
Abstract
Adenoid cystic carcinoma (ACC) is a rare but distinctive tumor. Oligonucleotide array comparative genomic hybridization has been applied for cataloging genomic copy number alterations (CNAs) in 17 frozen salivary or bronchial tumors. Only four whole chromosome CNAs were found, and most cases had 2–4 segmental CNAs. No high level amplification was observed. There were recurrent gains at 7p15.2, 17q21–25, and 22q11–13, and recurrent losses at 1p35, 6q22–25, 8q12–13, 9p21, 12q12–13, and 17p11–13. The minimal region of gain at 7p15.2 contained the HOXA cluster. The minimal common regions of deletions contained the CDKN2A/CDKN2B, TP53, and LIMA1 tumor suppressor genes. The recurrent deletion at 8q12.3–13.1 contained no straightforward tumor suppressor gene, but the MIRN124A2 microRNA gene, whose product regulates MMP2 and CDK6. Among unique CNAs, gains harbored CCND1, KIT/PDGFRA/KDR, MDM2, and JAK2. The CNAs involving CCND1, MDM2, KIT, CDKN2A/2B, and TP53 were validated by FISH and/or multiplex ligation-dependent probe amplification. Although most tumors overexpressed cyclin D1 compared with surrounding glands, the only case to overexpress MDM2 had the corresponding CNA. In conclusion, our report suggests that ACC is characterized by a relatively low level of structural complexity. Array CGH and immunohistochemical data implicate MDM2 as the oncogene targeted at 12q15. The gain at 4q12 warrants further exploration as it contains a cluster of receptor kinase genes (KIT/PDGFRA/KDR), whose products can be responsive to specific therapies.
Similar content being viewed by others

Main
Adenoid cystic carcinoma (ACC) is a rare subtype of adenocarcinoma occurring in major and minor salivary glands of the upper aero-digestive tract.1 Other sites of origin include the tracheobronchial tree, lachrymal gland, breast, skin, and vulva. ACC is an infiltrative tumor with a marked tendency to invade nerves and adjacent bone. It consists of two kinds of cells: ductal cells and myoepithelial cells.
Owing to its slow growth, ACC has a relatively indolent but relentless course. When distant metastases occur, only low response rates have been reported with multi-drug regimens.2 Studies have reported contradictory results regarding the usefulness of compounds targeting KIT.3, 4 As a general rule, responsiveness to KIT inhibitors correlates with gene mutations, which are not found in ACC.5 Significant efforts should be directed to improve the knowledge of molecular genetic events in this malignancy to provide new therapeutic targets.
There is wide agreement that recurrent genomic aberrations contain genes that are important for tumor occurrence or progression.6 To date, few studies of ACC using chromosomal comparative genomic hybridization (CGH) have revealed recurrent segmental losses at 6q23–qter, 12q12, and 13q21–q22 and recurrent segmental gains at 16p, 17q, and 22q13.7, 8 Yet, the resolution of these techniques was limited, and loss-of-heterozygoty (LOH) studies have subsequently been performed to map deletions at 6q and 12q.9, 10 Consistent with previous cytogenomic analyses, a recent study using concurrent LOH and copy number analysis by SNP genotyping arrays has concluded that ACCs are characterized by limited number of chromosomal aberrations with the most frequent area of deletion present on 9p.11
Array CGH has been developed to increase the spatial resolution for the detection of chromosomal copy number alterations (CNAs). Vékony et al12, published the results of a study using BAC-based CGH array in a series of 18 paraffin-embedded primary ACCs, showing very frequent copy number gains at loci of growth factors and their receptors. Here, we report the results of a study using high-resolution oligonucleotide CGH array in frozen samples with the goal to increase the knowledge of CNAs in ACC and establish a catalog of candidate cancer genes residing in the altered regions.
MATERIALS AND METHODS
Cases
The 17 frozen samples used for array CGH were obtained from Institut de Cancérologie Gustave-Roussy, Institut Mutualiste Montsouris, Beaujon, Pitié-Salpêtrière, and Avicenne hospitals, (France), but were treated, purified, quantified, and labeled at Institut Gustave Roussy. The study complied with French laws regarding patient information and consent. There were 13 females and four males. The median age was 51 years (interquartile range 38–55). The primary sites were major salivary glands in seven cases, including the parotid gland (three cases), the submandibular gland (three cases), and the sublingual gland (one case), or minor salivary glands along the aerodigestive tract in eight cases. Two cases involved the bronchial glands. The tumor samples were taken from the primary tumor in 11 cases, a local recurrence in three cases, or a metastatic site in three cases.
