
Abstract
Gitelman syndrome (GS) is an autosomal recessive renal tubulopathy characterized by hypokalemic metabolic alkalosis with hypocalciuria and hypomagnesemia. GS clinical symptoms range from mild weakness to muscular cramps, paralysis or even sudden death as a result of cardiac arrhythmia. GS is caused by loss-of-function mutations in the solute carrier family 12 member 3 (SLC12A3) gene, but molecular mechanisms underlying such a wide range of symptoms are poorly understood. Here we report cryptic exon activation in SLC12A3 intron 12 in a clinically asymptomatic GS, resulting from an intronic mutation c.1669+297 T>G that created a new acceptor splice site. The cryptic exon was sandwiched between the L3 transposon upstream and a mammalian interspersed repeat downstream, possibly contributing to inclusion of the cryptic exon in mature transcripts. The mutation was identified by targeted next-generation sequencing of candidate genes in GS patients with missing pathogenic SLC12A3 alleles. Taken together, this work illustrates the power of next-generation sequencing to identify causal mutations in intronic regions in asymptomatic individuals at risk of developing potentially fatal disease complications, improving clinical management of these cases.
Similar content being viewed by others
Introduction
Gitelman syndrome (GS, OMIM 263800) is one of the most common autosomal recessive kidney tubulopathies, with an estimated prevalence of 1:40 000 in Caucasians.1, 2 GS is characterized by hypokalemia, hypomagnesemia, metabolic alkalosis and hypocalciuria, and usually manifests as mild weakness, cramps and general fatigue. However, some GS cases are clinically asymptomatic or are not diagnosed until late childhood or adulthood,3 but molecular mechanisms for the variable penetrance and age of onset are poorly understood.
GS is caused by mutations in the solute carrier family 12 member 3 (SLC12A3) gene4 that encodes the thiazide-sensitive sodium-chloride cotransporter.4 The loss of sodium-chloride cotransporter function leads to a decrease in sodium and chloride reabsorption in the distal convoluted tubule, causing salt-wasting tubulopathy. To date, more than 400 different SLC12A3 mutations have been identified in GS; however, as many as 20–41% patients were found only with a single pathogenic allele, suggesting that the mutation screening is unsatisfactory.3, 4, 5, 6, 7, 8 Although using reverse transcription-PCR (RT-PCR) may help identify deep intronic mutations resulting in RNA processing abnormalities,9, 10 this method may not identify all aberrant mRNAs. The reason for the large fraction of missing GS alleles is poorly understood.
Here we report a case of latent GS caused by a partial cryptic exon activation in SLC12A3 intron 12. Identification of the new exon was facilitated by next-generation sequencing (NGS) followed by in silico analysis and RT-PCR validation of aberrant transcripts. The case highlights the importance of detecting intronic variants in low-penetrance genetic conditions at risk of potentially fatal complications, permitting a more focused management of affected families.
Materials and methods
The proband was a 5-year-old girl diagnosed by chance with mild proteinuria (urinary protein/creatinine: 0.4 g gCr−1), hypokalemia (2.7 mEq l−1), hypomagnesemia (1.5 mg dl−1), metabolic alkalosis (HCO−3 : 26.7 mEq l−1) and hypocalciuria (urinary calcium/creatinine: 0.005 mg mg−1). The laboratory findings were indicative of GS, but she had not suffered any overt clinical symptoms.
All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine. Informed consent was obtained from all patients or their parents.
NGS samples were prepared using a HaloPlex Target Enrichment System Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions to capture 12 genes (Supplementary Table 1), including SLC12A3, CLCNKB, SLC12A1, KCNJ1 and other genes associated with hypokalemia. Amplified target libraries were sequenced using MiSeq (Illumina, San Diego, CA, USA), which was followed by variant analysis with SureCall (v.3.0; Agilent Technologies). The SLC12A3 reads were mapped to the human reference sequence NC_000015.9 and NM_000338.2. Exons were numbered according to a previous report.11 Rare variants with a frequency <1% were analyzed using the Human Splicing Finder v.3.0 software (http://www.umd.be/HSF3/).
