
Abstract
Epiregulin (EPI) and amphiregulin (AR) are epidermal growth factor receptor (EGFR) ligands implicated in mucosal repair and tumorigenesis. We have shown that Toll-like receptor 4 (TLR4) induces intestinal epithelial cell (IEC) proliferation by activating EGFR through AR expression. We examined whether TLR4 differentially regulates expression of EGFR ligands in response to mucosal injury. The human IEC line SW480 was examined expression of EGFR ligands, EGFR phosphorylation, and proliferation in response to lipopolysaccharide (LPS). Small-interfering RNA (siRNA) was used to block TLR4. Neutralizing antibodies to EGFR ligands were used to examine inhibition of LPS-dependent EGFR activation. Acute colitis and recovery were examined in the mice given 2.5% dextran sodium sulfate (DSS). Colonic secretion of EPI and AR was analyzed by enzyme-linked immunosorbent assay. LPS selectively induces EPI and AR but not other EGFR ligands. LPS induced early EPI mRNA expression between 30 min and 24 h. The neutralizing antibodies to EPI and AR prevented activation of EGFR by LPS. LPS induces IEC proliferation (200%, P=0.01) in 24 h but blocking EPI and AR significantly decreased proliferation. In vivo, mucosal EPI and AR expression are significantly decreased in TLR4−/− mice (P=0.02) compared to wild-type mice during acute colitis. EPI and AR exhibit different kinetics in response to mucosal damage: EPI expression is upregulated acutely at day 7 of DSS, but falls during recovery at day 14. By contrast, a sustained upregulation of AR expression is seen during mucosal injury and repair. We show that TLR4 regulates EPI and AR expression and that both these EGFR ligands are necessary for optimal proliferation of IEC. The diverse kinetics of EPI and AR expression suggest that they function in distinct roles with respect to acute injury vs repair. Our results highlight the role of bacterial sensing for IEC homeostasis and may lead to targeted therapy for mucosal healing and prevention of tumorigenesis.
Similar content being viewed by others


Main
The intestinal epithelium is juxtaposed with a high density of bacteria and other commensal microbiota along its apical border. Growing evidence supports a role for host bacterial recognition in the maintenance of intestinal homeostasis.1, 2 Data from germ-free animals show that intestinal epithelial cell (IEC) proliferation is substantially reduced, suggesting that bacterial signaling is linked to proliferation.3 The molecular link between luminal bacteria and IEC proliferation has not been fully characterized. Despite the requirement of such commensal bacteria for the normal function of the intestine, an abnormal host response to commensal bacteria has been implicated as a crucial factor in the pathogenesis of inflammatory bowel disease (IBD).4, 5 This highlights both the beneficial and pathophysiological roles of the gut's commensal bacteria in intestinal mucosal biology. In this regard, bacterial sensing along the surface of the mucosa must have an important role in managing the fine balance of fighting pathogenic bacterial invasion while maintaining the homeostasis of our intestinal mucosa.
We have found that Toll-like receptor (TLR) signaling triggers IEC proliferation, contributing to mucosal repair after injury.6, 7 TLRs are a family of receptors that sense pathogen-associated molecular patterns (PAMPs) expressed by microbes and induce innate immune responses to the pathogens.8 This pathogen sensing system is necessary to protect the host against pathogenic invasion in the event that mucosal barrier function is disrupted by epithelial injury. Also, TLR signaling may act as a key link between luminal bacteria and intestinal inflammation. The early events in IBD may include defects in the innate immune response to commensal bacteria, which ultimately result in chronic intestinal inflammation.9, 10, 11 Once TLRs sense PAMPs, the TLR signal culminates in induction of nuclear translocation of nuclear factor-κB (NF-κB),12 followed by an array of gene expression to aid in an immune response.
In addition to preventing bacterial invasion, TLR4 may have an important role in mucosal repair.7 We have previously described the link between TLR4 signaling and epidermal growth factor receptor (EGFR) activation in the setting of chronic inflammation.13 Activation of EGFR aids in growth, repair, and barrier integrity of the gastrointestinal mucosa.14, 15, 16, 17 It has been shown that the EGFR signal is necessary to maintain intestinal homeostasis in the setting of acute mucosal damage.18 In the intestinal mucosa, there are several members of the family of EGF-related growth factors that can activate EGFR signaling.19 Although each of these growth factors should have redundant functions, little is known about differences among these growth factors in the maintenance of mucosal homeostasis. Furthermore, uncontrolled epithelial proliferation may induce tumorigenesis so tonic signaling may be detrimental. One EGFR ligand is amphiregulin (AR). We have described that AR is induced by TLR4 signaling and may have a significant role in the transition from chronic inflammation to tumorigenesis in the setting of colitis.1 Therefore, the aim of this study was to explore the molecular mechanisms used by TLR4 to signal epithelial repair. In particular, we have focused on the link between TLR4 and activation of EGFR signaling during acute mucosal injury.
In this study, we examined expression of EGF-related growth factors in response to TLR4 signaling. We found that TLR4 induces epiregulin (EPI) expression in IECs. We show that TLR4 signaling differentially regulates the expression of EPI and AR in the IEC response to mucosal damage both in vitro and in vivo. Our results implicate a distinct role of TLR signaling as a bridge between luminal bacteria and mucosal healing in the setting of acute colitis.
