI, CVB3 may limit the host response, for example, by cleaving critical host proteins such as MAVS and TRIF.59 Nevertheless, resulting damage to cells may ultimately lead to additional host gene activation at later time points (48 h PI).
Further, developmentally regulated and CNS-specific genes including Mbp were found to be altered following both GFP-CVB3 and LCMV infection. Of note, CVB3 and LCMV have both been associated with white matter damage following infection.9, 60 Identification of genes altered in expression following early viral infection may be essential in devising early treatment regimes, which may prevent possible pathology and detrimental effects on the host CNS. We expect that other neurotropic enteroviruses that cause human disease, such as enterovirus-71, may induce a host response similar to our results described here for CVB3.
References
Norris CM, Danis PG, Gardner TD . Aseptic meningitis in the newborn and young infant. Am Fam Physician 1999;59:2761–2770.
Kimberlin D . Herpes simplex virus, meningitis and encephalitis in neonates. Herpes 2004;11 (Suppl 2):65A–76A.
Tyler KL . Emerging viral infections of the central nervous system: part 1. Arch Neurol 2009;66:939–948.
Chamberlain RN, Christie PN, Holt KS et al. A study of school children who had identified virus infections of the central nervous system during infancy. Child Care Health Dev 1983;9:29–47.
Kang SS, McGavern DB . Lymphocytic choriomeningitis infection of the central nervous system. Front Biosci 2008;13:4529–4543.
Berger JR, Fee DB, Nelson P, Nuovo G . Coxsackie B meningoencephalitis in a patient with acquired immunodeficiency syndrome and a multiple sclerosis-like illness. J Neurovirol 2009;15:282–287.
Gau SS, Chang LY, Huang LM et al. Attention-deficit/hyperactivity-related symptoms among children with enterovirus 71 infection of the central nervous system. Pediatrics 2008;122:e452–e458.
Verboon-Maciolek MA, Groenendaal F, Cowan F et al. White matter damage in neonatal enterovirus meningoencephalitis. Neurology 2006;66:1267–1269.
Cree BC, Bernardini GL, Hays AP et al. A fatal case of coxsackievirus B4 meningoencephalitis. Arch Neurol 2003;60:107–112.
Kemball CC, Harkins S, Whitmire JK et al. Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLoS Pathog 2009;5:e1000618.
Whitmire JK . Induction and function of virus-specific CD4+ T cell responses. Virology 2011;411:216–228.
Yi JS, Cox MA, Zajac AJ . T-cell exhaustion: characteristics, causes and conversion. Immunology 2010;129:474–481.
Feuer R, Whitton JL . Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr Top Microbiol Immunol 2008;323:149–173.
Tabor-Godwin JM, Ruller CM, Bagalso N et al. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J Neurosci 2010;30:8676–8691.
Sawyer MH . Enterovirus infections: diagnosis and treatment. Semin Pediatr Infect Dis 2002;13:40–47.
Bonthius DJ . Lymphocytic choriomeningitis virus: a prenatal and postnatal threat. Adv Pediatr 2009;56:75–86.
McGavern DB, Homann D, Oldstone MB . T cells in the central nervous system: the delicate balance between viral clearance and disease. J Infect Dis 2002;186 (Suppl 2):S145–S151.
Whitton JL, Cornell CT, Feuer R . Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol 2005;3:765–776.
Sharma A, Valadi N, Miller AH et al. Neonatal viral infection decreases neuronal progenitors and impairs adult neurogenesis in the hippocampus. Neurobiol Dis 2002;11:246–256.
Feuer R, Pagarigan RR, Harkins S et al. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J Neurosci 2005;25:2434–2444.
Baldridge JR, Pearce BD, Parekh BS et al. Teratogenic effects of neonatal arenavirus infection on the developing rat cerebellum are abrogated by passive immunotherapy. Virology 1993;197:669–677.
Tsueng G, Tabor-Godwin JM, Gopal A et al. Coxsackievirus preferentially replicates and induces cytopathic effects in undifferentiated neural progenitor cells. J Virol 2011;85:5718–5732.
Feuer R, Mena I, Pagarigan R et al. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 2002;76:4430–4440.
Rhoades RE, Tabor-Godwin JM, Tsueng G et al. Enterovirus infections of the central nervous system. Virology 2011;411:288–305.
Bonthius DJ, Perlman S . Congenital viral infections of the brain: lessons learned from lymphocytic choriomeningitis virus in the neonatal rat. PLoS Pathog 2007;3:e149.
Bonthius DJ, Mahoney J, Buchmeier MJ et al. Critical role for glial cells in the propagation and spread of lymphocytic choriomeningitis virus in the developing rat brain. J Virol 2002;76:6618–6635.
Pearce BD, Steffensen SC, Paoletti AD et al. Persistent dentate granule cell hyperexcitability after neonatal infection with lymphocytic choriomeningitis virus. J Neurosci 1996;16:220–228.
Christensen JE, de Lemos C, Moos T et al. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol 2006;176:4235–4243.
Kunz S, Rojek JM, Roberts AJ et al. Altered central nervous system gene expression caused by congenitally acquired persistent infection with lymphocytic choriomeningitis virus. J Virol 2006;80:9082–9092.
Feuer R, Ruller CM, An N et al. Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J Virol 2009;83:9356–9369.
Sasik R, Calvo E, Corbeil J . Statistical analysis of high-density oligonucleotide arrays: a multiplicative noise model. Bioinformatics 2002;18:1633–1640.
Feuer R, Mena I, Pagarigan RR et al. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol 2003;163:1379–1393.