An additional cohort of patients with primary salivary ACC, for which only paraffin-embedded samples were available, had been treated at Pitié-Salpêtrière hospital from 1968–2002. The 21 samples were chosen to directly compare the immunohistochemical expression of selected proteins in the tumor cells and in surrounding salivary cells.
Array CGH
Samples at least 3 × 3 × 3 mm in size were cut in a cryostat at −20°C after removing most of OCT. Beginning, middle and end sections of each sample were stained with hematoxylin and eosin to assess the proportions of tumor cells. The average percentage of tumor cells was above 70% in each case. Thirty 40 μm sections were placed in a tube and kept frozen. The following procedures are detailed in the supplementary material. The samples were homogenized, and genomic DNA was extracted according to Quiagen protocols with modifications. Restriction of DNA, quality control of restricted DNA by microanalysis, pooling, and clean up, were performed following Agilent instructions. The samples were hybridized in single vs a standard DNA (male or female, Promega) with oligo 44 k CGH Agilent micro arrays (G4410B). The scanning and image analysis were performed with Agilent technology using default settings. Raw data txt files from the Agilent Feature Extraction software 8.5.1.1 were then imported for analysis into CGHAnalytics 3.3.1. Aberrations were obtained with ADM1 algorithm and filtering options of a minimum of five probes and abs (log 2 ratio) >0.3. Aberration segments were manually reviewed. Those that were localized to regions with high-copy repetitive or GC-rich DNA sequences including telomeric regions were excluded.
Multiplex Ligation-Dependent Probe Amplification
The multiplex ligation-dependent probe amplification (MLPA) method for relative quantification of nucleic acids was performed according to manufacturer's instructions (MRC-Holland, Amsterdam, The Netherlands). The P006 MLPA probe mix included two probes for CCND1, two probes for TP53, and one probe for CDKN2B, all labeled with 6-FAM fluorescent dye. The PCR products were analyzed in duplicate and in two dilutions (1/1 and 1/20) using an ABI-3730 DNA Analyser (Applied Biosystems). Preliminary tests showed an excellent reproducibility with less than 5% in the variation of peak area. After normalization, a difference of 30% in peak area of the sample relative to the reference DNA sample was required to call a deletion or a gain.
Fluorescent In Situ Hybridization
Tumor touch preps were prepared by imprinting slightly thawed tissues onto glass slides and air-drying them. The slides were fixed in methanol/acetic acid. For KIT and CDKN2A/2B, FISH were performed with commercial probes (Kreatec, Nl). For CCND1 and MDM2, BAC clones were selected from Pieter De Jong libraries (http://www.chori.org/bacpac/) according to their position on UCSC human genome (http://www.genome.ucsc.edu/). Purified BAC DNA was labeled by random priming in the presence of Alexa 488-dUTP (green) and Alexa 594-dUTP (red) (Abott). The quality of the probes was verified by hybridization to metaphases spreads of normal peripheral lymphocytes.
Immunohistochemistry
Standard immunohistochemistry protocols were used to detect cyclin D1 (pre-diluted mab P2D11F11, Ventana, Tucson, AZ, USA ), MDM2 (mab IF2, Zymed, San Francisco, CA, USA), and VEGF (rabbit polyclonal antibody A-20, Santa Cruz Biotechnology, Santa Cruz, CA, USA). For cyclin D1, after epitope retrieval in 10 mM, pH 7.3 citrate buffer for 20 min, all steps were performed in a Ventana automated stainer. For MDM2, after epitope retrieval for 30 min, the primary antibody was incubated overnight at 4°C at 1:25, then revealed using the Histostain kit (Zymed) For VEGFA, after epitope retrieval for 30 min, the primay antibody was incubated at room temperature at 1:200 for 1 h, then revealed using the ABC Elite kit (Vector, Burlingame, CA, USA). DAB was used as a substrate.
Statistical Analysis
The Fischer's exact test was used to compare primary tumors vs recurrences or metastases according to whether they had a unique or multiple large segmental losses as well as to compare expression of cyclin D1 in tumor and salivary gland cells.