MATERIALS AND METHODS
Cell Lines and Reagents
Human IEC line SW480 (1 × 106 cells per well) (American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco's modified Eagle's medium supplemented with 2% heat-inactivated fetal calf serum, 2 mmol/l L-glutamine, 5% penicillin/streptomycin, and were incubated in six-well plates overnight at 37°C in a 5% CO2 humidified incubator. Cells were incubated with Ultrapure lipopolysaccharide (LPS), Escherichia coli 0111: B4 (InvivoGen, San Diego, CA, USA) for varying time periods at 2 μg/ml, recombinant human EPI (R&D Systems, Minneapolis, MN, USA) at 40 ng/ml or recombinant human AR (R&D Systems) at 40 ng/ml. For experiments using neutralizing antibodies, we pre-incubated cells with an anti-human EPI (R&D Systems) or anti-human AR neutralizing antibody (R&D Systems) or anti-EGFR neutralizing antibody (Upstate, Lake Placid, NY, USA) at a concentration of 10 μg/ml, 2 h before LPS stimulation. Normal mouse IgG (10 μg/ml) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a negative control.
Reverse-Transcription PCR
Total RNA was isolated from SW480 cells using RNA Bee (Tel-Test, Friendswood, TX, USA) according to the manufacturer's instructions. RNA (1 μg) was used as the template for single-strand complementary DNA (cDNA) synthesis using the Transcriptor First Strand cDNA synthesis kit (Roche, Indianapolis, IN, USA) according to the manufacturer's instructions. PCR was performed for EPI, AR, betacellulin, transforming growth factor-α (TGF-α), heparin-binding EGF, EGF, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primers used in this study are listed in Table 1. All primers were designed using Beacon Designer 3.0 (Premier Biosoft International). The cDNA was amplified using Titanium Taq DNA polymerase (Clontech, Mountain View, CA, USA) on an Eppendorf Thermal cycler, programmed for 95°C for 5 min and then 30 cycles of 95°C for 30 s, 57°C (EPI, betacellulin, TGF-α, heparin-binding EGF, and GAPDH) and 46°C (AR and EGF) for 30 s, and 74°C for 30 s followed by 74°C for 10 min. Amplification products were detected by agarose gel electrophoresis (1.5% agarose gel), ethidium bromide staining, and UV transillumination.
Real-Time PCR
A total of 1 μg RNA was used as the template for single-strand cDNA synthesis using the Transcriptor First-Strand cDNA Synthesis Kit (Roche) according to the manufacturer's instructions. Quantitative real-time PCR was performed for EPI, AR, and β-actin using the primers and probes listed in Table 1. The cDNA was amplified using SYBR Premix Ex Taq (Takara Bio, Shiga, Japan) with the standard SYBR Green setting of 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA). The amplification results were analyzed using sequence detection systems (SDS) 2.2.1 software (Applied Biosystems) and the gene of interest was normalized to the corresponding β-actin results. Data were expressed as fold induction relative to the lowest gene product amplified.
RNA Interference
SW480 cells were plated at a density of 1.5 × 105 cells per well in 12-well plates 24 h before the first transfection. TLR4 small-interfering RNA (siRNA) oligonucleotide corresponding to the sequence GGUAAGGAAUGAGCUAGUAUU was purchased from Dharmacon (Chicago, IL, USA). A total of 50 nmol/l of siRNA was transfected twice every 24 h with X-trim gene siRNA transfection reagent (Roche) as per the manufacturer's instructions. At 48 h after the first transfection, cells were stimulated with LPS (2 μg/ml) for 4 h. Negative control siRNA (50 nmol/l), which has no significant homology to any known gene sequences from mouse, rat, or human being, was used as a control (Ambion, Austin, TX, USA).
Western Blot Analysis
Whole-cell lysates were prepared from SW480 cells after treatment with different stimuli using a lysis buffer containing 50 mmol/l Tris-HCl, 50 mmol/l NaF, 1% Triton X-100, 2 mmol/l EDTA, and 100 mmol/l NaCl, with a proteinase inhibitor cocktail (Calbiochem, San Diego, CA, USA). The protein concentration was determined by the Bradford method using Bio-Rad Protein Assay Dye and SmartSpec 3000 (Bio-Rad Laboratories, Hercules, CA, USA). A total of 25 μg of the lysates were subjected to 10% SDS–polyacrylamide gel electrophoresis and transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). The membrane was blocked in 5% skim milk and was immunoblotted with the primary antibodies for 1 h, followed by horseradish peroxidase-conjugated secondary antibodies rabbit anti-mouse or goat anti-rabbit IgG (Zymed Laboratories, South San Francisco, CA, USA). The membrane was exposed on radiographic film using an enhanced chemiluminescent substrate SuperSignal West Pico Trial Kit (Pierce Biotechnology, Rockford, IL, USA). Anti-phosphorylated EGFR antibody (Tyr1068; 1H12) was purchased from Cell Signaling Technology (Danvers, MA, USA) and anti-EGFR antibody was purchased from Upstate (Temecula, CA, USA). Anti-TLR4, anti-β-actin, and anti-EPI were purchased from eBiosciences (San Diego, CA, USA), Sigma-Alrdich (St Louis, MO, USA), and R&D systems, respectively.