Results
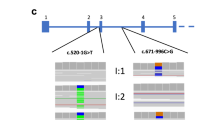
Conventional mutation screening of SLC12A3 in the proband revealed only a single pathogenic allele on the maternal chromosome (c.2927C>T, p.Ser976Phe), which was previously reported in two patients.12, 13 We next used the HaloPlex target enrichment system analysis of candidate genes for tubulopathies/pseudo-GS, including SLC12A3. NGS detected a total of 88 heterozygous intronic SLC12A3 variants (Figure 1a). Analysis of variants with a minor allele frequency of <1% using the Human Splicing Finder (v.3.0) software14 revealed a single heterozygous variant predicted to create a new 3′ splice site, c.1567+297 T>G, located in intron 12. This change had a potential of activating a cryptic exon with a high-score cryptic 5′ splice site further downstream (Supplementary Figures 1A and B). Validation of the putative cryptic exons using RT-PCR with primers in exons 12 and 13 revealed a 108-bp insertion of the new coding sequence in the patient’s SLC12A3 mRNA (Figures 1c and d), confirming the in silico prediction. The new exon was sandwiched between the L3 transposon and a mammalian interspersed repeat located upstream and downstream, respectively (Figures 1c and d and Supplementary Figures 2 and 3). The presence of each mutation was confirmed in parental samples (Figure 1e). These results indicated that the GS proband was a compound heterozygote for SLC12A3 mutations, one resulting in amino-acid substitution S976F in the C-terminal domain of sodium-chloride cotransporter and the other in protein truncation through cryptic exon activation.

Cryptic exon activation in solute carrier family 12 member 3 (SLC12A3) in Gitelman syndrome (GS). (a) Flow chart illustrates the step-wise identification of the intronic mutation in our proband. MAF, minor allele frequency. (b) Genomic sequence of SLC12A3 intron 12 in the proband. Mutation c.1567+297 T>G was detected by next-generation sequencing and confirmed by Sanger sequencing. (c) Transcript analysis by reverse transcription-PCR (RT-PCR). Left panel: S, size marker with fragment sizes shown to the left; C, control; P, patient. The fraction of aberrant transcripts was ~12% of the total signal from polyadenylated RNAs. Right panel: Direct sequencing of the larger transcript showed a 108-bp insertion between exons 12 and 13. (d) Schematics of the cryptic exon activation. Exons are shown as boxes, introns as lines and aberrant transcripts as dotted lines. Transposed elements are schematically shown by green rectangles and correspond to alignments in Supplementary Figure 3. (e) Pedigree of the GS family. The proband is denoted by an arrow. Each GS allele was inherited from the parents. A full color version of this figure is available at the Journal of Human Genetics journal online.
We then used NGS in two more GS cases with previously reported deep intronic splicing mutations. In Case 1, NGS detected a total of 30 heterozygous intronic SLC12A3 variants, including mutation c.1670−191C>T, which created a new 5′ splice site (Supplementary Figures 4A–C) and activated the cryptic exon, as reported.10 In Case 2, NGS detected a total of 27 heterozygous intronic SLC12A3 variants, including variant c.2548+253C>T, which creates a new 3′ splice site (Supplementary Figures 5A–C).9
Discussion
Previously, we reported two deep intronic SLC12A3 mutations, both of which were hotspots for Japanese and Taiwanese GS patients, suggesting that these cases might be observed more frequently.9, 10 In this report, we used NGS to detect missing alleles in a suspected GS. Our results clearly demonstrate that NGS can facilitate their identification in asymptomatic cases that may develop potentially life-threatening complications later in life. As an example, arrhythmia as a result of hypokalemia can develop in GS children as young as 6 years of age.15 To confirm the utility of this method, NGS successfully identified known variants in two other GS cases possessing deep intronic variants that affected splicing.
Interestingly, the novel cryptic exon was activated in a region closely flanked by a long interspersed repeat upstream and a mammalian interspersed repeat downstream (Supplementary Figure 3). Mammalian interspersed repeats have a propensity to exonize by a single mutation, and have very high exonization levels compared with other transposable elements,16 although it remains to be seen if this element facilitated usage of the new 3′ splice site.
Although NGS is still more expensive compared with conventional direct sequencing, NGS of DNA samples may complement RT-PCR approaches and facilitate identification of aberrant transcripts missed by conventional techniques. Definite genetic diagnosis of GS will permit better management of patients and improve their quality of life.