RESULTS
Cytogenomic Analysis
The results of our analysis for each case are provided in Table 1. The profile of gains and losses for the study population is displayed in Figure 1. The number of CNAs varied from 17 (case 3) to one (cases 9 and 16) with most cases being 2–4 (Figure 1, Table 1). Only four whole chromosome CNAs, linked to mitotic non-disjunction, were found. This compares to the 66 segmental CNAs, linked to structural chromosomal rearrangements, which were observed. Losses were much more frequent than gains in the large segmental CNAs (>10 Mb), while they were in the same proportions in smaller segmental CNAs (<10 Mb) (P<0.01). Among the three tumors with more than a unique large segmental CNA loss, one was a local recurrence (case 1), and two were a metastasis (cases 5 and 3) compared to 11 primary tumors and three recurrences or metastases among cases having no or a unique large segmental CNA loss (P=0.03). Case 3 had the highest number of segmental CNAs and complex structural rearrangements on 4q, 11q and 15.

Profile of gains and losses after exclusion of repetitive DNA sequences in 17 frozen adenoid cystic carcinomas using high-resolution oligonucleotide CGH array. Shown are log 2 ratios for each chromosome.
Recurrent CNAs
Segmental CNAs individually spanning at least 10 probes were compared across samples to identify overlapping groups of positive- or negative-value CNAs (CNA clusters). This step identified 12 CNA clusters that were defined by at least two positive- or negative-value overlapping CNAs. The number of overlapping CNAs per cluster ranged from two to four.
The 12 clusters were localized on chromosomes 1p, 6q, 7p, 8q, 9p, 12q, 17p, 17q, and 22q. Two distinct clusters that were separated by at least 1 Mb were present on 1p, 6q, and 12q, whereas the chromosomes 7p, 8q, 9p, 17p, and 22q harbored only 1 cluster each. On 17q, there were actually six distinct regions of overlapping positive-value CNAs, which were considered together as a single complex cluster extending from 17q21.32 to 17q25.3.
The boundaries of segmental CNAs in each cluster were used in an effort to identify the minimal regions of overlap or minimal common regions (MCRs). Eleven clusters included only 1 MCR. In the highly complex cluster on 17q, 2 MCRs could be identified, while there was no clear pattern of recurrence in other 17q regions. The data for the 13 MCRs are shown in Table 2. There were 4 MCR of gains and 9 MCR of losses, which were localized to chromosomes 7p, 17q, 22q, and to chromosomes 1p, 6q, 8q, 9p, 12q, 17p, respectively. The median MCR size was 1365 kb (range 90–5447 kb). The maximum ratio ranged from 1.31 to 2.04 for gains, while the minimum ratio ranged from 0.52 to 0.61 for losses.
Putative Oncogenes or Tumor Suppressor Genes Present within MCRs
Genes were localized to MCRs using the UCSC Genome Browser (HG 17, build 35). Among MCR genes, coding genes that have been associated with cancer were identified using the list of genes possibly implicated in cancer in the Atlas of Genetics and Cytogenetics in Oncology and Haematology.13
As shown in Table 2, the LIMA1, CDKN2A/CDKN2B, and TP53 cancer-associated genes were present within MCRs of losses. Interestingly, although the MCR at 9p21 also involved the MTAP and TUSC1 putative tumor suppressor genes, an additional eight probes loss (41 kb) targeted exactly the CDNK2A/CDKN2B locus. The MCR at 7p15.2 contained the HOXA cluster.
Several regions included gene encoding microRNAs. The loss extending 2193 kb at 8q12.3–13.1, which was noted in three cases, involved the MIRN124A2 microRNA gene.
Putative Oncogenes or Tumor Suppressor Genes Present within Unique CNAs
Several unique CNAs were sufficiently narrow (16–79 probes) to point to possibly relevant genes. These events include gain of JAK2, high level gain of CCND1, MDM2, and KIT (Table 1).
Validation of Aberrations with MPLA
The deletions involving TP53 at 17p13.2. and CDKN2B at 9p21 were confirmed by a reduction of more than 30% of peak areas in cases 8 and 10 and in cases 1 and 10, respectively. An increase more than 30% in relative peak area for both the CCND1 and the adjacent FGF3 probes at 11q13 confirmed the gain in case 3.