Immunofluorescent Staining
Intestinal samples were fixed in 10% neutral buffered formalin and embedded in paraffin wax. Sections (4-μm-thick) were incubated in 10% normal rabbit serum for 1 h and stained with goat anti-AR antibody (1:200; Santa Cruz Systems, Santa Cruz, CA) overnight at 4°C, followed by FITC-conjugated anti-goat IgG (1:400; Zymed Laboratories) for 1 h at room temperature. For EPI staining, frozen sections (8-μm-thick) were fixed with –20°C cold acetone for 10 min, and stained with goat anti-murine EPI antibody (1: 400; R&D systems) after blocking with 10% normal rabbit serum for 1 h. The specificity of staining was confirmed by omitting the primary antibody. The same secondary antibody was used as AR staining. Slides were viewed on a Nikon eclipse TS100 immunofluorescence microscope and photographs were taken with a digital camera using NIS Elements D 3.0 software program (Nikon, Melville, NY, USA).
Cell Proliferation Assay
SW480 cells (5 × 104 cells per well) were cultured in 96-well plates in the absence or presence of neutralizing antibody to EPI (10 μg/ml), neutralizing antibody to AR (10 μg/ml), or isotype IgG control antibody in low serum condition (1% fetal calf serum), and stimulated with LPS (2 μg/ml), human recombinant EPI (40 ng/ml), or human recombinant AR (40 ng/ml). After 24, 48, or 72 h of stimulation, cell proliferation was analyzed using CellTiter 96 aqueous nonradioactive cell proliferation assay kit (Promega, Madison, WI, USA). Cell proliferation was detected by measurement of formazan product in each well at the absorbance of 490 nm after incubation with tetrazolium/phenazine methosulfate for 1 h at 37°C. The cell proliferation index was calculated as a percentage of the absorbance in relation to the untreated control cells.
Enzyme-Linked Immunosorbent Assay
For the AR enzyme-linked immunosorbent assay (ELISA), SW480 (1 × 106 cells per well) were plated in six-well plates. Cells were treated with LPS (2 μg/ml) for the indicated periods. For ex vivo colonic tissue cultures, 100 mg of tissue from each part of the colon (not including the polypoid lesions) were cultured for 24 h in 12-well flat-bottom plates in serum-free RPMI 1640 supplemented with penicillin and streptomycin. Supernatants were harvested for measurement of 1% penicillin/streptomycin. An ELISA (R&D Systems) was performed per the manufacturer's instructions.
Mice and Interventions
TLR4−/− mice were purchased from Oriental Bio Service (Kyoto, Japan) and were back-crossed to C57Bl/6 mice more than eight times. C57Bl/6 mice were obtained from Jackson Laboratory as controls (Bar Harbor, ME, USA). Sex-matched mice (7- to 10-week-old) were given 2.5% dextran sodium sulfate (DSS) (molecular weight, 36–50 kDa; ICN, Aurora, OH, USA) in their drinking water for 7 days followed by normal water as recovery. Mice were killed either at the end of 4 or 7 days of DSS treatment or days 11, 14, or 56 during recovery. At the kill, the cecum and proximal and distal halves of the colon were removed and fixed in 10% buffered formalin, paraffin-embedded, sectioned, and stained with H&E for histological evaluation. All experiments were performed according to the Mount Sinai School of Medicine Animal Experimental Ethics Committee guidelines.
Assessment of In Vivo Epithelial Proliferation
The number of proliferating cells was detected by immunoperoxidase staining for the thymidine analog bromodeoxyuridine (BrdU). At 1.5 h before the kill, mice were injected intraperitoneally with 5-bromo-2′-deoxyuridine (Sigma-Alrdich) at a concentration of 100 mg/kg. Sections (4 μm) of paraffin-embedded colonic tissue were deparaffinized and incubated with 3% H2O2 in methanol for 15 min. Sections were incubated with 2 N HCl for 1 h, washed in PBS, and then incubated in 0.1% trypsin for 15 min at 37°C. Sections were stained for BrdU incorporation using BrdU staining kit (Zymed Laboratories) according to the manufacturer's instructions. The number of BrdU-positive cells per well-oriented crypt were calculated in every three crypts for each colon segment at high magnification under light microscopy.
Reporter Gene Assay
SW480 cells were plated in 12-well plates at a density of 1.5 × 105 cells per well. The following day, cells were transfected with pRL-TK (0.05 μg) and ELAM-NF-κB-luciferase (0.4 μg) using FuGENE 6 transfection reagent (Roche) as per manufacturer's instructions. After overnight transfection, cells were stimulated with LPS and then lysed and firefly luciferase activity was measured with a Dual-Luciferase Reporter Assay (Promega, San Luis Obispo, CA, USA). Luciferase measurements were normalized to the relative light units from Renilla luciferase from pRL-KT.
Statistical Analysis
Calculation of the Kruskal–Wallis followed by Mann–Whitney U-test, standard error (s.e.m.) and standard deviation (s.d.) were performed using the PRISM version 5.0b. Student's t-test was performed to compare two independent variables by using the statistics package within Microsoft Excel (Microsoft, Redmond, WA, USA). P-values <0.05 were considered significant.
RESULTS
TLR4 Induces Expression of EPI
Our previous studies have shown that LPS induces AR expression in IECs.1 However, there are multiple EGF-related growth factors that can contribute to EGFR-mediated cell proliferation, including EGF,20 AR,21 heparin-binding EGF,22 TGF,23 EPI,24 and betacellulin.25 We hypothesized that TLR4 signaling regulates not only AR expression but also the expression of other EGFR ligands. To address this question, we examined the mRNA expression of the six EGFR ligands in LPS-treated SW480 cells. SW480 cells are human IECs that express TLR4 and respond to LPS.26 Of the six EGFR ligands we examined, only AR and EPI expression were upregulated by LPS stimulation at 24 h (Figure 1a). Quantitative measurement of gene expression by real-time PCR confirmed significant increases of AR (P=0.024) and EPI (P=0.033) expression in response to LPS stimulation (Table 2). Because we previously showed that AR is regulated by TLR4,1 we focused on EPI in this study to explore the hypothesis that TLR4 has specific, regulated effects on trophic growth factors in response to intestinal injury. We found early induction of EPI by LPS stimulation by 4 h (Figure 1b).

