
Abstract
Nephrotic syndrome arising from monogenic mutations differs substantially from acquired ones in their clinical prognosis, progression, and disease management. Several pathogenic mutations in the COQ8B gene are known to cause nephrotic syndrome. Here, we used the whole-exome sequencing (WES) technology to decipher the genetic cause of nephrotic syndrome (CKD stage-V) in a large affected consanguineous family. Our study exposed a novel missense homozygous mutation NC_000019.9:g.41209497C > T; NM_024876.4:c.748G > A; NP_079152.3:p.(Asp250Asn) in the 9th exon of the COQ8B gene, co-segregated well with the disease phenotype. Our study provides the first insight into this homozygous condition, which has not been previously reported in 1000Genome, ClinVar, ExAC, and genomAD databases. In addition to the pathogenic COQ8B variant, the WES data also revealed some novel and recurrent mutations in the GLA, NUP107, COQ2, COQ6, COQ7 and COQ9 genes. The novel variants observed in this study have been submitted to the ClinVar database and are publicly available online with the accessions: SCV001451361.1, SCV001451725.1 and SCV001451724.1. Based on the patient's clinical history and genomic data with in silico validation, we conclude that pathogenic mutation in the COQ8B gene was causing kidney failure in an autosomal recessive manner. We recommend WES technology for genetic testing in such a consanguineous family to not only prevent the future generation, but early detection can help in disease management and therapeutic interventions.
Similar content being viewed by others


Introduction
Approximately 25% of CKD cases reveal a familial background in their inheritance1,2. Child and adolescent CKD cases mostly exhibit congenital anomalies of the kidneys and urinary tract (CAKUT), chronic glomerulonephritis, steroid-resistant nephrotic syndrome (SRNS), renal cystic ciliopathies, focal segmental glomerulosclerosis (FSGS) and nephrolithiasis or nephrocalcinosis. SRNS in children is one of the leading causes of progression to CKD stage-V or ESRD. Monogenic mutations account for about ~ 20% of individuals with early-onset CKD (before 25 years‑of‑age)3. CKDs with mendelian causes often differ substantially from acquired ones in their clinical prognosis, progression, and critical disease management, imposing a challenge in routine diagnostic processes. Moreover, CKD patients with stage-I to stage-III often remain asymptomatic and undetected until the patient reaches an advanced-stage disease (i.e., stage-IV to V).
Coenzyme Q8B (OMIM * 615567) glomerular nephropathy (COQ8B-GN) or nephrotic syndrome type 9 (NPHS9, OMIM # 615573) is an autosomal recessive type of CKD, inherited through allelic homogeneous variant or compound heterozygous variants. Recent studies have evidenced the monogenic mutations in COQ8B-GN predominantly manifested through SRNS, with proteinuria and typical FSGS renal histopathology4,5,6. The COQ8B is one of the several genes encoding enzymes required for the Coenzyme Q (ubiquinone; CoQ10) pathway. CoQ10, an essential cellular antioxidant, acts as an electron shuttle in the mitochondrial respiratory chain and a permeability transition pore regulator. Pathogenic mutations in any of the genes encoding enzymes of the CoQ10 biosynthetic pathway have been associated with nephrotic syndrome or CKD7,8,9,10.
Massive parallel sequencing (MPS) or next-generation sequencing (NGS) technologies help us conveniently to unravel the genetic basis of the disease, which could be used as a primary diagnostic tool for medical genetic testing. No detailed report of CKD at the genetic level available from Jammu & Kashmir, Northern India. Here, we employed the whole-exome sequencing (WES) approach to precisely identify the functional pathogenic mutations in a large consanguineous family affected with nephrotic syndrome (CKD stage-V or ESRD).