Validation of Aberrations with FISH
FISH validated the gains in regions involving KIT/PDGFRA/KDR, MMD2, and CCND1 as well as the loss in the region involving CDKN2A/CDKN2B.
Immunohistochemistry
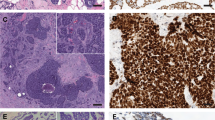
Cyclin D1 was diffusely expressed within tumor nuclei in case 3—a metastatic deposit to the liver—that had a CCND1 gain (Figure 2). Tissue from the primary site in the palate was not available for comparison. In an independent series of 21 ACCs with paraffin-embedded samples with paired surrounding salivary glands, cyclin D1 was expressed in every sample in 30–100% tumor cells, while none of the salivary glands expressed the protein (P<0.001).

Immunoperoxydase staining for cyclin D1, MDM2, and VEGFA. (a and b) Cyclin D1: diffuse reactivity within tumor nuclei in case 3 (a); diffuse reactivity in tumor nuclei on the right side and lack or reactivity in the adjacent salivary gland on the left side in one paraffin-embedded case (case 18) (b). (c and d) MDM2: strong reactivity in scattered nuclei (arrows) in case 7 (c); moderate cytoplasmic staining, but lack of nuclear reactivity in the adjacent epithelial structures in the cavum in the same case (d). (e and f) VEGFA: diffuse cytoplasmic reactivity in case 7 (e); another field of the same case showing on the left side minor salivary glands with negative mucous cells and positive serous cells (arrow) (f).
Case 7 that harbored a gain involving MDM2 exhibited strong MDM2 nuclear expression, which was not seen in the adjacent glands in the cavum (Figures 2 and 3). Nuclear MDM2 expression was not seen in any other tumor, including the 21 independent paraffin-embedded samples. None of the surrounding bronchial or salivary glands expressed MDM2 within nuclei; light or moderate cytoplasmic MDM2 staining could occasionally be observed. Moderate or strong expression of VEGFA was noted within the cytoplasm of 20–100% of tumor cells in every tumor sample. In salivary glands strong expression was seen in serous acinar cells and some duct cells. Mucous cells were consistently negative.

Validation of aCGH results using FISH on touch imprint of frozen tumors. (a) Case 3. Four copies of KIT gene were noted on the nuclei (white arrows). (b) Case 1. A single CDKN2A/2B locus (red signal) was observed while the control FISH probe (9q21) in green was normal. The nuclei with deletion are indicated with arrows. (c) Case 3. The BAC clone RP11-195M19 (red) in the region of CCND1 gene was present in three copies on tumor nuclei. (d) Case 7. The BAC RP11-159C11 (red) and RP11-1143G9 (green) were chosen at the locus and near MDM2 gene respectively. Some nuclei showed a typical amplification pattern seen with double minute amplification.
DISCUSSION
A genome-wide analysis of 17 frozen samples of ACC has been performed using long oligonucleotide CGH 44k array. Previous chromosomal CGH studies have revealed recurrent segmental losses at 6q23-qter, 12q12, and 13q21–q22 and recurrent segmental gains at 16p, 17q, and 22q13.7, 8 The comparison with our results show that the majority of the most frequent losses or gains that were previously reported by chromosomal CGH are listed in this study in a limited cohort, but often delineated with a better precision. Notably, we report MCRs of deletions on 6q and on 12q, which overlap with previously identified regions of common allelic losses that were mapped by LOH studies, an observation that further supports the validity of our methodology.9, 10 Using oligonucleotide CGH array, at least five probes are required for a reliable call for loss or gain.14, 15 In this study, we made a definite call only using at least 10 probes. Moreover, aberrations were subjected to manual review. An important aspect of our analysis is that we chose to discard recurrent calls in regions characterized by GC-rich and high-copy repetitive sequences. As in chromosomal CGH, less specific hybridization to arrays can occur in GC-rich regions, probably in relationship with not perfectly effective blocking by Cot-1 DNA even when the quality of genomic DNA preparations is carefully controlled.16, 17 To validate our results, we used FISH and MLPA for six selected aberrations. We were able to validate five of the six aberrations assayed, including those containing CCND1, MDM2, KIT, CDKN2A/2B, and TP53. However, we could not validate one recurrent low amplitude gain, which is not shown in this paper. Therefore, although our approach was conservative, caution should be advised for the regions that we did not validate with secondary techniques.