TLR4 induces expression of epiregulin. (a) mRNA expression of EGFR ligands from human IEC SW480 in response to LPS. Agarose gel electrophoresis of real-time (RT)–PCR products from IECs (SW480 cells) stimulated with LPS (100 ng/ml) for 18 h. Expression of EGFR ligands is indicated. The graph (right) shows NF-κB activation by LPS stimulation as signaling control. Certain response is shown. (b) SW480 cells were stimulated with LPS (2 μg/ml) for indicated times. SYBR Green RT–PCR shows LPS-induced expression of epiregulin mRNA with a peak at 4 h of stimulation and a significant drop after 24 h when compared to 4 h. Data are represented as mean±s.e.m. of relative values of expression in three individual experiments of triplicate samples (*P<0.05). (c) TLR4-dependent induction of epiregulin in response to LPS. SW480 cells were stimulated in the presence or absence of LPS (2 μg/ml) for 4 h and co-transfected with either TLR4 siRNA or negative control siRNA. RT–PCR showed LPS-induced expression of epiregulin in negative control siRNA samples treated with LPS. This induction of epiregulin by LPS was largely abolished in the cells in which TLR4 was blocked with siRNA, indicating a TLR4-dependent pathway. Data are represented as mean±s.e.m. of relative values of expression in three individual experiments with triplicate samples (*P<0.05). Extent of TLR4 suppression by siRNA (upper right panel). SW480 cells were transiently transfected with siRNA against TLR4 or Negative siRNA, which has no significant homology to any gene sequences, was applied as a control. The knockdown efficiency of siRNA against TLR4 was assessed by western blot analysis. The siRNA decreased TLR4 protein expression. Negative siRNA did not affect TLR4 protein expression. (d) LPS induced epiregulin and amphiregulin protein expression in different kinetics. (Top panel) SW480 cells were stimulated in the presence LPS (2 μg/ml) for 30 min, 4 h, and 24 h. Blots of whole-cell lysates (25 μg per lane) were probed sequentially for the active form for epiregulin. β-Actin was used as an internal control for protein loading. The data are one representative experiment of three with similar results. (Bottom panel) SW480 cells were stimulated in the presence LPS (2 μg/ml) for 4, 24, and 48 h. Cell supernatants were measured concentration of amphiregulin by ELISA. (e) Epiregulin and amphiregulin mRNA expression in response to TLR ligands by RT–PCR. SW480 cells were stimulated with Pam3CSK4 (TLR2 ligand), polyI:C (TLR3 ligand), and LPS at a concentration of 100 ng/ml for 24 h. Expression of epiregulin, amphiregulin, and GADPH was examined by RT–PCR. The data are one representative experiment of three with similar results.
To determine if EPI expression is dependent on TLR4 signaling, we knocked down TLR4 expression using siRNA and stimulated cells with LPS. The cells with decreased TLR4 expression showed decreased EPI expression similar to baseline control levels (Figure 1c), whereas cells transfected with control siRNA upregulated EPI. These data implicate TLR4 signaling in expression of specific trophic growth factors important in mucosal repair.
We have previously shown that TLR4 regulates AR expression in the setting of mucosal injury.1 Because there may be biological differences between AR and EPI, we asked whether these EGFR ligands are regulated differently by TLR4. To address this question, we examined the kinetics of expression of these growth factors in LPS-stimulated SW480 cells. Real-time PCR showed a peak in EPI gene expression within 4 h of LPS stimulation (Figure 1b). However, after 24 h of stimulation, there is a significant decrease in EPI expression. By contrast, our previous studies show that AR mRNA expression in LPS-stimulated SW480 cells rises significantly at 4 h and continues to peak at 24 h.1 Consistent with the different kinetics at the RNA level between these two TLR4-inducible EGFR ligands, protein expression mirrored their gene expression. LPS induces EPI protein expression at 30 min to 4 h; at 24 h, EPI expression is almost undetectable (Figure 1d). AR protein expression, on the other hand, is upregulated at 4 h and continues to increase in intensity up to 24 h (Figure 1d). We also wanted to examine specificity of TLR4 in regulation of these growth factors. To address this issue, we stimulated SW480 cells with different TLR ligands for TLR2 and TLR3 and compared these to TLR4. The expression of the corresponding TLRs has been shown in SW480.27 In addition to TLR4, signaling through TLR3 can also induce EPI and AR mRNA expression, whereas TLR4 still induces the most robust induction (Figure 1e). These results suggest that AR and EPI gene and protein expression are upregulated differentially by TLR4 signaling. Although EPI expression peaks early, AR shows a gradual and sustained activation in vitro.