Results
Case presentation
This 25 years old lady (proband: MC4) was first detected to be having renal dysfunction during the third trimester of pregnancy with a serum creatinine of 13 mg/dL and hemoglobin level of 4 g/dL. Though the patient was hypertensive in the first trimester as well, she was not investigated further. On further evaluation, she was detected to be having normal size kidneys. However, it was decided not to go ahead with renal biopsy at that gestation and she continued on hemodialysis. She delivered a 1.5 kg baby and subsequently, her creatinine settled to around 3–4 mg/dL. She was lost to follow up and again presented with azotemic symptoms and serum creatinine of 21 mg/dL, five months down the line. The patient underwent live-related renal transplantation at the age of 23 years, with the donor being her father of age 45 years in 2018. Post-transplantation, the patient had an uneventful course except for an occasional episode of UTI. She has normal allograft function two years post-transplant with no complications and normal renal function. Her previous pregnancy was at the age of 21 years and she delivered at full term by cesarean section. She did not have a bad obstetric history. She underwent all the routine investigations during her first pregnancy and no abnormality was detected at that time. The detailed (follow up) report of estimated glomerular filtration rate (eGFR) and proteinuria in the MC4 (V-1) case has been displayed in Fig. 1.

Pathophysiological characteristics of the proband (MC4, V-1). (A) Presenting the eGFR levels before dialysis treatment. (B) eGFR level after kidney transplant, showing the improvement in eGFR to the normal range. (C) Bar graph presenting the proteinuria (mean ± SE) before and after kidney transplant. The protein levels (mg/24 h) were considered only before the patient undergone dialysis. Mean (average protein loss) was calculated from three readings (intervals) in each category. ***p < 0.0001 statistically significant, using Student’s t-test.
The proband (MC4) had 05 siblings with 04 sisters and 01 brother. There is a family history of renal disease in two of her younger sisters (MC6 and MC7). Both of them were detected to have renal failure at the age of 15 and 16 years, respectively and subsequently became dialysis-dependent. Renal biopsy was not done in any of the siblings. Unfortunately, both of them succumbed to complications related to renal failure. The exact cause of death in both of them could not be ascertained.
Identification of a pathogenic mutation using whole-exome sequencing
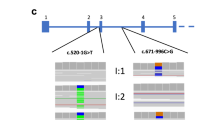
The whole-exome sequencing produced approximately 73,000 overall pass-on-target variants for all seven samples (Supplementary Table S1). Based on the filtering strategy, the variant statistics and a summary of probable disease-causing have been presented in Supplementary Tables S2–S5. Figure 2B illustrates the flowchart of the variant filtering strategy used to find out the most promising causative mutation. Using online GenIO (based on RefGene, NHLBI-ESP, 1000Genomes, dbSNP, ClinVar, COSMIC, gnomAD, OMIM and M-CAP databases)11 and mutation distiller (based on HPO, OMIM, OrphaNet and ACMG actionable genes databases)12 integrated pipelines, we observed 72 likely pathogenic variants with MAF < 0.01 (Supplementary Table S5). Upon exclusion of non-coding and overlapping variants with control subjects and considered those conserved in evolution (GERP score > 0), the pathogenic variant (COQ8B gene, chr19:41209497:C > T; NM_024876.4:c.748G > A; NP_079152.3:p.Asp250Asn) with reference genome GRCh37/hg19 was observed in the homozygous condition and co-segregated with the disease phenotype in the family (Fig. 3A). The mutation was confirmed using BAM and VCF files in IGV 2.5.3 software13 (Fig. 3B and Supplementary Fig. S1). Figure 4 shows the distribution of runs of homozygosity (ROH) among all individuals of the family. Upon delineating the region of interest (chr19:41209497:C > T) on chromosome 19, we found the two cases (MC4 and MC7) exhibit the ROH > 13 Mb at position chr19:40373891-54106547, while the rest of the individuals do not present ROH regions covering the candidate mutation. The ROH data provides evident detail for disease-causing mutation harboring in the homozygous region of the two children of a consanguineous union, inherited through an autosomal recessive manner.

Family pedigree and Variant filtering strategy. (A) Family pedigree showing the inheritance pattern of nephrotic syndrome. Cases are shown in red color, and blue circles represent the subjects undergone WES (B) Variant filtering strategy. A narrowing down approach to identify the most promising causative mutation.