Every case reported here had CNAs with an average number of three. Large chromosomal band size segmental CNAs were significantly more frequently losses than gains, to the contrary of small segmental CNAs. No high level amplification was observed. Whole CNAs were observed on small chromosomes, that is, chr16, chr21 and chr22 in two cases. Those results are in accordance with most previous reports, which claim that ACC does not have very extensive chromosomal aberrations, and they suggest that global genomic instability is not a mechanism.7, 8, 11 A potential technical problem could have been the dilution of tumor cell DNA into non tumor cell DNA. However, the percentage of tumor cells was above 70% in each of our samples. Consistent with the accumulation of genetic events as tumors progress toward more advanced stages, the only tumor that had a relatively high number of genomic aberrations was a metastatic lesion, and recurrent or metastatic lesions had significantly more large segmental losses than primary tumors.
The low level of structural complexity and the resolution of the oligonucleotide CGH array may facilitate the identification of regions that may be of functional importance. An interesting gain at 12q15 involves MDM2. A previous study has previously reported an amplicon of 6.8 Mbps at 12q14.3–q21.1,12 containing many genes. Here, the gain that was validated by FISH as a double minute type amplification extended only along 800 kB. Furthermore, we showed that the case with the 12q15 gain was the only one to express MDM2 within tumor nuclei and that none of the salivary or bronchial glands expressed the protein within nuclei. Together, our data strongly support that MDM2 is the targeted oncogene. CDKN2A and CDKN2B are the prime suspects in the loss at 9p21 in two cases. An additional 8-probes loss involved exactly CDKN2A and CDKN2B, further suggesting that the locus is the target of the deletion. Several studies have reported deletion or methylation of CDKN2A in ACC.18, 19 In addition to genetic changes, epigenetic modifications have an important impact on gene expression and the phenotypes of cancer cells. CDKN2A/CDKNB also encodes p14ARF, a member of the MDM2/TP53 pathway. Interestingly, TP53 was present in a recurrent loss, and the losses of CDKN2A/CDKNB, the losses of TP53, and the gain of MDM2 were mutually exclusive aberrations, suggesting that the pathway is involved in ACC.
Another interesting aberration is the gain at 4q12. Freier et al,8 using classic CGH has previously reported a gain at 4q12–q13 in one case, but our study is the first to have narrowed the gain to a region that contains the cluster of the transmembrane receptor kinase genes KIT, PDGFRA, and KDR. Inhibitors of KIT are used in the treatment of advanced ACC,2, 3, 4 although no KIT mutation has been found so far.5 KIT is consistently expressed by luminal cells in ACCs.5 Before the removal of the liver deposit that was analyzed by array CGH, the patient had not been treated with KIT inhibitors, a chronology that indicates that the 4q12 gain was not secondary to such treatment. The KIT oncoprotein interacts with PDGFRA, whose gene is localized in the vicinity. The region also harbors KDR, which encodes the receptor for VEGF. Serous cells in normal salivary glands constitutively express high levels of VEGF,20 a fact that we confirmed by means of immunohistochemistry. Moreover, we showed that most tumors expressed high VEGF levels. The target gene in the 4q12 region remains to be determined.
CCND1 was gained in one case, in which cyclin D1 was diffusely expressed. As reported by Frierson et al, CCND1 is one of the highest ranked overexpressed genes in ACC compared with normal salivary gland.21 We confirmed that ACC expressed cyclin D1 by means of immunohistochemistry in an independent series, while the surrounding glands did not (P<0.001). Unlike MDM2, cyclin D1 expression within tumor nuclei is not restricted to cases with a gain in the corresponding locus, a fact indicating that several mechanisms are responsible for cyclin D1 overexpression.
The MCR at 12q12–q13.13 overlaps with one of the major consensus region of allele loss reported by Rutherford et al,9 though the boundaries of the changes were not coincident. Several cancer genes reside in the loss, including the LIMA1 putative tumor suppressor gene.22 The ANP32D and LETMD1 genes, also present within the consensus region of loss, are oncogenic. DUSP6, another candidate tumor suppressor gene within the LOH consensus deletion loci, is located outside the array CGH defined MCRs on 12q that is reported here.