TLR4 Induces EGFR Phosphorylation and Cell Proliferation Through EPI Expression
Our previous studies have shown that TLR4 induces EGFR phosphorylation and cell proliferation by AR in vitro.7 Given that TLR4 signaling also induces EPI expression, we hypothesized that LPS-induced EPI would also lead to EGFR transactivation and cell proliferation. When we stimulated the SW480 cells with exogenous EPI, we observed EGFR phosphorylation within 30 min (Figure 2a). MTT assay showed that exogenous addition of EPI also induced cell proliferation to a level seen with TLR4 stimulation (Figure 2b). To characterize the individual contribution of EPI in TLR4-dependent EGFR signaling, we used neutralizing antibodies to EPI and measured EGFR activation. We previously showed that neutralizing antibodies to AR blocked LPS-induced EGFR activation.1 When the bioactivity of EPI is inhibited, EGFR phosphorylation is reduced when compared to cells treated with an IgG isotype control (Figure 2a). This result and our previous studies suggest that both ligands contribute to TLR4-dependent EGFR transactivation.

TLR4 induces EGFR phosphorylation and cell proliferation through epiregulin expression. (a) LPS-mediated activation of EGFR is epiregulin and amphiregulin dependent. SW480 cells were stimulated with LPS (2 μg/ml) for 30 min or treated with exogenous epiregulin (40 ng/ml). SW480 cells were pretreated with neutralizing antibodies to the ligand-binding site of EGFR, and/or amphiregulin, or control immunoglobulin G for 2 h. Blots of whole-cell lysates (25 μg per lane) were probed sequentially for phospho-EGFR or EGFR. β-Actin was used as an internal control for protein loading. The data are one representative experiment of three with similar results. (b) LPS, epiregulin, and amphiregulin induce cell proliferation. SW480 cells were stimulated with LPS (2 μg/ml), epiregulin (40 ng/ml), or amphiregulin (40 ng/ml) for 24 h. Data are shown as the mean percentage of absorbance and s.d. in comparison with untreated control cells from three independent experiments (*P<0.05). (c) LPS-induced cell proliferation is epiregulin and amphiregulin dependent. SW480 cells were pretreated with neutralizing anti-amphiregulin antibody, anti-epiregulin antibody, antibodies to both, or control immunoglobulin G for 2 h. Subsequently, cells were stimulated with LPS (2 μg/ml) for 24 h. Data are shown as the mean percentage of absorbance and s.d. in comparison with untreated control cells from three independent experiments (*P<0.05). (d) Epiregulin- and amphiregulin-induced cell proliferation show diverse kinetics. SW480 cells were stimulated with epiregulin (40 ng/ml) or amphiregulin (40 ng/ml) for 24, 48, or 72 h. Data are shown as the mean percentage of absorbance and s.d. in comparison with untreated control cells from three independent experiments (*P<0.05).
We next wanted to determine the functional consequences of EGFR phosphorylation downstream of TLR4-dependent EGFR ligand expression. To do this, we examined the effect of blocking EPI, AR, or both, on TLR4-dependent IEC proliferation. Using individual blocking antibodies against either EPI or AR that do not cross-react, cell proliferation drops approximately 60% (Figure 2c). Importantly, blocking both AR and EPI in response to TLR4 activation reduces proliferation to baseline levels (Figure 2c). As a control, isotype IgG control treatment does not affect TLR4-induced cell growth. These data support a role for both AR and EPI in the proliferative effects of TLR4.
Our expression data (Figure 1) and previous studies1 suggest that TLR4 activation of IEC results in expression of AR and EPI but with distinct kinetics. We thus hypothesized that AR and EPI may influence cell proliferation with distinct kinetics. To address this question, we measured proliferation in response to exogenous AR and EPI and compared their kinetics with LPS stimulation of IECs. We used concentrations of AR and EPI that are seen with LPS stimulation. Our data show that EPI induces early cell proliferation, which peaks at 48 h at 222% proliferation and falls by 72 h (P<0.05) (Figure 2d). By contrast, AR-induced proliferation increases steadily over 24 and 48 h to 182% proliferation (P=0.04) and peaks at 72 h at 247% proliferation (P<0.05). LPS-induced proliferation showed a steady rise over time. These data suggest that these two EGF-related growth factors, EPI and AR, induce cell proliferation differentially and may thus have different functional roles during mucosal injury and repair.
TLR4 Differentially Regulates EPI and AR Expression in a Murine Model of Acute Colitis
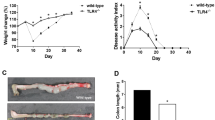
Our previous in vivo studies have shown that AR production is significantly lower in TLR4-deficient mice when compared to wild-type (WT) mice.1 Going along with our in vitro studies, we hypothesized that EPI expression is also decreased in TLR4-deficient mice in the setting of acute colitis. After DSS-induced mucosal injury, WT mice have a large increase in proliferating cells that persists even a week after DSS is discontinued. By contrast, TLR4-deficient mice have significantly fewer proliferating cells.7 We used real-time PCR to examine expression of EPI after DSS-induced injury in WT vs TLR4−/− mice (Figure 3a). Our data show that mucosal EPI mRNA expression is significantly lower in TLR4−/− mice when compared to WT mice after 7 days of DSS treatment (Figure 3a).