In conclusion, we identified a novel SLC12A3 allele in GS that activates a cryptic exon flanked by interspersed repeats deep in intron 12. Our study illustrates the power of NGS to fully define the mutation pattern in GS and also improve our understanding of the phenotypic variability of this condition. This approach can be used more widely to identify individuals at risk of fatal complications in many other genetic disorders, providing a better support for their clinical management.
References
Seyberth, H. W. An improved terminology and classification of Bartter-like syndromes. Nat. Clin. Pract. Nephrol. 4, 560–567 (2008).
Seyberth, H. W. & Schlingmann, K. P. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr. Nephrol. 26, 1789–1802 (2011).
Cruz, D. N., Shaer, A. J., Bia, M. J., Lifton, R. P., Simon, D. B., Yale, G. S. et al. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 59, 710–717 (2001).
Simon, D. B., Nelson-Williams, C., Bia, M. J., Ellison, D., Karet, F. E., Molina, A. M. et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 12, 24–30 (1996).
Colussi, G., Bettinelli, A., Tedeschi, S., De Ferrari, M. E., Syren, M. L., Borsa, N. et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin. J. Am. Soc. Nephrol. 2, 454–460 (2007).
Lemmink, H. H., Knoers, N. V., Karolyi, L., van Dijk, H., Niaudet, P., Antignac, C. et al. Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. 54, 720–730 (1998).
Lin, S. H., Shiang, J. C., Huang, C. C., Yang, S. S., Hsu, Y. J. & Cheng, C. J. Phenotype and genotype analysis in Chinese patients with Gitelman's syndrome. J. Clin. Endocrinol. Metab. 90, 2500–2507 (2005).
Monkawa, T., Kurihara, I., Kobayashi, K., Hayashi, M. & Saruta, T. Novel mutations in thiazide-sensitive Na-Cl cotransporter gene of patients with Gitelman's syndrome. J. Am. Soc. Nephrol. 11, 65–70 (2000).
Lo, Y. F., Nozu, K., Iijima, K., Morishita, T., Huang, C. C., Yang, S. S. et al. Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome. Clin. J. Am. Soc. Nephrol. 6, 630–639 (2011).
Nozu, K., Iijima, K., Nozu, Y., Ikegami, E., Imai, T., Fu, X. J. et al. A deep intronic mutation in the SLC12A3 gene leads to Gitelman syndrome. Pediatr. Res. 66, 590–593 (2009).
International Human Genome Sequencing, C Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Jang, H. R., Lee, J. W., Oh, Y. K., Na, K. Y., Joo, K. W., Jeon, U. S. et al. From bench to bedside: diagnosis of Gitelman's syndrome—defect of sodium-chloride cotransporter in renal tissue. Kidney Int. 70, 813–817 (2006).
Nakamura, A., Shimizu, C., Nagai, S., Yoshida, M., Aoki, K., Kondo, T. et al. Problems in diagnosing atypical Gitelman's syndrome presenting with normomagnesaemia. Clin. Endocrinol. (Oxf) 72, 272–276 (2010).
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Beroud, G., Claustres, M. & Beroud, C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Bettinelli, A., Tosetto, C., Colussi, G., Tommasini, G., Edefonti, A. & Bianchetti, M. G. Electrocardiogram with prolonged QT interval in Gitelman disease. Kidney Int. 62, 580–584 (2002).
Vorechovsky, I. Transposable elements in disease-associated cryptic exons. Hum. Genet. 127, 135–154 (2010).
Acknowledgements
This study was supported by a grant from the Ministry of Health, Labour and Welfare (Japan) for Research on Rare Intractable Diseases in the Kidney and Urinary Tract (H24-nanchitou (nan)-ippan-041 to Kazumoto Iijima) in the ‘Research on Measures for Intractable Diseases’ Project, and a Grant-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (subject ID: 25893131 to KN and 26293203 to KI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Nozu, K., Nozu, Y., Nakanishi, K. et al. Cryptic exon activation in SLC12A3 in Gitelman syndrome. J Hum Genet 62, 335–337 (2017). https://doi.org/10.1038/jhg.2016.129
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.129