Candidate COQ8B variant co-segregation, variant validation using WES data and COQ8B protein conservation. (A) Pedigree showing the co-segregation of COQ8B variant (NM_024876.4:c.748G > A; NP_079152.3:p.D250N) in the family. Cases are presented in red color. (B) Whole-exome sequencing data (using IGV 2.5.3 software) reflects exactly the phenotypic data with homozygous cases (V-1 and V-4), heterozygous normal parents (IV-1 and IV-2) and three normal with homozygous reference allele (V-2, V-5 and V-6). (C) Comparing COQ8B protein for 14 vertebrates showed a highly conserved region at the mutation site (Asp250Asn).

Runs of homozygosity (ROH). (A) Showing the distribution of homozygous regions (blue blocks) in the autosomes. (B) Exposition of ROH on chromosome 19. The histograms highlighted in red color represent the estimated ROH.
The in silico evaluation of the candidate variant (c.748G > A/p.D250N) using VEP tools (CADD, FATHMM, MetaLR, MetaSVM, MutationTaster, PolyPhen-2 and SIFT) confirmed its deleterious impact and predicted to be disease-causing in all of its coding transcripts (ENST00000324464.3, ENST00000243583.6, ENST00000450541.1, ENST00000595254.1). Moreover, the observed rare variant (NM_024876.4:c.748G > A, NP_079152.3:p.D250N, rs769834604) with the homozygous condition has no previous records in the genomAD, ExAC, 1000Genome and ClinVar databases. Our study provides the first report of COQ8B:c.748G > A (p.D250N) pathogenicity in the ClinVar database (accession: SCV001451361.1).
In addition to the pathogenic COQ8B variant, we surprisingly found some novel variants in GLA (NM_000169.2:c.473C > T; p.T158I) and NUP107 (NM_020401.2:c.1781G > A; p.C594Y) genes and have been submitted to ClinVar database with accessions SCV001451725.1 and SCV001451724.1 respectively. Our study has also revealed some known missense mutations in the COQ2, COQ6, COQ7 and COQ9 genes involved in the CoQ10 biosynthesis pathway.
Discussion
Our study exposed the adolescence onset COQ8B nephropathy with clinical features characterized by absence of hematuria, moderate to severe proteinuria, edema and chronic renal failure making it unique to the congenital glomerulopathies. The studied patients remained asymptomatic at an early age and peculiar symptoms of the advanced CKD appeared at the time of diagnosis. The genetic study of the present consanguineous family revealed the heterozygous mutation in COQ8B among parents leading to the homozygous offspring in an autosomal recessive inheritance. The homozygous mutation (p.D250N) in cases co-segregated well with the nephrotic phenotype in the family. Several other studies have also reported different genetic substitutions at locus chr19:41209497 of the COQ8B gene causing nephrotic syndrome (Table 1).
Most of these studies have observed c.748G > C (p.D250H) and a few c.748G > T (p.D250Y)variations linked with SRNS and proteinuria with clinical FSGS/ or glomerular abnormalities. Our study provides the first insight into the homozygous condition of c.748G > A (p.D250N) with similar pathophysiological observations.
The COQ8B (OMIM * 615567), previously termed as ADCK4 (encoding the aarF domain‐containing protein kinase 4), belongs to the protein kinase superfamily containing a conserved domain with the catalytic activity of protein kinases. The co-enzyme Q8B encoded by the COQ8B gene (chromosomal position: 19q13.2a) is a mitochondrial matrix protein tangentially associated with the inner membrane. The protein contains 544aa in humans and is characterized by a single-pass transmembrane with a molecular weight of 60,069 Da. The COQ8B encoded protein plays a regulatory function in the biosynthesis of CoQ10. There have been 29 mutations reported in Human Gene Mutation Database (HGMD), including missense, nonsense and indels, mostly showing the autosomal recessive inheritance pattern for nephrotic disorders. Reports of several mutations in the COQ8B gene have been involved in damaging the kidney function4,7,17,18,19. The expression of COQ8B in the mitochondria of podocytes, renal tubules and collecting ducts is clear with a well-established corresponding link toward renal homeostasis hallmarks. A recent study provides the genetic and pathophysiologic evidence for the COQ8B in podocytes function. The in vitro experiments revealed a significant reduction of CoQ10 concentration in the COQ8B-knockout podocytes, leading to decreased respiratory chain activity, mitochondrial potential and consequently showing increased dysmorphic mitochondria20.