TLR4 differentially regulates epiregulin and amphiregulin expression in a murine acute colitis model. (a) Mucosal epiregulin expression is decreased in TLR4−/− mice at day 7 in the DSS model. Real-time PCR was used to compare mucosal expression of epiregulin mRNA in WT and TLR4−/− mice in the DSS model (n=6 each). Data are represented as mean±s.d. of relative values of expression (*P<0.05). (b) Mucosal amphiregulin and epiregulin expression display different kinetics in WT mice over 4, 7, 11, 14, and 56 days of DSS treatment. Real-time PCR was used to compare mucosal expression of epiregulin and amphiregulin mRNA in WT and TLR4−/− mice in the DSS model (n=6 each). Data are represented as mean±s.d. of relative values of expression (*P<0.05). (c) Representative immunofluorescent photographs of epiregulin and amphiregulin in colitic mucosa of day 7 and 14 of DSS colitis, respectively. The control slide stained only with secondary antibody. (d) IEC proliferation and histological changes of colonic mucosa during DSS colitis. IEC proliferation was assessed by counting the number of BrdU-positive cells per well-oriented crypt at high magnification under light microscopy. Data show average BrdU-positive cell number (±s.d.) in every three crypts for each colon segment. (e and f) Mucosal amphiregulin and epiregulin secretion display different kinetics in WT mice at day 7 vs day 56 of DSS treatment. The production of amphiregulin and epiregulin was measured in colonic mucosa from WT (n=6) mice. Data are expressed as mean±s.d. (*P<0.05).
A previous study has suggested that EPI is required for protection against intestinal damage, but not for tumorigenesis.28 We have described that TLR4-dependent AR expression may be involved in colitis-associated tumorigenesis through EGFR activation in the setting of chronic mucosal inflammation.1 Given our in vitro data, we hypothesized that the two TLR4-inducible growth factors may be expressed differentially in a murine model of acute vs chronic colitis. We subjected mice to two cycles of DSS to represent an acute phase (4 or 7 days of DSS treatment) or a chronic phase (14 or 56 days after DSS treatment) of inflammation. Histological severity of colitis was similar between WT and TLR4−/− mice in the phase of chronic colitis (WT 13.1±3.1 vs TLR4−/− 10.5±4.4, P=0.09, maximum score: 24). After 7 days of DSS treatment, both AR and EPI mRNA expression in WT mice were significantly upregulated compared to day 0 and 4 expression levels (Figure 3b). EPI mRNA expression reaches its peak at day 7 and quickly decays in spite of ongoing inflammation and repair. Conversely, AR mRNA expression levels in WT mice show a sustained upward trend through day 14 and continue to be elevated during the chronic phases of inflammation at day 56 (Figure 3b). Conversely, TLR4−/− mice display very low expression levels of AR and EPI mRNA (Figure 3b), highlighting the importance of TLR4 signaling in mucosal expression of growth factors. Immunofluorescent detection of mucosal EPI at day 7 and AR at day 14 shows that the main source of EPI and AR expression is epithelial cells (Figure 3c). Although EPI and AR showed distinct peaks of mucosal expression during acute and chronic colitis, epithelial proliferation as measured by BrdU incorporation peaked at day 14 (Figure 3d). These data indicate that mucosal induction of EPI and AR expression during DSS-induced colitis is dependent on TLR4 signaling, supporting a significant role in vivo for TLR4 in expression of EGFR ligands. They also show distinct functional roles for each of these ligands.
We next examined mucosal EPI and AR protein secretion in the mouse colitis model. We found that EPI and AR protein expression mirrors gene expression. EPI released from the colonic mucosa is significantly increased in the acute phase of colitis (day 7) when compared to day 0, but not in the chronic phase corresponding to day 56 of DSS treatment (Figure 3d). AR, however, exhibits inverted levels: low in acute injury, but higher in chronic injury (Figure 3e). Given the differences in kinetics of EPI and AR expression, these data suggest that TLR4 orchestrates a complex program of epithelial repair that is tailored to acute mucosal injury vs chronic injury.
DISCUSSION
This study clarifies the mechanism by which TLR4 signaling modulates EGFR activation in the setting of mucosal injury. In our previous work, we have shown that epithelial damage permits exposure of IECs to an array of PAMPs and activates a TLR4-mediated signaling cascade that results in enhanced transcription of cyclooxygenase 2 (Cox-2), increased production of PGE2, and EGFR activation.7 In this study, we focus on TLR4-mediated regulation of EGFR ligands. We show that TLR4 differentially regulates the expression of two EGF-related growth factors, AR and EPI, in the setting of intestinal damage. Although EPI expression is upregulated immediately in acute mucosal injury, AR has a more significant role during chronic inflammation. In the absence of TLR4, neither is expressed. These results suggest a novel function of the TLR4 signaling pathway in mucosal repair and IEC proliferation to maintain mucosal homeostasis.
TLRs function within the innate immune system as caretakers of the intestinal mucosa. We have shown that TLR4−/− mice in the face of DSS-induced mucosal injury have increased bleeding, increased weight loss, and bacterial translocation to mesenteric lymph nodes.6 Epithelial injury permits bacterial invasion of the lamina propria by commensal bacteria. TLR4 is critical at multiple levels to clear intramucosal bacteria before they have a chance to escape to mesenteric lymph nodes and beyond.2 But the role of TLR4 in the intestinal mucosa is much more complex than just bacterial clearance. TLR4 activates a program that is critically involved in epithelial repair. LPS, a TLR4 ligand, induces epithelial restitution in a dose-dependent way.29 We have previously shown that TLR4 regulates repair and cell proliferation by modulating the EGFR signaling pathway.7 EGFR has a vital role in intestinal mucosal restitution. Animals with mutations in EGFR or animals unable to produce TGF-α have increased susceptibility to epithelial damage after DSS-induced injury.18, 30 Therefore, to maintain intestinal homeostasis, TLR4 has two distinct and interrelated roles during epithelial injury: a gatekeeper to prevent and fight off bacterial invasion and a switch to modulate repair signals.