Ten genes coding for microRNAs were located to regions of recurrent gain or loss, a finding consistent with the fact that this class of small, regulatory RNA genes frequently resides in cancer-associated genomic regions.23 Evidence is accumulating that microRNas contribute to tumorigenesis, which indicates that aberrations should be examined for the presence of potentially cancer-associated microRNA genes. The deletion at 8q12.3, where no straightforward tumor suppressor gene has been located, harbors the MIRN124A2 gene, which regulates MMP2, which is involved in invasion and metastasis, and the oncogene CDK6.24 Two regions of gains at clusters of HOX genes, several of which are oncogenic, contain MRIN196 genes that participate in the post-transcriptional regulation of HOX messages.25
While we were in the final phase of writing this paper, Vékony et al12, published the results of a study using BAC-based CGH 4.2 k array in a series of 18 paraffin-embedded primary ACCs, showing very frequent (∼60–80%) copy number gains at loci of growth factors and their receptors.12 Here, we analyzed 17 samples, but the number of aberrations, particularly gains, in our report is clearly lower than that found by Vékony. Choosing to use conservative criteria with a deliberate bias against high-copy repetitive or GC-rich sequences, it is likely that we underestimated the number of aberrations. Reviewing our primary data we noticed, albeit at a far lower frequency, abnormalities consistent with 15 of the 21 gains that were reported by Vekony et al12. Strikingly, 12 of these 15 gains are localized to telomeres (1p36.33, 4p16.3, 8q24.3, 9q34.3, 11p15.5, 13q34, 16p13.3, 16q24.3, 17p13.3, 19p13.3, 20q13.33, and 22q13.33), and were discarded in our study as telomere-associated sequences are markedly GC-rich. While telomeric regions may contain important cancer genes, our opinion is that further evidence is required to establish the occurrence of widespread gains at telomeric sites in ACC.
In conclusion, although the present study is only a first-pass screening approach, the results presented here suggest that it provides an efficient entry point to catalog genomic CNAs in ACC. We were able to find most regions of recurrent gains and loss previously reported. Together with expression data, our results strongly implicate MDM2. The gain at 4q12 warrants further exploration as it contains a cluster of receptor kinase genes, whose products can be responsive to specific therapies. Further efforts to identify the target genes will probably involve systematic screens to produce orthogonal data sets (mutational, epigenetic, expression and loss of function phenotypes).
Accession codes
References
El-Naggar A, Huvos A . Adenoid cystic carcinoma. In: Barnes L, Eveson J, Reichart P, Sidransky D (eds). Pathology and Genetics of Head and Neck Tumours (World Health Organization Classification of Tumours), 1st edn. IARC Press: Lyon, France, 2005, pp 221–222.
Dodd R, Slevin N . Salivary gland adenoid cystic carcinoma: a review of chemotherapy and molecular therapies. Oral Oncol 2006;42:759–769.
Faivre S, Raymond E, Casiraghi O, et al. Imatinib mesylate can induce objective response in progressing, highly expressing KIT adenoid cystic carcinoma of the salivary glands. J Clin Oncol 2005;23:6271–6273.
Pfeffer M, Talmi Y, Catane R, et al. A phase II study of Imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral Oncol 2006;43:33–36.
Holst V, Marshall C, Moskaluk C, et al. KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol 1999;12:956–960.
Albertson D, Collins C, McCormick F, et al. Chromosome aberrations in solid tumors. Nat Genet 2003;34:369–376.
El-Rifai W, Rutherford S, Knuutila S, et al. Novel DNA copy number losses in chromosome 12q12–q13 in adenoid cystic carcinoma. Neoplasia 2001;3:173–178.
Freier K, Flechtenmacher C, Walch A, et al. Copy number gains on 22q13 in adenoid cystic carcinoma of the salivary gland revealed by comparative genomic hybridization and tissue microarray analysis. Cancer Genet Cytogenet 2005;159:89–95.
Rutherford S, Yu Y, Rumpel C, et al. Chromosome 6 deletion and candidate tumor suppressor genes in adenoid cystic carcinoma. Cancer Lett 2006;236:309–317.
Rutherford S, Hampton G, Frierson H, et al. Mapping of candidate tumor suppressor genes on chromosome 12 in adenoid cystic carcinoma. Lab Invest 2005;85:1076–1085.
Yu Y, Baras A, Shirasuna K, et al. Concurrent loss of heterozygosity and copy number analysis in adenoid cystic carcinoma by SNP genotyping arrays. Lab Invest 2007;87:430–439.