Our study revealed the novel and rare pathogenic mutation (NM_024876.4:c.748G > A (p.Asp250Asn) in the highly conserved region of exon 9 of the COQ8B gene (Fig. 3C and Supplementary Fig. S2), which is one of the 16 different regulatory genes (PDSS1, PDSS2, COQ2, COQ3, COQ4, COQ5, COQ6, COQ7, COQ8A, COQ8B, COQ9, COQ10A, COQ10B, FDX1L, FDXR, and ALDH3A1) ubiquitously required for the CoQ10 biosynthesis pathway in mammals4,8,9,21. The pathogenic mutations, including missense/non-sense, indels and frameshifts, could lead to loss of COQ8B kinase function, ultimately affecting the COQ10 biosynthesis pathway22.
Figure 5A,B provided the comprehensive report of the COQ8B gene with its expressed transcript (containing functional domains) and predicted the 3-dimensional structure of the protein with the precise exposition of the candidate variant impact. The protein–protein interaction with biological processes and domain functions (using STRING v11.0)23 of COQ8B has been displayed in Fig. 5C. A recent study unraveled the COQ8B interactions with mitochondrial proteins such as COQ5, as well as cytoplasmic proteins such as myosin and heat shock proteins. They observed a decreased COQ5 level in the COQ8B-knockout podocytes, but overexpression of COQ8B transfected with wild-type COQ8B rescued the COQ5 level20. We assume the candidate pathogenic mutation (COQ8B:p.Asp250Asn) identified in the present study disrupts the protein–protein interaction via COQ5 in the CoQ megacomplex, leading to progressive renal damage and consequently ESRD. In addition to the candidate variant, our study also revealed multiple known mutations in the STRING model (COQ2:p.Val66Leu, COQ6:p.Val406Met, COQ7:p.Thr103Met and COQ9:p.Ile121Thr) involved in the CoQ10 biosynthesis pathway (Fig. 5C).

COQ8B gene and its transcript, three-dimensional structure of COQ8B and STRING network. (A) Showing the details of the COQ8B gene (15 exons) and the expressed protein (using neXtProt, online database)24. The pathogenic mutation (NC_000019.9:g.41209497C > T) in the 9th exon leading to the replacement of Aspartic acid (negatively charged) by Asparagine (non-charged) amino acid at position 250. (B) Structural model of COQ8B with the exposition of candidate variant position (D250), using SuSPect prediction algorithm25. Multiple in silico algorithm (shown on the right) data reflect the candidate mutation to be deleterious and disease-causing. (C) Network showing the functional association of COQ8B protein using STRING v11.0 (https://stringdb.org/). The proximity of the proteins via threads predicts the association and their role in the COQ10 biosynthesis pathway. In addition to the candidate pathogenic variant in the COQ8B gene, we have also identified the variants in COQ2, COQ6, COQ7 and COQ9 genes for the current STRING model.
Multiple in silico tools confirmed the deleterious and possibly damaging impact of the candidate variant [NM_024876.4(COQ8B):c.748G > A (p.Asp250Asn)]. The observed candidate variant harbor the most conserved region of the COQ8B gene across different vertebrates (Supplementary Fig. S2a). Using NCBI database, the expression profiling of the COQ8B gene of 16 human tissue samples provided substantial evidence for its active role in kidney function (Supplementary Fig. S2b). Multiple in silico programs confirmed the pathogenicity candidate variant. However, functional studies using in-vivo models will be carried out in the future.
In podocytes, the expressed COQ8B interacts with other members of the COQ10 biosynthesis pathway and the mutations in the gene have been linked with steroid-resistant nephrotic syndrome (SRNS)4. The biological functions and domains feature depicts the importance of COQ8B as one of the significant components of the COQ10 biosynthesis pathway. The kinase activity of COQ8B allows it to interact selectively and non-covalently with ATP, involved in various metabolic processes22. The COQ10 (coenzyme Q10) a lipid-soluble electron carrier, specifically involved in the electron transport chain that exerts antioxidant, anti-inflammatory and present favorable cardiac, hypertension, lipid and glucose metabolic functions26. In the progression of CKD, the oxidative stress, inflammation and endothelial dysfunction biomarkers are easily detectable27. COQ10 supplementation has also been used to restrain oxidative stress in CKD patients28. In the present study, the delayed genetic diagnosis hindered the possible use of CoQ10 supplementation to the patients.