In this study, we show that TLR4 signaling not only induces the expression of two different growth factors, but also differentially regulates their expression. We examined several EGFR ligands, but these two seem to be particularly regulated by TLR4 signaling. In the setting of acute mucosal injury, TLR4 strongly upregulates EPI expression. By contrast, TLR4-mediated AR expression is seen predominantly during chronic inflammation. Because EPI can bind to multiple ErbB receptors—which consists of four closely related receptor tyrosine kinases, including EGFR (ErbB-1),19 ErbB-2,31, 32 ErbB-3,33, 34 and ErbB-434, 35—EPI seems to be a more potent and broader regulator of ErbB-mediated cell proliferation.36, 37 Furthermore, EPI has the ability to cross-induce the expression of other EGF-related growth factors.38, 39 These distinct characteristics of EPI might help to explain why TLR4 induces its expression dramatically in the early stages of acute epithelial damage. Furthermore, expression of EPI has been shown to be localized to epithelial and stromal cells after DSS-induced tissue injury. A potential risk of promiscuous growth factor activity is an overactive repair signal, leading to uncontrolled cell proliferation and cancer. However, EPI has been shown to be required for repair and protection against acute mucosal injury induced by DSS treatment, but does not have a role in tumorigenesis.28 AR, on the other hand, has been implicated in tumorigenesis.40, 41, 42
Although EPI acts broadly and can cross-induce other growth factors to aid in early mucosal recovery from injury, AR may serve as a backup system for EGFR-mediated repair. We show that AR is significantly upregulated in the setting of DSS-induced chronic colitis but its rise is gradual and sustained. Although AR can serve as a substitute for EGF, it acts as a much weaker growth factor compared to other EGF family growth factors,21 and its only target within the ErbB family is EGFR itself.19 In fact, our studies show that AR induces lower levels of cell proliferation than EPI does, at least in a human IEC line after 24 and 48 h of stimulation. Although TLR4-mediated expression of EPI is quick and potent, AR expression may serve as a sustained, backup mechanism for mucosal restitution that aids in long-term homeostasis such as in the setting of chronic inflammation. In vivo, when we examined mucosal activation of EGFR by western blot analysis at different time points after DSS treatment, mucosal activation of EGFR was similar between days 7, 14, and 56 of 7 days DSS treatment although normal mucosa had no EGFR activation (data not shown).
How can TLR4 regulate expression of EGFR ligands with different kinetics under similar conditions? The TLR4 signaling pathway results in the activation of the transcription factor, NF-κB, which has an important role in inflammation, apoptosis suppression, and cell proliferation.43, 44 Interestingly, the promoter region of the EPI gene contains an NF-κB transcriptional binding site.45 Therefore, TLR4 may signal immediate repair through the NF-κB-mediated upregulation of EPI. By contrast, NF-κB activation has yet to be shown to directly regulate AR transcriptional activity. In our previous studies, we show that AR expression is induced by TLR4 signaling through Cox-2. Therefore, Cox-2 expression may regulate AR expression in a more indirect manner. Furthermore, AR possesses autocrine activity to induce a positive feedback mechanism,46 helping to explain the slow, gradual, and ultimately sustained expression of AR we have seen.
In this study, we have found a novel function of TLRs that helps to clarify the molecular link between TLR4 signaling and EGFR activation during acute mucosal damage. Our findings indicate that TLR4 is important for the expression of two distinct EGF-related growth factors, AR and EPI, but more importantly, that TLR4 has a crucial role in mucosal healing and repair. Our results serve to highlight the complex regulation by our host-microbial innate immune response in fighting infection while aiding in repair. An imbalance in this system of bacterial recognition for repair of injury could result in too little repair (ie, bacterial invasion) or too vigorous repair (ie, cancer). This study highlights the potential to target and modulate this novel innate immune response to control intestinal mucosal inflammation and carcinogenesis.
References
Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007;133:1869–1881.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004;118:229–241.
Abrams GD, Bauer H, Sprinz H . Influence of the normal flora on mucosal morphology and cellular renewal in the ileum. A comparison of germ-free and conventional mice. Lab Invest 1963;12:355–364.
Sartor RB . Clinical applications of advances in the genetics of IBD. Rev Gastroenterol Disord 2003;3 (Suppl 1):S9–S17.
Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005;128:891–906.
Fukata M, Michelsen KS, Eri R, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 2005;288:G1055–G1065.
Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology 2006;131:862–877.
Pasare C, Medzhitov R . Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol 2005;560:11–18.
Duchmann R, Kaiser I, Hermann E, et al. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol 1995;102:448–455.
Xavier RJ, Podolsky DK . Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427–434.
Yamamoto-Furusho JK, Podolsky DK . Innate immunity in inflammatory bowel disease. World J Gastroenterol 2007;13:5577–5580.
Sato S, Sanjo H, Takeda K, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 2005;6:1087–1095.
Goodlad RA, Wright NA . Epidermal growth factor (EGF). Baillieres Clin Gastroenterol 1996;10:33–47.
Barnard JA, Beauchamp RD, Russell WE, et al. Epidermal growth factor-related peptides and their relevance to gastrointestinal pathophysiology. Gastroenterology 1995;108:564–580.
Beck PL, Podolsky DK . Growth factors in inflammatory bowel disease. Inflamm Bowel Dis 1999;5:44–60.
Konturek PK, Brzozowski T, Konturek SJ, et al. Role of epidermal growth factor, prostaglandin, and sulfhydryls in stress-induced gastric lesions. Gastroenterology 1990;99:1607–1615.
Playford RJ, Wright NA . Why is epidermal growth factor present in the gut lumen? Gut 1996;38:303–305.