Vékony H, Ylstra B, Wilting S, et al. DNA copy number gains at loci of growth factors and their receptors in salivary gland adenoid cystic carcinoma. Clin Cancer Res 2007;13:3133–3139.
Huret JL, Dessen P, Bernheim A . Atlas of genetics and cytogenetics in oncology and haematology. Nucleic Acids Res 2003;31:272–274.
Ylstra B, van den Ijssel P, Carvalho B, et al. BAC to the future! or oligonucleotides: a perspective for micro array comparative genomic hybridization (array CGH). Nucleic Acids Res 2006;34:445–450.
Wicker N, Carles A, Mills I, et al. A new look towards BAC-based array CGH through a comprehensive comparison with oligo-based array CGH. BMC Genomics 2007;8:84.
Pinkel D, Albertson D . Array comparative genomic hybridization and its applications in cancer. Nat Genet 2005;37:S11–S17.
Tan DS, Lambros MB, Natrajan R, et al. Getting it right: designing microarray (and not ‘microawry’) comparative genomic hybridization studies for cancer research. Lab Invest 2007;87:737–754.
Maruya S, Kurotaki H, Shimoyama N, et al. Expression of p16 protein and hypermethylation status of its promoter gene in adenoid cystic carcinoma of the head and neck. ORL J Otorhinolaryngol Relat Spec 2003;65:26–32.
Li J, El-Naggar A, Mao L . Promoter methylation of p16INK4a, RASSF1A, and DAPK is frequent in salivary adenoid cystic carcinoma. Cancer 2005;104:771–776.
Pammer J, Weninger W, Mildner M, et al. Vascular endothelial growth factor is constitutively expressed in normal human salivary glands and is secreted in the saliva of healthy individuals. J Pathol 1998;186:186–191.
Frierson H, El-Naggar A, Welsh J, et al. Large scale molecular analysis identifies genes with altered expression in salivary adenoid cystic carcinoma. Am J Pathol 2002;161:1315–1323.
Maul R, Chang D . EPLIN, epithelial protein lost in neoplasm. Oncogene 1999;18:7838–7841.
Calin G, Sevignani C, Dumitru C, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 2004;101:2999–3004.
Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 2007;67:1424–1429.
Yekta S, Shih I, Bartel D . MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004;304:594–596.
Acknowledgements
We are indebted to the following investigators who participated in the collection of tissue samples: J-C Bertrand (GH Pitié-Salpêtrière), F Janot (Institut Gustave-Roussy), A Martin (CH Avicennes), and D Cazal-Hatem (CH Bichat). We thank V Lazar for help with the PCR assays and A Valent for the generous gift of metaphase spreads of normal peripheral lymphocytes (both investigators at Institut Gustave-Roussy). This work was supported by Institut Gustave-Roussy (taxe professionnelle) and PNES Poumon (INCa).
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure
All authors deny any ethical or financial conflict of interest.
Data availability: The microarray data related to this paper have been submitted to the Array Express data repository at the European Bioinformatics Institute (http://www.ebi.ac.uk/arrayexpress/) under the accession number E-TABM-267. They are available for reviewing purpose at the following URL: www.ebi.ac.uk/aerep/login
Username: Reviewer_E-TABM-267
Password: 1179220984917
Rights and permissions
About this article
Cite this article
Bernheim, A., Toujani, S., Saulnier, P. et al. High-resolution array comparative genomic hybridization analysis of human bronchial and salivary adenoid cystic carcinoma. Lab Invest 88, 464–473 (2008). https://doi.org/10.1038/labinvest.2008.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2008.18
Keywords
This article is cited by
-
Prognostic significance of 1p36 locus deletion in adenoid cystic carcinoma of the salivary glands
Virchows Archiv (2018)
-
miR-21 regulates tumor progression through the miR-21-PDCD4-Stat3 pathway in human salivary adenoid cystic carcinoma
Laboratory Investigation (2015)
-
Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2–MALT1 in MALT lymphoma
Nature Communications (2015)
-
Breast- and Salivary Gland-Derived Adenoid Cystic Carcinomas: Potential Post-Transcriptional Divergencies. A Pilot Study Based on miRNA Expression Profiling of Four Cases and Review of the Potential Relevance of the Findings
Pathology & Oncology Research (2015)
-
Adenoid Cystic Carcinoma: Clinical and Molecular Features
Head and Neck Pathology (2013)