The genotypic data of COQ8B among offspring present all the homozygous genotypes. Interestingly, there were no heterozygous children, although the chance of this was supposed to be twice of a homozygous state in each pregnancy. The lack of heterozygous genotypes in the offspring and the presence of only homozygous genotypes (WT/WT or MUT/MUT) among the offspring seems most likely due to chance in this family.
Conclusion
Our study provides the first report of COQ8B homozygous variant NM_024876.4:c.748G > A (p.Asp250Asn) in a family affected with idiopathic end-stage kidney disease. The variant p.Asp250Asn was co-segregated with the disease phenotype and the in silico evaluations confirmed its pathogenicity. Based on the clinical and genetic findings, we assume SRNS (NPHS9) would have led these patients to the most advanced CKD (stage-V). Due to consanguineous marriage between parents, the autosomal recessive effect of COQ8B appeared in their generation. Our study provides a piece of safeguard advice for clinicians to make people familiar with the genetic cause and its increased risk in the context of consanguineous marriages. The familial and premarital screening will help in controlling the COQ8B-related nephrotic syndrome in future generations. Identification of such mutations supports better management of such disorders through genetic counseling and appropriate diagnosis could open new gateways for precision medicine. Early detection of COQ8B pathogenic mutations in such cases could better help disease management by adopting personalized therapy using CoQ10 supplements.
Materials and methods
Ethics statement
The study was approved by Institutional Ethics Committee (IEC), Dayanand Medical College and Hospital (DMCH), Ludhiana, India. The informed written consent was also obtained from the subjects involved in this study. On behalf of children under the age of 18 years, we obtained the informed written consent from the parents. The study was performed following the guidelines and regulations of the Indian Council of Medical Research (ICMR), New Delhi.
Study design
Earlier studies have evidenced a high prevalence of consanguinity among Muslim populations of Jammu and Kashmir, North India29,30,31,32,33. A large consanguineous Muslim family was identified from the Poonch District of Jammu and Kashmir. The family comprised of 08 members and a detailed pedigree up to three generations back provided the evidence for no prior history of kidney disease (Fig. 2A).
Out of 06 children, 03 females were diagnosed with advanced CKD/ ESRD (stage-V) at 16–23 years. Two females died within a year on dialysis treatment. One eldest female survived after a kidney transplant from her father. The medical records of all family members were examined. We collected blood samples from 7 members (02 cases and 05 controls) only, while one female (MC6) died before we could plan this study. The detailed history and disease manifestation based on medical records has been presented in Supplementary Fig. S3.
Sample collection, DNA isolation and quantification
The blood samples were collected and DNA was extracted using QIAamp DNA Mini Kit 50 (Cat# 51304, Qiagen). The DNA samples were was subjected to QIAXPERT for quantifying the amount of DNA and the purity was checked by measuring the 260/280 nm ratio. DNA samples were also subjected to agarose gel electrophoresis and, after passing through DNA quality check (Supplementary Table S6 and Supplementary Fig. S4), were proceeded for Library protocol.
Whole-exome sequencing and bioinformatic analysis
The whole-exome sequencing library was prepared using Agilent-Sure Select XT Reagent Kit, Illumina (ILM) platforms. The sequencing was carried out in Illumina HiSeq X10 to generate 2X-150 bp sequence reads at an average 100 × sequencing depth to cover the maximum genomic variations. The quality check of WES-generated reads has been presented in Supplementary Tables S7, S8 and Supplementary Fig. S5. The alignment statistics and coverage analysis and average depth have been presented in Supplementary Tables S9, S10 and Supplementary Fig. S6, respectively. The bioinformatics analyses were achieved for variant calling and filtering with the major databases as GnomAD, ExAC, 1000G and ClinVar. The bioinformatic pipelines, alignment variant calling (Supplementary Fig. S7) and variant annotation (Supplementary Fig. S8) were processed and analyzed using the BROAD Institute’s GATK-Toolkit34. The variant calling was performed using the complete human reference genome (GRCh37/hg19). ClinVar database was used to check the previously reported mutations and associated phenotypes. Exclusion of intronic, synonymous, inframe insertions/deletions (InDels) and mutations in untranslated regions whereas the missense, nonsense variations and frameshift InDels located in exons or splice sites were prioritized. The remaining variants were then verified in dbSNP and NCBI databases. The WES methodology and detailed report have been presented in Supplementary material S1. The runs of homozygosity (ROH) was estimated using AutoMap35 and HomozygosityMapper36.