Egger B, Buchler MW, Lakshmanan J, et al. Mice harboring a defective epidermal growth factor receptor (waved-2) have an increased susceptibility to acute dextran sulfate-induced colitis. Scand J Gastroenterol 2000;35:1181–1187.
Riese 2nd DJ, Stern DF . Specificity within the EGF family/ErbB receptor family signaling network. Bioessays 1998;20:41–48.
Carpenter G, Cohen S . Epidermal growth factor. J Biol Chem 1990;265:7709–7712.
Shoyab M, Plowman GD, McDonald VL, et al. Structure and function of human amphiregulin: a member of the epidermal growth factor family. Science 1989;243 (4894 Part 1):1074–1076.
Higashiyama S, Abraham JA, Miller J, et al. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science 1991;251:936–939.
Derynck R . The physiology of transforming growth factor-alpha. Adv Cancer Res 1992;58:27–52.
Toyoda H, Komurasaki T, Uchida D, et al. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem 1995;270:7495–7500.
Shing Y, Christofori G, Hanahan D, et al. Betacellulin: a mitogen from pancreatic beta cell tumors. Science 1993;259:1604–1607.
Vora P, Youdim A, Thomas LS, et al. Beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. J Immunol 2004;173:5398–5405.
Otte JM, Cario E, Podolsky DK . Mechanisms of cross hyporesponsiveness to toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology 2004;126:1054–1070.
Lee D, Pearsall RS, Das S, et al. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol Cell Biol 2004;24:8907–8916.
Koff JL, Shao MX, Kim S, et al. Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. J Immunol 2006;177:8693–8700.
Egger B, Procaccino F, Lakshmanan J, et al. Mice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitis. Gastroenterology 1997;113:825–832.
Coussens L, Yang-Feng TL, Liao YC, et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985;230:1132–1139.
Yamamoto T, Ikawa S, Akiyama T, et al. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature 1986;319:230–234.
Kraus MH, Issing W, Miki T, et al. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc Natl Acad Sci USA 1989;86:9193–9197.
Plowman GD, Whitney GS, Neubauer MG, et al. Molecular cloning and expression of an additional epidermal growth factor receptor-related gene. Proc Natl Acad Sci USA 1990;87:4905–4909.
Riese 2nd DJ, Komurasaki T, Plowman GD, et al. Activation of ErbB4 by the bifunctional epidermal growth factor family hormone epiregulin is regulated by ErbB2. J Biol Chem 1998;273:11288–11294.
Komurasaki T, Toyoda H, Uchida D, et al. Epiregulin binds to epidermal growth factor receptor and ErbB-4 and induces tyrosine phosphorylation of epidermal growth factor receptor, ErbB-2, ErbB-3 and ErbB-4. Oncogene 1997;15:2841–2848.
Shelly M, Pinkas-Kramarski R, Guarino BC, et al. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem 1998;273:10496–10505.
Komurasaki T, Toyoda H, Uchida D, et al. Mechanism of growth promoting activity of epiregulin in primary cultures of rat hepatocytes. Growth Factors 2002;20:61–69.
Shirakata Y, Komurasaki T, Toyoda H, et al. Epiregulin, a novel member of the epidermal growth factor family, is an autocrine growth factor in normal human keratinocytes. J Biol Chem 2000;275:5748–5753.
Shao J, Lee SB, Guo H, et al. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res 2003;63:5218–5223.
De Luca A, Arra C, D'Antonio A, et al. Simultaneous blockage of different EGF-like growth factors results in efficient growth inhibition of human colon carcinoma xenografts. Oncogene 2000;19:5863–5871.
Ciardiello F, Kim N, Saeki T, et al. Differential expression of epidermal growth factor-related proteins in human colorectal tumors. Proc Natl Acad Sci USA 1991;88:7792–7796.
Baldwin Jr AS . The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649–683.
Ghosh S, May MJ, Kopp EB . NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998;16:225–260.
Li X, Massa PE, Hanidu A, et al. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem 2002;277:45129–45140.
Streicher KL, Willmarth NE, Garcia J, et al. Activation of a nuclear factor kappaB/interleukin-1 positive feedback loop by amphiregulin in human breast cancer cells. Mol Cancer Res 2007;5:847–861.
Acknowledgements
This work was supported by NIH Grant 3R56AI05226607S1 (MTA), NCI Grant 5R01CA137869-2 (MTA), and Career Development Award from CCFA (MF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hsu, D., Fukata, M., Hernandez, Y. et al. Toll-like receptor 4 differentially regulates epidermal growth factor-related growth factors in response to intestinal mucosal injury. Lab Invest 90, 1295–1305 (2010). https://doi.org/10.1038/labinvest.2010.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2010.100
Keywords
This article is cited by
-
Targeting autocrine amphiregulin robustly and reproducibly inhibits ovarian cancer in a syngeneic model: roles for wildtype p53
Oncogene (2021)
-
Epithelial Toll-like receptors and their role in gut homeostasis and disease
Nature Reviews Gastroenterology & Hepatology (2020)
-
Amphiregulin in intestinal acute graft-versus-host disease: a possible diagnostic and prognostic aid
Modern Pathology (2019)
-
Pharmacological activation of epidermal growth factor receptor signaling inhibits colitis-associated cancer in mice
Scientific Reports (2018)
-
Role of LPS-elicited signaling in triggering gastric mucosal inflammatory responses to H. pylori: modulatory effect of ghrelin
Inflammopharmacology (2017)