Lee KJ, Novella IS, Teng MN et al. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J Virol 2000;74:3470–3477.
Emerich DF, Skinner SJ, Borlongan CV et al. The choroid plexus in the rise, fall and repair of the brain. Bioessays 2005;27:262–274.
Doetsch F . A niche for adult neural stem cells. Curr Opin Genet Dev 2003;13:543–550.
Doetsch F . The glial identity of neural stem cells. Nat Neurosci 2003;6:1127–1134.
Helfand BT, Chang L, Goldman RD . The dynamic and motile properties of intermediate filaments. Annu Rev Cell Dev Biol 2003;19:445–467.
Kleeberger W, Bova GS, Nielsen ME et al. Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res 2007;67:9199–9206.
Marshall CA, Novitch BG, Goldman JE . Olig2 directs astrocyte and oligodendrocyte formation in postnatal subventricular zone cells. J Neurosci 2005;25:7289–7298.
Asensio VC, Campbell IL . Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol 1997;71:7832–7840.
Reigstad CS, Lunden GO, Felin J et al. Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLoS ONE 2009;4:e5842.
Reigstad CS, Backhed F . Microbial regulation of SAA3 expression in mouse colon and adipose tissue. Gut Microbes 2010;1:55–57.
Ransohoff RM, Kivisakk P, Kidd G . Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 2003;3:569–581.
Cornell CT, Kiosses WB, Harkins S et al. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J Virol 2007;81:6785–6797.
McLaren FH, Svendsen CN, Van der Meide P et al. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J Neuroimmunol 2001;112:35–46.
Ruller CM, Tabor-Godwin JM, Van Deren DAJ et al. Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am J Pathol 2012;180:1107–1120.
Jakovcevski I, Zecevic N . Olig transcription factors are expressed in oligodendrocyte and neuronal cells in human fetal CNS. J Neurosci 2005;25:10064–10073.
Bonthius DJ, Nichols B, Harb H et al. Lymphocytic choriomeningitis virus infection of the developing brain: critical role of host age. Ann Neurol 2007;62:356–374.
Truong P, Heydari S, Garidou L et al. Persistent viral infection elevates central nervous system MHC class I through chronic production of interferons. J Immunol 2009;183:3895–3905.
Wellen J, Walter J, Jangouk P et al. Neural precursor cells as a novel target for interferon-beta. Neuropharmacology 2009;56:386–398.
Li L, Walker TL, Zhang Y et al. Endogenous interferon gamma directly regulates neural precursors in the non-inflammatory brain. J Neurosci 2010;30:9038–9050.
Gonzalez-Perez O, Jauregui-Huerta F, Galvez-Contreras AY . Immune system modulates the function of adult neural stem cells. Curr Immunol Rev 2010;6:167–173.
Palha JA, Fernandes R, de Escobar GM et al. Transthyretin regulates thyroid hormone levels in the choroid plexus, but not in the brain parenchyma: study in a transthyretin-null mouse model. Endocrinology 2000;141:3267–3272.
Fleming CE, Nunes AF, Sousa MM . Transthyretin: more than meets the eye. Prog Neurobiol 2009;89:266–276.
Rantakallio P, Jones P, Moring J et al. Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol 1997;26:837–843.
Koponen H, Rantakallio P, Veijola J et al. Childhood central nervous system infections and risk for schizophrenia. Eur Arch Psychiatry Clin Neurosci 2004;254:9–13.
Bonapace G, Iuliano F, Molica S et al. Cytosolic carbonic anhydrase activity in chronic myeloid disorders with different clinical phenotype. Biochim Biophys Acta 2004;1689:179–181.
Dodgson SJ . The Carbonic Anhydrases: Cellular Physiology And Molecular Genetics. New York, NY, USA: Plenum Press, 1991.
Mukherjee A, Morosky SA, Delorme-Axford E et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 2011;7:e1001311.
Humbertclaude V, Tourtet S, Semprino M et al. Acute myelitis of an unusual cause in a child: the lymphocytic choriomeningitis virus. Arch Pediatr 2001;8:282–285.
Acknowledgements
This work was supported by National Institutes of Health (NIH) awards R01 NS054108 (to RF), RO1 NS051247 (to KSD), an NIH Research Supplement to Promote Diversity in Health-Related Research Award 3R01NS054108-01A2S1 (to RF and SMR), an National Institutes of Mental Health (NIMH) Minority Research Infrastructure Support Program (M-RISP) R24 Faculty Fellow Award MH065515 (to RF), an SDSU University Grants Program Award (to RF), and an Achievement Rewards for College Scientists (ARCS) Foundation Scholarship (to JMP). Scott M Robinson is a recipient of the Rees-Stealy Research Foundation and the San Diego State University Heart Institute Fellowship. Microarray analysis was performed at the Biogem Core Facility of the University of California San Diego—Director Gary Hardiman.
Disclaimer
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Supplementary information
Rights and permissions
About this article
Cite this article
Puccini, J., Ruller, C., Robinson, S. et al. Distinct neural stem cell tropism, early immune activation, and choroid plexus pathology following coxsackievirus infection in the neonatal central nervous system. Lab Invest 94, 161–181 (2014). https://doi.org/10.1038/labinvest.2013.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.138
Keywords
This article is cited by
-
Sodium Butyrate Induced Neural Stem/Progenitor Cell Death in an Experimental Model of Japanese Encephalitis
Metabolic Brain Disease (2023)
-
Treatment of perinatal viral infections to improve neurologic outcomes
Pediatric Research (2017)