In silico evaluation for the pathogenicity of candidate variant
The altered amino acid was checked for its evolutionary conservation across different species, including the primates and mammals, using the genome browser of the University of California at Santa Cruz (UCSC)37. Multiple in silico prediction algorithms, including CADD, FATHMM, MetaLR, MetaSVM, MutationTaster, PolyPhen-2 and SIFT were used to predict the possible impact of the detected variants.
Data availability
Web resources: ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/. GenIO, http://bioinformatics.ibioba-mpsp-conicet.gov.ar/GenIO/index.php/. Mutation taster, http://www.mutationtaster.org/. NCBI dbSNP, https://www.ncbi.nlm.nih.gov/snp/. neXtProt, www.nextprot.org. OMIM, https://www.omim.org/. PolyPhen-2 http://genetics.bwh.harvard.edu/pph2/. SIFT, http://sift.jcvi.org/. STRING, https://stringdb.org/. SuSPect, http://www.sbg.bio.ic.ac.uk/~suspect/. The Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php/. UniProtKB, https://www.uniprot.org/. Genome Aggregation Database (gnomAD) v2.1.1, https://gnomad.broadinstitute.org/. University of California Santa Cruz Genomics Institute Genome Browser, https://genome.ucsc.edu/.
References
Bello, A. K. et al. Assessment of global kidney health care status. JAMA J. Am. Med. Assoc. 317, 1864–1881 (2017).
Inker, L. A. et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am. J. Kidney Dis. 63, 713–735 (2014).
Vivante, A. & Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 12, 133–146 (2016).
Vazquez Fonseca, L. et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum. Mutat. 39, 406–414 (2018).
Feng, C. et al. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation. Medicine (United States) 96, e8880 (2017).
Park, E. et al. Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations. Pediatr. Nephrol. 32, 1547–1554 (2017).
Ashraf, S. et al. ADCK4 mutations promote steroid-Resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 123, 5179–5189 (2013).
Doimo, M. et al. Genetics of coenzyme Q10 deficiency. Mol. Syndromol. 5, 156–162 (2014).
Desbats, M. A., Lunardi, G., Doimo, M., Trevisson, E. & Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J. Inherit. Metab. Dis. 38, 145–156 (2014).
Heeringa, S. F. et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 121, 2013–2024 (2011).
Koile, D., Cordoba, M., de Sousa Serro, M., Kauffman, M. A. & Yankilevich, P. GenIO: A phenotype–genotype analysis web server for clinical genomics of rare diseases. BMC Bioinform. 19, 25 (2018).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Robinson, J. T., Thorvaldsdóttir, H., Wenger, A. M., Zehir, A. & Mesirov, J. P. Variant review with the integrative genomics viewer. Can. Res. 77, e31–e34 (2017).
Wang, F. et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. (Berlin, Germany) 32, 1181–1192 (2017).
Yang, Z. et al. Mutation analysis of a family affected with isolated proteinuria. Zhonghua yi xue yi chuan xue za zhi Zhonghua yixue yichuanxue zazhi Chin. J. Med. Genet. 36, 598–601 (2019).
Lolin, K. et al. Early-onset of ADCK4 glomerulopathy with renal failure: A case report. BMC Med. Genet. 18, 28 (2017).
Yang, J., Yang, Y. & Hu, Z. A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy. Biochem. Biophys. Res. Commun. 506, 444–449 (2018).
Zhai, S. B., Zhang, L., Sun, B. C., Zhang, Y. & Ma, Q. S. Early-onset COQ8B (ADCK4) glomerulopathy in a child with isolated proteinuria: A case report and literature review. BMC Nephrol. 21, 406 (2020).
Song, X. et al. COQ8B nephropathy: Early detection and optimal treatment. Mol. Genet. Genom. Med. 8, e1360 (2020).
Widmeier, E. et al. ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. 31, 1191–1211 (2020).
Payet, L. A. et al. Mechanistic details of early steps in coenzyme Q biosynthesis pathway in yeast. Cell Chem. Biol. 23, 1241–1250 (2016).
Reidenbach, A. G. et al. Conserved lipid and small-molecule modulation of COQ8 reveals regulation of the ancient kinase-like UbiB family. Cell Chem. Biol. 25, 154-165.e11 (2018).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Zahn-Zabal, M. et al. The neXtProt knowledgebase in 2020: Data, tools and usability improvements. Nucleic Acids Res. 48, D328–D334 (2020).
Yates, C. M., Filippis, I., Kelley, L. A. & Sternberg, M. J. E. SuSPect: Enhanced prediction of single amino acid variant (SAV) phenotype using network features. J. Mol. Biol. 426, 2692–2701 (2014).
Xu, Y., Liu, J., Han, E., Wang, Y. & Gao, J. Efficacy of coenzyme Q10 in patients with chronic kidney disease: Protocol for a systematic review. BMJ Open 9, e029053 (2019).
Matsuyama, Y., Terawaki, H., Terada, T. & Era, S. Albumin thiol oxidation and serum protein carbonyl formation are progressively enhanced with advancing stages of chronic kidney disease. Clin. Exp. Nephrol. 13, 308–315 (2009).
Gutierrez-Mariscal, F. M. et al. Coenzyme q10 supplementation for the reduction of oxidative stress: Clinical implications in the treatment of chronic diseases. Int. J. Mol. Sci. 21, 1–19 (2020).
Fareed, M. & Afzal, M. Evidence of inbreeding depression on height, weight, and body mass index: A population-based child cohort study. Am. J. Hum. Biol. 26, 784–795 (2014).
Fareed, M. & Afzal, M. Estimating the inbreeding depression on cognitive behavior: A population based study of child cohort. PLoS ONE 9, e109585 (2014).
Fareed, M. & Afzal, M. Increased cardiovascular risks associated with familial inbreeding: A population-based study of adolescent cohort. Ann. Epidemiol. 26, 283–292 (2016).
Fareed, M., Kaisar Ahmad, M., Azeem Anwar, M. & Afzal, M. Impact of consanguineous marriages and degrees of inbreeding on fertility, child mortality, secondary sex ratio, selection intensity, and genetic load: A cross-sectional study from Northern India. Pediatr. Res. 81, 18–26 (2017).
Fareed, M. et al. Whole-exome sequencing reveals a rare variant of OTOF gene causing congenital non-syndromic hearing loss among large Muslim families favoring consanguinity. Front. Genet. 12, 641925 (2021).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Quinodoz, M. et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 12, 518 (2021).
Seelow, D., Schuelke, M., Hildebrandt, F. & Nürnberg, P. HomozygosityMapper—An interactive approach to homozygosity mapping. Nucleic Acids Res. 37, W593–W599 (2009).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Acknowledgements
We thank the Department of Science & Technology (DST), New Delhi, for financial assistance (File No. DST/INSPIRE/04/2017/001350) to Dr. Mohd Fareed. Special thanks to the participants of the study for their utmost support and cooperation. Thanks to Mr. Shaghaf M. Ansari, Scientist at the Information Technology Division of CSIR-IIIM, for downloading the WES data. Thanks to the IPR committee (CSIR-IIIM/IPR/00295) for reviewing and approving the manuscript.
Author information
Authors and Affiliations
Contributions
M.F. designed the study. V.M. and R.A. contributed to the clinical presentation of the patients. M.F., V.M. and M.A. analyzed and interpreted the data. M.F., V.M., R.A., M.A. and G.S. involved in drafting the manuscript. All authors critically reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fareed, M., Makkar, V., Angral, R. et al. Whole-exome sequencing reveals a novel homozygous mutation in the COQ8B gene associated with nephrotic syndrome. Sci Rep 11, 13337 (2021). https://doi.org/10.1038/s41598-021-92023-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92023-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
